

Abstract
T-cell development and activation are usually accompanied by expansion and production of numerous proteins that require active translation. The eukaryotic translation initiation factor 4E (eIF4E) binds to the 5' cap structure of mRNA and is critical for cap-dependent translational initiation. It has been hypothesized that MAPK-interacting kinase 1 and 2 (Mnk1/2) promote cap-dependent translation by phosphorylating eIF4E at serine 209 (S209). Pharmacological studies utilizing inhibitors have suggested that Mnk1/2 play important roles in T-cells. However, genetic evidence supporting such conclusions is lacking. Moreover, the signaling pathways that regulate Mnk1/2 in T-cells remain unclear. We demonstrated here that T-cell receptor (TCR) engagement activates Mnk1/2 in primary T-cells. Such activation is dependent on Ras-Erk1/2 signaling and is inhibited by diacylglycerol kinases α and ζ. Mnk1/2 double deficiency in mice abolishes TCR-induced eIF4E S209 phosphorylation, indicating their absolute requirement for eIF4E S209 phosphorylation. However, Mnk1/2 double deficiency does not affect the development of conventional αβ T-cells, regulatory T-cells, or NKT-cells. Furthermore, T-cell activation, in vivo primary and memory CD8 T-cell responses to microbial infection, and NKT-cell cytokine production were not obviously altered by Mnk1/2 deficiency. Although Mnk1/2 deficiency causes decreased IL-17 and IFNγ production by CD4 T-cells following immunization of mice with myelin oligodendrocyte glycoprotein peptide in complete Freud's adjuvant, correlating with milder experimental autoimmune encephalitis scores, it does not affect T helper cell differentiation in vitro. Together, these data suggest that Mnk1/2 play a minimal role in T-cell development and activation but may regulate non-T-cell lineages to control Th1/Th17 differentiation in vivo.
Introduction
T-cells play a critical role in adaptive immune responses. Activation of T-cells is critical for mounting immune responses against foreign antigens and to protect the host from infection (1). However, tight regulation of this process is important for the maintenance of self-tolerance (2). The signal from the T-cell receptor (TCR), via multiple intracellular signaling pathways such as the RasGRP1-Ras-Erk1/2-AP1, PKCθ-NFκB, PI3K-Akt, and Ca++-calcineurin-NFAT pathways, plays critical roles for T-cell maturation and activation (3–5). Orchestrated actions of these signaling cascades ensure proper T-cell maturation and T-cell activation.
In addition to TCR engagement, various other extracellular stimuli such as growth factors, cytokines, and stress also can induce activation of MAPKs. Based on the signals that trigger their activation, MAPKs are categorized as Extracellular signal-Regulated Kinases (Erk1/2), p38 kinases, and C- Jun N-terminal kinase/stress-activated protein kinases (JNK)(6). MAPKs control a wide range of functions including proliferation, differentiation, survival and apoptosis through direct phosphorylation and activation of substrates (7). These substrates, called MAPK-activated protein kinases (MAPKAPKs), are divided into four families based on the type of phosphorylating MAPK (8, 9). For example, the p90 ribosomal S6 kinase (Rsk) family includes Rsk1, Rsk2, and Rsk3 that are specifically phosphorylated and activated by ERKs (10); MAPK-activated protein kinases (MKs) such as MK2/3 and MK5 are activated by JNKs (11); and mitogen- and stress-activated kinases (MSKs) including MSK1 and MSK2 are phosphorylated by p38 MAPKs (12). Different from these MAPKAPKs, MAPK-interacting kinases 1 and 2 (Mnk1/2) are serine/threonine kinases and are phosphorylated by both ERKs and p38 kinases (13). Murine Mnk1 and Mnk2 are phosphorylated at threonine 197 and 202 (T197 and T202) or T244 and T249 respectively, which leads to their activation (14–18). Activated Mnk1/2 directly phosphorylate the eukaryotic translation initiation factor 4E (eIF4E) at S209 downstream of growth factor receptors (18). eIF4E binds to 5' methyl guanosine (m7GpppN) cap structure found in all eukaryotic mRNAs and this binding is obligatory for initiation of cap-dependent translation (19, 20). Cap-dependent translational is the primary mode of eukaryotic translation by which 95% of total cellular mRNAs are translated (21). It has been hypothesized that Mnk1/2 are key protein kinases that can promote cap-dependent translation through eIF4E phosphorylation (22).
The roles of Mnk1/2 were originally studied in Drosophila, whose Mnk ortholog is called LK6. Deficiency of this gene was found to impair growth and development, leading to a shortened life span (23). However, in mice, Mnk1/2 double deficiency did not grossly affect development and growth, although eIF4E phosphorylation at S209 was abolished (18). While dispensable for murine development, Mnk1/2 have been demonstrated to play an oncogenic role in mice, and their deficiency delays tumor development in a murine tumor model (24). Studies using pharmacological inhibitors and eIF4E phosphorylation mutants have shown that eIF4E phosphorylation plays an important role in cell survival and cancer progression (25).
Using chemical inhibitors, several previous studies have reported that Mnk1/2 may play an important role in immune cells. For example, chemical inhibition of Mnk1/2 was found to decrease translation of IL-17 in CD4+ T-cells (26), IFNγ and IL-4 in iNKT-cells (27), and inflammatory cytokines in macrophages (28). Though these studies provide preliminary evidence that Mnk1/2 activity may play a critical role in immune cell function, possible off-target effects of the chemical inhibitors used cannot be overlooked. In this report, we demonstrate that TCR engagement induces activation of Mnk1/2 and phosphorylation of eIF4E, which is enhanced by Ras signaling, and inhibited by diacylglycerol (DAG) kinases α and ζ that terminate DAG-mediated signaling (29). By using mice deficient in both Mnk1 and Mnk2 (Mnk1/2DKO), we show that Mnk1/2 are essential for TCR induced phosphorylation of eIF4E. However, deficiency of both Mnk1/2 does not affect gross T-cell development, activation, proliferation, or cytokine production. Furthermore, Mnk1/2 activities are dispensable during CD8 T-cell-mediated immune responses against Listeria monocytogenes and Lymphocytic choriomeningitis virus (LCMV) and for iNKT-cell development and cytokine production.
Materials and methods
Mice
C57BL6/J mice and TCR-OT1 transgenic mice were purchased from the Jackson Laboratory. Mice expressing a conditional constitutively active form of Ras in a T-cell specific manner (caKRas-CD4Cre), and Mnk1/2DKO mice on a C57BL6/J background were previously described (18, 30–32). DGKα and ζ double knockout mice were previous reported (33, 34). All mice were used according to a protocol approved by the Duke University Institute Animal Care and Use Committee.
Flowcytometry
Thymocytes, splenocytes, and lymph node (LN) cells were prepared following standard procedures. Cells were stained with flurochrome-conjugated antibodies for CD4, CD8, CD62L, CD44, CD25, TCRβ, CD24, NK1.1, and CD69 (Biolegend) as well as CD1d-Tetramer (kindly provided by NIH Tetramer Facility) in 2% FBS-PBS at 4°C for 30 minutes. Additionally, Live/Dead fixable crystal violet dead cell stain (Invitrogen) was used to identify the viable cells. The stained cells were collected using a BD FACS Canto II flow cytometer. The collected data was analyzed using Flowjo software. Isolation of liver mononuclear cells and staining of iNKT-cells were performed as previously described (32, 35)
Activation, anergy, and proliferation assays
Splenocytes from wild type (WT) or Mnk1/2DKO were left unstimulated or stimulated with α-CD3 (1μg/mL; 2C-11) overnight in the presence or absence of either anti-CD28 (0.5 μg/ml) or CTLA4-Ig (10 μg/ml, BioXcell) to assess the upregulation of early activation markers by FACS. For proliferation assays, splenocytes were labeled with CFSE as previously described (36), left unstimulated or stimulated with α-CD3 for 72h. After staining for CD4 and CD8, cells were subjected to FACS analysis. In some experiments, CGP57380 (TOCRIS Bioscience, a Mnk1/2 inhibitor) was added in the culture at the indicated concentrations. To examine T cell anergy, WT and Mnk1/2DKO splenocytes were stimulated with anti-CD3 in the presence or either anti-CD28 (0.5 μg/ml) or CTLA4-Ig (10 μg/ml) for at 37°C 48 hours. Cells were then washed three times and rested in IMDM-10 at 37°C for 24 hours. Live cells enriched after Lympholyte (CEDARLANE) gradient separation were restimulated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (0.5 μg/ml) in the presence of 5 μM monensin at 37°C for 24 hours. Cells were surface-stained for CD4 and CD8 and intracellularly stained for IFNγ for FACS analysis.
In vitro stimulation of iNKT
Thymocytes were cultured in vitro in 10% FBS-complete IMDM with or without α-GalCer (125ng/ml) stimulation at 37°C for 72h. During the last 5 hours of stimulation, PMA (phorbol 12-myristate 13-acetate, 50 ng/ml), ionomycin (500 ng/ml, Sigma) and Golgi plug were added. After surface staining with anti-TCR antibody and the PBS-57-loaded mouse CD1d tetramer (CD1d-Tet), cells were intracellularly stained for IFNγ and IL-17 followed by FACS analysis. iNKT-cell proliferation was similarly assessed except that thymocytes were labeled with CFSE and PMA and ionomycin stimulation was not added.
Immunoblot
Immunoblots were prepared as previously described (31). Thymocytes or splenocytes were washed with phosphate-buffered saline (PBS). Cells were suspended in PBS with calcium and left unstimulated or stimulated with with 5μg/mL of anti-CD3ε (500A2; BD Pharmingen) for different times. Following stimulation cells were centrifuged and lysed in 1% Nonidet P-40 buffer (1% Nonidet-40, 150mM NaCl, and 50mM Tris, pH 7.4) supplemented with protease and phosphatase inhibitor cocktail (Sigma). Total proteins were separated by SDS-PAGE and were transferred to a Trans-Blot Nitrocellulose membrane (Bio-Rad). To examine protein phosphorylation, the membranes were incubated overnight with antibodies specific for phospho Erk1/2, phospho p38, phospho-4E-BP1 (T37/46), phospho-eIF4E (S209), phospho-Mnk1 (T197/202), total Mnk1 (Cell Signaling technology), total Mnk2 (18), and β-actin (Santa Cruz Biotechnology). Later, the membranes were incubated with the appropriate secondary, peroxidase-conjugated antibodies. The blots were developed using the ECL System from Perkin-Elmer. The same blots were stripped and re-probed using control antibodies.
T helper (Th) differentiation assay
Naïve CD4+ T-cells were purified from LN cells and cultured with plate-bound anti-CD3 (2C11) 5μg/ml, soluble anti-CD28 (1μg/ml) and indicated skewing conditions. Skewing conditions were as follows: TH1, IL-12 (5 ng/ml), IFN-γ (100 ng/ml) and anti-IL-4 (100 μg/ml), with IL-2 (100U/ml) during the rest period; TH2, IL-4 (1 ng/ml), anti-IL-12 (100 μg/ml) and anti-IFN-γ (100 μg/ml), with IL-2 (100U/ml) during the rest period; TH17, TGF-β (10 ng/ml), IL-6 (10 ng/ml), anti-IFN-γ (100 μg/ml) and anti-IL-4 (100 μg/ml);. TH9, IL-4 (20 ng/ml) anti-IL-12 (10 μg/ml) and TGF-β (2 ng/ml) during rest period. TH0, IL-2 (100U/ml) during the rest period. After culturing for 5 days, cells were stimulated with PMA and ionomycin in the presence of Golgi plus at 37°C for 5 hours. Following surface staining, cells were intracellularly stained for indicated cytokines, followed by FACS analysis.
Adoptive transfer and Lm-OVA infection to assess CD8+ T-cell response in vivo
Naïve OT1 T-cells (Vα2+CD8+ 7AAD−CD44−) were sorted from LN cells from Thy1.1+ WT-OT1 and Thy1.2+ Mnk1/2DKO OT1 mice. Five thousand sorted WT OT1 cells were mixed with an equal number of sorted Mnk1/2DKO OT1 cells in 200μl of serum-free IMDM and adoptively transferred by retro-orbital injection into sex-matched WT Thy1.1+Thy1.2+ recipients. After 24 hours, recipient mice were intravenously injected with 1×104 colony-forming units of Listeria monocytogenes expressing recombinant ovalbumin (Lm-OVA) (37). Peripheral blood samples (collected in PBS with 5mM EDTA) and splenocytes were analyzed at 1 and 2 weeks post infection. After lysis of red blood cells, samples were labeled with fluorochrome-conjugated antibodies and analyzed by flow cytometry. Frequencies of Thy1.1+ WT-OT1 and Thy1.2+ Mnk DKO populations were corrected to account for the deviation of the input ratio from 50:50.
Lymphocytic choriomeningitis virus (LCMV) infection
LCMV Armstrong stocks were propagated on BHK-21 cells and quantitated as described previously (38). LCMV infection and assessment of viral specific CD8 T-cell responses were performed as previously described (39). Briefly, mice were infected with 2 × 105 pfu of virus i.p, and monitored by serial bleeding and tetramer staining. For memory experiments, viable CD8+ CD44+ memory cells were sorted from donor mice 8 weeks after LCMV infection. While donor mice were Thy1.1−Thy1.2+, recipients were Thy1.1+Thy1.2+. Frequency of H-2Db tetramer loaded with LCMV gp33–41 (TetG) positive cells in the sorted population was determined by flow cytometry, and an appropriate number of total memory cells was transferred such that each recipient mouse received 5000 TetG+ memory cells. Recipients were infected with 2 × 105 pfu of LCMV i.p the next day, and taken 7 days later to assess the memory response.
Induction and scoring of experimental autoimmune encephalitis (EAE)
EAE was induced in 6–10 week old female mice by subcutaneous injection of myelin oligodendrocyte glycoprotein (MOG35–55) peptide (100 μg/mouse) emulsified in complete Freund's adjuvant containing 2 mg/ml M. tuberculosis (100 μl/mouse). Mice were also injected i.p. with 200 ng of pertussis toxin on day 0 (day of immunization) and day 2. Mice were monitored for about 40 days to assess the development of clinical score based on the following criteria: 1=tail limpness, 2= impaired righting reflex, 3=hind limb paralysis, 4=complete paralysis, 5=death. Some mice were sacrificed on day 7 to assess CD4 T-cell differentiation in response to the immunization. Draining lymph node cells were stimulated with MOG35–55 peptide for 3 days to expand the pool of antigen-specific CD4 T-cells, followed by stimulation for 5 hours with PMA (50 ng/ml) and ionomycin (500 ng/ml) in the presence of GolgiPlug. After stimulation, cells were stained for cell surface CD4 and intracellularly stained for IL-17A and IFNγ.
Statistical analysis
Statistical significance was determined using the ANOVA or Student's t-test. p-Values are defined as follows: *p<0.05;**p<0.01; ***p<0.001.
Results
Regulation of Mnk1/2 activation and expression in T-cells
TCR signaling has been shown to trigger several intracellular signaling pathways leading to phosphorylation and activation of p38 and Erk1/2. We assessed anti-CD3 induced Mnk1 activation in freshly isolated thymocytes or splenic T-cells from WT mice. As shown in figure 1A, TCR engagement induced Mnk1 and eIF4E phosphorylation in both thymocytes and splenic T-cells, correlated with Erk1/2 and p38 activation. In T-cells, DAG binds to and activates RasGRP1, which in turn activates the Ras-Mek1/2-Erk1/2 pathway. In the presence of a constitutively active form of Ras (kRas), TCR-induced Erk1/2, Mnk1, and eIF4E phosphorylation were significantly increased, indicating that Ras signaling promotes Mnk1/2 activation in T-cells (Fig. 1B). We have previously demonstrated that DGKα and ζ inhibit the activation of the Ras-Erk1/2 signaling (33, 34, 40, 41). In DGKα and ζ double deficient thymocytes (α/ζDKO), both Erk1/2 and eIF4E phosphorylation were enhanced (Fig. 1C). Furthermore such phosphorylation was greatly inhibited by U0126 (U0), a MEK1/2 inhibitor. Together, these observations indicate that TCR engagement induces Mnk1/2 activation, and that such activation is mediated by the Ras-Erk1/2 pathway and is inhibited by DGK activity.
Figure 1. Regulation of Mnk1/2 activation in T-cells.

Thymocytes and/or splenocytes of indicated genotypes were rested in PBS at 37°C for 30 min and were then left unstimulated or stimulated with 5 μg/mL anti-CD3 (500A2) for the indicated times. Lysates were subjected to immunoblot analysis with the indicated antibodies. (A) TCR engagement activates Mnk1/2 in WT thymocytes (left) and splenocytes (right). (B) Constitutively active KRas promotes Mnk1/2 activation. Thymocytes from WT and ca.kRas-CD4Cre mice were subjected to similar analysis as in (A). (C) DGKα and ζ inhibit TCR-induced Mnk1/2 activation in a MEK1/2 dependent manner. WT and in DGKαζDKO (azDKO) thymocytes were examined as in (A) with the addition of groups treated with the MEK1/2 inhibitor U0126 (10μM). (D, E) Differential expression of Mnk1/2 in naïve and activated T-cells. Mnk1 and Mnk2 mRNA (D) and protein (E) levels in sorted WT naïve and in vitro activated CD4 and CD8 T-cells were examined by real-time quantitative PCR and western blotting analysis, respectively. *, p<0.05; **, p<0.01; ***, p<0.001.
Although Mnk1/2 are ubiquitously expressed, their expression is varied in different tissues (17). Using real-time qPCR, we assessed Mnk1/2 mRNA levels in naïve and activated CD4+ and CD8+ T-cells. Both Mnk1 and 2 mRNA levels were expressed at higher levels in naïve T-cells than in activated T-cells. Mnk1 was decreased to 45% and 30%, while Mnk2 expression was reduced to 50% and 20% in activated CD4 and CD8 T-cells respectively as compared to naïve T-cells (Fig. 1D). The decreased expression of Mnk1/2 proteins in activated T-cells was further confirmed by immunoblotting analysis (Fig. 1E). Naive and activated T-cells are drastically different in metabolism and in protein synthesis. Given the proposed role of Mnk1/2 in cytokine production, it is intriguing that Mnk1/2 expression is decreased in activated T-cells, in which proteins including cytokines are actively translated.
Effect of combined Mnk1/2 deficiency on T-cell development
To investigate the role of Mnk1/2 in T-cells, we analyzed mice with germline deletion of these two genes. Since individual deficiency of Mnk1 or Mnk2 did not affect T-cell development and activation (data not shown), we examined Mnk1/2DKO mice. The percentages and absolute numbers of CD4 and CD8 subsets in the thymus and spleen from Mnk1/2DKO mice were similar to those from WT control mice (Fig. 2A–2D). The overall thymic and splenic cellularities in Mnk1/2DKO mice were also comparable to WT mice (Fig. 2E). To further examine the role of Mnk1/2 in T cell development, we generated Mnk1/2DKO mice carrying the OT1 TCR transgene, which directs CD8 T-cell development. As shown in Figure 2F, thymocyte numbers were not obviously different between Mnk1/2DKO OT1 mice and WT OT1, supporting a minimal role of Mnk1/2 for intrathymic T-cell development. Mnk1/2DKO mice did not display obvious alteration in natural regulatory T-cell numbers as compared to WT controls (Fig. 2G). Furthermore, CD44 and CD62L staining showed similar naïve and effector T-cell populations in WT and Mnk1/2DKO mice (Fig. 2H, 2I). Together, these observations indicate that Mnk1/2 double deficiency does not cause obvious defects in T-cell development or homeostasis.
Figure 2. T-cell development in Mnk1/2DKO mice.

(A, B) CD4 and CD8 expression in WT and Mnk1/2DKO (DKO) thymocytes (A) and splenocytes (B). Representative dot-plots of CD4 and CD8 staining are shown. (C, D) Absolute numbers of thymic (C) and splenic (D) T-cell populations in WT and Mnk1/2DKO mice (n=6). (E) Total thymic and splenic cellularities in WT and Mnk1/2DKO mice. (F) Absolute numbers of thymic T-cell populations in WT-OT-I and Mnk1/2DKO-OT-I mice (n=3). (G) Treg staining in the thymus and spleen. CD25 and Foxp3staining in CD4+ T-cells are shown. (H) CD44 and CD62L staining of gated WT and Mnk1/2DKO CD4+ and CD8+ T-cells. (I) Mean ± SEM presentation of cell numbers of indicated T-cell populations (n=5). Data shown are representative of at least three experiments.
Mnk1/2 are required for TCR induced eIF4E phosphorylation
As mentioned above, TCR engagement induced eIF4E phosphorylation at S209. To determine if such phosphorylation is dependent on Mnk1/2, we compared TCR-induced eIF4E phosphorylation in WT and Mnk1/2DKO T-cells. Although eIF4E total protein was similar between WT and Mnk1/2DKO T-cells, TCR-induced eIF4E phosphorylation was virtually abolished in Mnk1/2DKO T-cells (Fig. 3A). On the contrary, TCR-induced phosphorylation of Erk1/2, Rsk1 (Erk1/2 substrate), and p38 were not affected by Mnk1/2 deficiency (Fig. 3B), suggesting that Mnk1/2 deficiency does not cause global signaling defects, and that there is no obvious negative feedback regulation of Erk1/2 and p38 by Mnk1/2 in T-cells. Binding of eIF4E to mRNA is inhibited by its association with 4E-BP1. mTOR phosphorylates 4E-BP1, leading to the release of eIF4E from 4E-BP1 to initiate translation (42). Neither 4E-BP1 protein levels nor its phosphorylation was altered in Mnk1/2 deficient T-cells as compared with WT T-cells (Fig. 3C), suggesting that Mnk1/2-mediated eIF4E phosphorylation does not affect 4E-BP1-mediated suppression of eIF4E or mTOR activity. Together, these results suggest that TCR-induced eIF4E phosphorylation is mediated by Mnk1/2.
Figure 3. Effect of Mnk1/2 deficiency on TCR-induced signaling.

WT and Mnk1/2DKO thymocytes and splenocytes were similarly stimulated and analyzed by immunoblot as in figure 1A. (A) Mnk1/2 are critical for TCR-induced eIF4E phosphorylation at S209. (B) Mnk1/2 deficiency does not alter Erk1/2 and p38 phosphorylation. (C). Mnk1/2 deficiency does not impair mTOR-mediated 4E-BP1 phosphorylation. Data shown are representative of three experiments.
Normal in vitro T-cell activation in the absence of Mnk1/2
To investigate if Mnk1/2 deficiency affects T-cell activation, we first examined the upregulation of early activation markers CD69 and CD25 following overnight anti-CD3 stimulation in the presence or absence or CD28-mediated costimulation. Mnk1/2DKO T-cells upregulated CD69 and CD25 similarly to WT controls in response to a wide range of anti-CD3 stimulation in the presence of anti-CD28 (Fig 4A). The presence of CTLA4-Ig to block CD28-mediated costimulation decreased CD25 and CD69 upregulation in WT T-cells. However, absence of Mnk1/2 did not cause further reduction of CD25 and CD69 expression in T-cells.
Figure 4. Mnk1/2 are not essential for in vitro T-cell activation.

(A) Upregulation of early activation markers in Mnk1/2DKO T-cells. WT and Mnk1/2DKO splenocytes were left un-stimulated or stimulated overnight with an anti-CD3 antibody (2C11) at the indicated concentrations in the presence or absence of an anti-CD28 antibody (37.51, 0.5 μg/ml) or CTLA4-Ig (10 μg/ml). Overlaid histograms show CD69 and CD25 expression on gated CD4+ and CD8+ cells. (B) Mnk1/2 deficiency does not affect T-cell proliferation. CFSE-labeled WT and Mnk1/2DKO splenocytes were un-stimulated or stimulated with an anti-CD3 antibody for 72h. Cultured cells were stained for CD4 and CD8 and analyzed by flow cytometry. Histograms show CFSE intensity on CD4+ and CD8+ cells. (C) Effect of Mnk1/2 deficiency on cytokine production by T-cells. Splenocytes from WT or Mnk1/2DKO mice were left unstimulated (top) or stimulated with an anti-CD3 antibody (bottom) for 48h, followed by PMA (50 ng/ml) and ionomycin (500 ng/ml) stimulation in the presence of Golgi plug for 5h. Cells were stained for surface CD4 and CD8 and intracellular cytokines followed by FACS analysis. IFNγ and TNFα expression in gated CD4+ and CD8+ T-cells are shown. (D) Mnk1/2 deficiency does not affect OT1 T-cell proliferation. Splenocytes from WT OT1 and Mnk1/2DKO OT1 mice were either labeled or not labeled with CFSE, and then treated with SIINFEKL peptide at indicated concentrations for 18 h or 72 h to assess early T-cell activation and proliferation respectively. Overlaid histograms show CD25 and CD69, and CFSE intensity on live-gated CD8+Vα2+ T-cells. (E,F) WT and Mnk1/2DKO splenocytes were stimulated with anti-CD3 in the presence or either anti-CD28 (0.5 μg/ml) or CTLA4-Ig (10 μg/ml) at 37°C for 48 hours. After resting for 24 hours, live T cells were restimulated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (0.5 μg/ml) in the presence of 5 μM monensin at 37°C for 24 hours. Cells were surface-stained for CD4 and CD8 and intracellularly stained for IFNγ. FACS plots show IFNγ expression in live gated CD4 and CD8 T cells (E). Bar graph is mean ± SEM presentation of percentages of IFNγ+ cells in the indicated populations of cells (n=3). Data shown are representative of three experiments.
Mnk1/2DKO T-cells also showed comparable proliferation to WT T-cells following α-CD3 stimulation for 72 hours as demonstrated by a CFSE dilution assay (Fig 4B). Moreover, Mnk1/2DKO CD4 and CD8 T-cells produced similar levels of IFNγ and TNFα after stimulation for 48 hours, as compared with WT controls, based on intracellular staining (Fig 4C). In order to study the effect of Mnk1/2 deficiency on antigen-specific T-cell activation, we utilized the OT1 T-cells, which express the Vα2+Vβ5+ TCR and recognizes the OVA257-264 (SIINFEKL) epitope of ovalbumin presented on H-2Kb. When stimulated with different concentrations of OVA257-264 peptide, Mnk1/2DKO OT1 T-cells upregulated CD25 and CD69, and proliferated similarly to WT OT1 T-cells (Fig 4D). Together, these data indicate that Mnk1/2 double deficiency does not obviously affect T-cell activation in vitro.
As mentioned earlier, mTORC1 phosphorylates 4E-BP1 to promote eIF4E-mediated translation initiation. Decreased mTORC1 activity causes T-cell anergy while enhanced mTORC1 activity leads to resistance to anergy (43–45). We asked further whether Mnk1/2-mediated phosphorylation of eIF4E plays a role in T-cell anergy. We stimulated WT and Mnk1/2DKO splenocytes with anti-CD3 in the presence of CTLA4-Ig to block CD28 mediated costimulation for 48 hours. After an additional 24 hours of resting, live T-cells were restimulated by plate bound anti-CD3 and soluble anti-CD28 overnight, followed by intracellular staining for IFNγ. As shown in Figure 4E and 4F, similar IFNγ levels were detected in WT and Mnk1/2DKO T-cells under anergic conditions, suggesting that Mnk1/2 deficiency does not obviously affect T-cell sensitivity to anergy induction in vitro.
Minimal effect of Mnk1 and Mnk2 deficiency on Th differentiation in vitro
Using a Mnk1/2 inhibitor, a recent study has implicated Mnk1/2 in IL-17 production by T-cells (26). We examined if deficiency of Mnk1/2 affected Th differentiation. Sorted naïve WT and Mnk1/2DKO CD4 T-cells were subjected to in vitro Th1, Th2, Th17 and Th9 differentiation. Intracellular staining was used to assess the production of IFNγ, IL-17 and IL-9 under different skewing conditions, while IL-4 levels were measured by ELISA. As shown in Figure 5, no obvious difference was observed between WT and Mnk1/2DKO T-cells in the production of these cytokines. These observations suggest that Mnk1/2 and Mnk1/2-mediated eIF4E phosphorylation are dispensable for Th differentiation in vitro.
Figure 5. Combined Mnk1/2 deficiency does not affect Th differentiation in vitro.
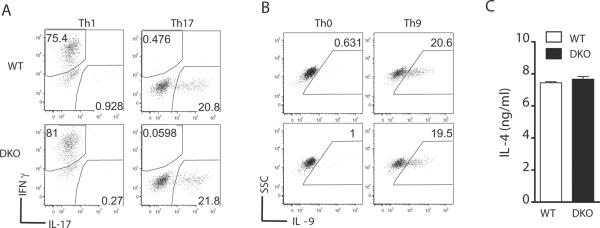
CD44−CD62L+ naïve CD4 T-cells sorted from WT and Mnk1/2DKO spleen and LNs were subjected to in vitro Th skewing conditions. PMA, ionomycin, and Golgi plug were added to the culture during the last 5 hours of differentiation. At the end of culture, cells were stained with anti-CD4, Live/Dead, and indicated cytokines. Dot plots show IFNγ and IL-17 staining on gated live CD4 T-cells under Th1 or Th17 skewing conditions (A) and IL-9 staining under Th0 and Th9 skewing conditions (B). IL-4 production by T-cells that were under a Th2 skewing condition was determined by ELISA of culture supernatants (C). All data shown are representative of at least three experiments.
Mnk1/2 deficiency may impair Th1 and Th17 differentiation in vivo in the EAE model
To determine if Mnk1/2 deficiency could affect CD4 cell differentiation in vivo, we used the EAE model since the differentiation of CD4 cells into Th1 and particularly into Th17 lineage is known to play an important role in the pathogenesis of the disease (46). Spleens and draining lymph nodes from WT and Mnk1/2DKO mice immunized with MOG35-55 peptide emulsified in CFA showed comparable total cellularity and frequency of CD4 cells one week after immunization (Fig 6A). When antigen-specific cells were expanded by ex vivo stimulation of lymph node cells with MOG35-55 for 3 days, stimulation with PMA and ionomycin revealed a marked reduction in the frequency of Mnk1/2DKO CD4 T-cells that were able to produce IL-17A or IFNγ (Fig 6B). Bearing in mind our previous results that Mnk1/2 deficient T-cells survive and proliferate similar to WT counterparts (Fig 4), the reduction in the IFNγ-producing and IL-17A-producing pools of antigen-specific cells suggests that the absence of Mnk1/2 may somehow impair the differentiation of CD4 T-cells into Th1 and Th17 cells in response to antigen stimulation in vivo. Correlating with the smaller pool of IFNγ-producing and IL-17A-producing cells, mice deficient in Mnk1/2 developed milder EAE disease than WT counterparts (Fig 6C). Taken together, these results suggest that Mnk1/2 deficiency may impair Th1 and Th17 differentiation in vivo to perturb disease development in the EAE model.
Figure 6. Mnk1/2 deficiency may impair Th1 and Th17 differentiation in vivo.

(A) Total cell numbers in the spleen and LNs and percentages of CD4 T-cells in these organs in WT and Mnk1/2DKO mice 7 days after immunization with MOG and CFA. (B) IL-17A and IFNγ producing cells within CD4 T-cells following MOG peptide stimulation for 3 days ex vivo. Dot plots show IFNγ and IL-17A expression in in gated CD4 T-cells. Bar graph represents mean ± SEM of IFNγ and IL-17A producing cells with in CD4 T-cells from multiple mice (n=6). (C) EAE score of immunized WT and Mnk1/2DKO mice monitored at indicated time points and scored as indicated in Materials and Methods (n=5). Mean ± SEM are calculated for the indicated number of mice per group. Data shown are representative of two or more independent experiments.
Combined Mnk1/2 deficiency does not affect CD8 T-cell response to Listeria monocytogenes infection in vivo
The data described above have revealed that Mnk1/2 are dispensable for T-cell activation in vitro. We used the Listeria monocytogenes infection model to determine if Mnk1/2 is required for T-cell responses in vivo. Equal numbers of sorted naïve WT (Thy1.1+) and Mnk1/2DKO (Thy1.2+) Vα2+CD8+ OT1 cells were mixed and co-injected intravenously into WT Thy1.1+Thy1.2+ recipient mice. Recipients were subsequently infected with Listeria expressing recombinant ovalbumin (Lm-Ova) (Fig 7A, 7B). Expansion of Ova-specific OT1 T-cells was monitored in the peripheral blood and spleen on days 7 and 14 after Lm-Ova infection. No significant difference was observed in the frequency of WT (Thy1.1+) and Mnk1/2DKO (Thy1.2+) OT1 cells in the recipient mice 7 and 14 days after infection (Fig 7C, 7D). These results suggest that Mnk1 and Mnk2 may not play a critical role in the expansion and early contraction phases of antigen-specific CD8 T-cell responses, at least in the Lm-Ova model.
Figure 7. Combined Mnk1/2 deficiency does not affect antigen-specific primary CD8 T-cell responses in vivo.

(A) Schematic representation of experimental design showing competitive adoptive transfer of WT Thy1.1+ and Mnk1/2DKO Thy1.2+ OT1 T-cells, and the Lm-Ova infection model. (B) Thy1.1 and Thy1.2 staining of mixture of sorted naïve WT and Mnk1/2DKO OT1 cells before injection. (C) Representative FACS analysis of peripheral blood and splenocytes detecting Vα2+ CD8 T-cells (top panels), and Thy1.2/Thy1.2 congenic markers within the gated Vα2+CD8+ T-cell population at indicated time points after infection. (D) Percentages of WT OT1 (Thy1.1+) and Mnk1/2DKO OT1 (Thy1.2+) cells among total Vα2+CD8+ cells in the peripheral blood and spleen. Mean ± SEM were calculated after correcting for the input ratio. Each dot represents one mouse. Data shown are representative of three independent experiments.
Mnk1/2 deficient mice mount normal primary and memory CD8 responses to LCMV infection
We next sought to better understand the effects of Mnk1/2 deficiency on polyclonal primary and memory CD8 T-cell responses to viral infection. To this end, we infected WT and Mnk1/2DKO mice with the Armstrong strain of LCMV that causes acute infection (Fig 8A). We then examined the frequency of CD8 cells in the peripheral blood that could recognize the LCMV GP33–41 peptide presented on H-2Db tetramers (TetG+ cells) at 1, 2 and 4 weeks after infection by flow cytometry. Our results showed that the frequencies of TetG+ cells were comparable in WT and Mnk1/2DKO mice at these time points (Fig 8B,8C). Similar results were obtained when we determined the frequency of CD8 T-cells that could recognize the LCMV NP 396–404 peptide presented on H-2Db tetramers (data not shown). Together, these results suggest that Mnk1/2 deficiency may not alter polyclonal primary CD8 responses, at least in the LCMV model.
Figure 8. Mnk1/2 deficiency does not affect primary or memory anti-viral CD8 responses in vivo.

(A) Schematic representation of experimental design showing primary infection with LCMV Armstrong, adoptive transfer of memory cells, and re-challenge. (B,C) Primary response. (B) Representative FACS plots of peripheral blood samples obtained at indicated time points and stained with anti-CD8 antibody and TetG. (C) Mean ± SEM presentation of percentages of CD8+TetG+ cells in the peripheral blood at the indicated time points (n=6 WT, n=7 Mnk1/2DKO). (D, E) Recall response. (D) Representative FACS plots of peripheral blood and spleen samples from recipient mice that received WT or Mnk1/2DKO memory cells. Top panels: CD8 and TetG staining of peripheral blood and splenocytes. Bottom panels: Thy1.1 expression in the gated CD8+TetG+ population. (E) Mean ± SEM presentation of percentages of CD8+TetG+Thy1.1− cells in peripheral blood and spleen samples from recipient mice (n=5). Data shown are representative of two independent experiments.
To understand if Mnk1/2 deficient memory CD8 T-cells generated after primary LCMV infection can respond robustly to pathogen re-challenge, we sorted out CD8+CD44+ memory cells from WT (Thy1.1−Thy1.2+) and Mnk1/2DKO (Thy1.1−Thy1.2+) mice 8 weeks after infection. These memory cells were adoptively transferred into WT Thy1.1+Thy1.2+ congenic recipients after normalizing the number of TetG+ cells. Recipient mice were challenged the next day with LCMV Armstrong, and the memory response was examined in the peripheral blood and spleen 7 days after infection. Results from flow cytometric analysis indicated a comparable expansion of adoptively transferred (Thy1.1−) WT and Mnk1/2DKO TetG+ memory cells in the peripheral blood and spleen (Fig 8D, 8E), suggesting that Mnk1/2-deficient memory CD8 cells can respond robustly to re-challenge. Taken together, these results support and extend those from the Lm-Ova model, suggesting that Mnk1/2 function may be dispensable during primary and memory CD8 responses to intracellular pathogens.
Effects of Mnk1/2 deficiency on iNKT-cell development and function
The invariant NKT (iNKT) cells are a rare subset of T-cells with the ability to bridge innate and adaptive immunity by rapidly producing and secreting copious amounts of cytokines The mechanisms regulating cytokine production in iNKT-cells are not well understood. We have recently demonstrated that proper iNKT-cell development requires tight regulation of DAG-mediated signaling. Deficiency of RasGRP1 or enhanced activation of DAG-mediated signaling due to DGKα and ζ deficiency can both lead to defects in iNKT-cell development (32, 35). Since Mnk1/2 are downstream effector molecules of the DAG-RasGRP1-Ras-Erk1/2 pathway, we investigated if Mnk1/2 play a role in regulating iNKT-cell development. Like conventional T-cells, individual or combined deficiency of Mnk1/2 did not affect the development of iNKT-cells in the thymus and spleen (Fig 9A). iNKT-cell percentages and absolute numbers were similar between WT and Mnk1/2DKO mice (Fig 9B). Further analysis of iNKT-cell developmental stages based on CD44 and NK1.1 expression did not reveal obvious differences between WT and Mnk1/2DKO mice (Fig 9A), suggesting normal development of iNKT-cells in the absence of both Mnk1 and Mnk2.
Figure 9. Effect of combined Mnk1/2 deficiency on iNKT development and activation.

(A) Normal iNKT-cell development in the absence of Mnk1/2. Thymocytes, splenocytes, and liver mononuclear cells from age and sex-matched Mnk1/2DKO mice and WT controls were stained with CD1d-Tet, anti-TCRβ, anti-CD24, anti-CD44, and anti-NK1.1 followed by FACS analysis. Top panels, CD1d-Tet and anti-TCRβ staining on live cells. Bottom panels, NK1.1 and CD44 expression on gated CD1d-Tet+CD24− iNKT-cells. (B) Total NKT-cell numbers in the indicated organs. (C) Mnk1/2 are dispensable for IFNγ and IL-17 production by iNKT-cells. WT and Mnk1/2DKO thymocytes were stimulated with α-GalCer in vitro for 72 hours with the addition of PMA, ionomycin and Golgi plug for the last 5 hours. Cultured cells were then stained with CD1d-Tet and anti-TCRβ, and intracellularly stained with anti-IFNγ and anti-IL-17. Dot-plots show IFNγ and IL-17 expression in live gated CD1dTet+TCRβ+ iNKT-cells (bottom panels). (D) Assessment of iNKT-cell proliferation. CFSE-labeled WT and Mnk1/2DKO thymocytes were left unstimulated or stimulated with α-GalCer for 72 hours. Overlaid histograms show CFSE intensity in gated WT and Mnk1/2DKO CD1dTet+TCRβ+ iNKT-cells. Data shown are representative of three experiments.
It has been reported that inhibition of Mnk1/2 by CGP57380 decreased cytokine production from iNKT-cells following α-GalCer stimulation (27). We stimulated WT and Mnk1/2DKO iNKT-cells with α-GalCer in vitro for 72 hours and intracellularly stained for IFNγ and IL-17 production. The percentages of IFNγ and IL-17 positive Mnk1/2DKO iNKT-cells were similar to those of WT iNKT-cells (Fig 9C). Using a CFSE dilution assay, we also examined iNKT-cell proliferation following α-GalCer stimulation for 72 hours. Mnk1/2DKO iNKT-cells appeared to proliferate slightly better than WT iNKT-cells (Fig 9D). Together, these observations indicate that Mnk1/2 are dispensable for iNKT-cell development, production of cytokines, and proliferation.
Effects of Mnk1/2 inhibitor CGP57380 on activation of Mnk1/2 double deficient T cells
The discrepancies between our data from Mnk1/2 double deficient mice and those generated by chemical inhibition of Mnk1/2 with CGP57380 raise concerns about the selectivity of CGP57380 for Mnk1/2 and about the conclusions drawn from studies based on this inhibitor. To determine whether CGP57380 contains activities beyond inhibiting Mnk1/2, we used examined the effects CGP57380 on Erk1/2, Mnk1/2, and eIF4E phosphorylation following TCR engagement. As shown in Figure 10A, CGP57380 inhibited not only eIF4E phosphorylation but also Mnk1/2 phosphorylation following TCR engagement. Moreover, it inhibited both WT and Mnk1/2DKO T-cell proliferation in similar magnitudes (Fig 10B), and reduced IFNγ but not IL-17 production in both WT and Mnk1/2DKO iNKT-cells (Fig 10C). These observations are consistent with the findings that CGP57380 is able to inhibit other protein kinases such as MAPK kinase-1 (MKK1), casein kinase 1 (CK1), and brain-specific kinase 2 (BRSK2) (47). Thus, the effects of CGP57380 on T-cells may not solely be attributed to Mnk1/2.
Figure 10. Effects of Mnk1/2 inhibitor CGP57380 on T cells.

(A) Effects of Mnk1/2 inhibitor CGP57380 on TCR signaling. WT thymocytes were rested in PBS at 37°C for 30 minutes, then either left unstimulated or stimulated with an anti-CD3ε antibody (500A2, 5 μg/ml) in the presence of CGP57380 of the indicated concentrations (nM) at 37°C for 5 minutes. Cell lysates were subjected to immunoblotting analysis with the indicated antibodies. (B) CGP57380 inhibits WT and Mnk1/2DKO T cell proliferation. CFSE-labeled WT and Mnk1/2DKO splenocytes were unstimulated or stimulated with anti-CD3 (2C11, 0.1 μg/ml) in the presence or absence of CGP57380 (10 nM) at 37°C for 72 hours. Cells were then stained for CD4 and CD8. Histograms show CFSE intensity of live-gated CD4 and CD8 T cells. (C) Effects of CGP57380 on cytokine production by WT and Mnk1/2DKO iNKT cells. WT and Mnk1/2 thymocytes were stimulated with α-Galcer (125 ng/ml) in the presence or absence of CGP57380 (10 nM) for 72 hours. In the last 5 hours of stimulation, cells were also treated with PMA plus ionomycin and Golgi-plug. Cells were then stained for TCRβ and CD1d-tetramer followed by intracellular staining for IFNγ and IL-17A. Contour plots show IL-17A and IFNγ expression in gated TCRβ+CD1d-tetramer+ cells. Data shown represent two independent experiments.
Discussion
Mnk1/2 are downstream substrates for the Ras-Mek1/2-Erk1/2 and MKK3/MKK6-p38 kinase pathways. The importance of these pathways in T-cells, the ability of Mnk1/2 to phosphorylate eIF4E, and the extremely dynamic nature of T-cells during development and immune responses raise the possibility that Mnk1/2 may play important roles in T-cells by promoting protein translation via eIF4E regulation. Several recent studies lend credence to the idea that Mnk1/2 may be important regulators of the immune system. Inhibition of Mnk1/2 by CGP57380 has been found to reduce translation of pro-inflammatory cytokines in keratinocytes (48), macrophages (28), and dendritic cells (49). CGP57380 has also been shown to decrease cytokine production by iNKT-cells and IL-17 production in CD4+ T-cells during Th17 differentiation (26, 27). In this report, we we have demonstrated that TCR-induced Mnk1/2 activation is promoted by Ras-Erk1/2 signaling and is negatively controlled by DGKα and ζ. In addition, both Mnk1 and Mnk2 are expressed at high levels in naïve T-cells but down-regulated in activated T-cells. Using Mnk1/2 double deficient mice, we demonstrated here that Mnk1/2 are dispensable for the development of conventional αβ T-cells, natural Treg cells, and iNKT-cells. Moreover, Mnk1/2 double deficiency does not obviously affect activation of conventional T-cells and iNKT-cells, or Th differentiation in vitro. The conclusion of a minimal role for Mnk1/2 in T-cell activation is further strengthened by the observation that Mnk1/2 deficiency does not impair in vivo CD8 T-cell responses in a bacterial model and a viral model of infection. Furthermore, our data also raise concerns over CGP57380 as a Mnk1/2 specific inhibitor since CGP57380 not only reduces eIF4E but also Mnk1/2 phosphorylation following TCR engagement, inhibits both WT and Mnk1/2DKO CD4 T cell proliferation, and decreases both WT and Mnk1/2 iNKT cells to produce IFNγ.
Mnk1/2 deficiency does not affect Th1/17 differentiation in vitro, suggesting there might be no obvious intrinsic defect of Mnk1/2DKO CD4 T-cells in Th differentiation. However, Th1/17 differentiation is diminished in Mnk1/2DKO mice in the EAE model, suggesting the possibility that Mnk1/2 deficiency may affect in vivo Th lineage differentiation in a T-cell-extrinsic manner. Further experiments are required to explore these extrinsic mechanisms, including the possibility that Mnk1/2 may function in antigen-presenting cells to shape Th differentiation. This notion is supported by a recent report which demonstrated that Mnk1/2 regulate innate immune responses by modulating NFκB activity (50).
Signals generated from the TCR can trigger the activation of T-cells from naïve or resting conditions, resulting in significant increase in transcription, protein synthesis and DNA synthesis (51, 52). Translation is one of the early events in activated T-cells that can contribute to protein synthesis. Translational control is one of the key process by which cells can generate crucial gene products quickly from pre-existing mRNA without delay that results from mRNA transcription and RNA processing (53, 54). Translation is a complex process and involves at least 10 translation initiation factors called eukaryotic translational initiation factors (eIFs), scaffolding or adaptor proteins, and 40S ribosomes. Binding of eIF4E to the mRNA is the foremost regulatory step in formation of pre-initiation complex that further leads to formation of complete translational machinery (19). However, how eIF4E is regulated is poorly understood. It has been proposed that recruitment of eIF4E to 5′ cap region can be controlled by at least two intracellular signaling pathways such as the PI3K-mTOR pathway (55) and the Ras-Mek-Erk1/2-Mnk1/2 and/or MKK3/6-p38-Mnk1/2 pathways (56, 57). 4E-BP1 binds to eIF4E, preventing its association with the 5′ cap. Activation of mTOR leads to hyper-phosphorylation of 4E-BP1 that results in the dissociation of eIF4E from 4E-BP1 to allow eIF4E binding to the 5′ cap of mRNA to drive translation. Inhibition of mTOR results in T-cell anergy while deregulation of mTOR leads to resistance of T-cell anergy (43–45), suggesting that mTOR-mediated release of eIF4E from suppression by 4E-BP1 is critical for T cell activation. Several studies have put forth a notion that Mnk1/2 signaling can regulate cap-dependent translation through phosphorylation of eIF4E at S209 (22, 58). However, the role of eIF4E phosphorylation at 209 for translation initiation has been controversial (59, 60). Similarly, the germ-line deletion of Mnk1/2 in mouse models resulted in ablation in eIF4E phosphorylation without global effects on protein translation in mouse embryonic fibroblasts. Additionally, these mouse models display normal growth and development. Consistently, TCR induced eIF4E phosphorylation is abolished in Mnk1/2DKO T-cells, suggesting that eIF4E phosphorylation at S209 is not essential for T-cell development, proliferation, activation and cytokine expression, and may not globally control protein translation in T-cells. It had been demonstrated that mice that carry a nonphosphorylatable form of eIF4E (S209A) allele and Mnk1/2 double deficient mice are more resistant to tumorigenesis. This was shown to be via translational control of a specific subset of genes related to tumorigenesis, such as vascular endothelial growth factor C (VEGFC), baculoviral IAP repeat-containing protein 2 (BIRC2), and matrix metalloproteinase-3 (MMP3) (24, 61). Thus, although we have demonstrated that Mnk1/2 are dispensable for T-cell development and activation in general, we cannot rule out the possibility that Mnk1/2 may be selectively required for efficient translation of specific subsets of proteins that may affect specific T-cell responses.
Acknowledgments
We thank Jinhong Wu for help with iNKT-cell experiments, Nancy Martin at Duke Cancer Center Flow Cytometry Core Facility for providing sorting services, and the NIH tetramer facility for providing CD1d-Tet. The authors claim no competing financial interests. B.K.G. and S.K. were involved in experimental design and execution, data analysis, and preparation of the manuscript. J.S. designed and performed experiments. M.I. and M.L.S. were involved in the EAE experiments. J.G. and R.F. provided essential reagents. X.P.Z conceived the project and was involved in experimental design, data analysis and manuscript preparation.
This study was supported by funding from the National Institutes of Health (AI076357, AI079088, and AI101206), the Food Allergy and Anaphylaxis Network, and the American Cancer Society (RSG-08-186-01-LIB) to X-P.Z and NIH funding (R01AI068952) for JMG.
Abbreviations
- Mnk1/2
MAPK-interacting kinase 1 and 2
- eIF4E
eukaryotic translation initiation factor 4E
- DAG
Diacylglycerol
- DGK
DAG kinase
- iNKT
invariant NKT
- mTOR
mammalian target of rapamycin
- SP
single positive
- WT
wild-type
- DN
double negative
- DP
double-positive
- α-GalCer
α-galactosylceramide
- OVA
ovalbumin
- LM
Listeria monocytogenes
- LCMV
Lymphocytic choriomeningitis virus
- EAE
experimental autoimmune encephalitis
- MOG
myelin oligodendrocyte glycoprotein
References
- 1.Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- 2.Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol. 2002;20:371–394. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 3.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong XP, Shin J, Gorentla BK, O'Brien T, Srivatsan S, Xu L, Chen Y, Xie D, Pan H. Receptor signaling in immune cell development and function. Immunol Res. 2011;49:109–123. doi: 10.1007/s12026-010-8175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss A. The right team at the right time to go for a home run: tyrosine kinase activation by the TCR. Nature immunology. 2010;11:101–104. doi: 10.1038/ni0210-101. [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 7.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 8.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaestel M. Specificity of signaling from MAPKs to MAPKAPKs: kinases' tango nuevo. Front Biosci. 2008;13:6050–6059. doi: 10.2741/3136. [DOI] [PubMed] [Google Scholar]
- 10.Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- 11.Gaestel M. MAPKAP kinases - MKs - two's company, three's a crowd. Nat Rev Mol Cell Biol. 2006;7:120–130. doi: 10.1038/nrm1834. [DOI] [PubMed] [Google Scholar]
- 12.Arthur JS. MSK activation and physiological roles. Front Biosci. 2008;13:5866–5879. doi: 10.2741/3122. [DOI] [PubMed] [Google Scholar]
- 13.Buxade M, Parra-Palau JL, Proud CG. The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases) Front Biosci. 2008;13:5359–5373. doi: 10.2741/3086. [DOI] [PubMed] [Google Scholar]
- 14.Fukunaga R, Hunter T. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997;16:1921–1933. doi: 10.1093/emboj/16.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morley SJ, McKendrick L. Involvement of stress-activated protein kinase and p38/RK mitogen-activated protein kinase signaling pathways in the enhanced phosphorylation of initiation factor 4E in NIH 3T3 cells. J Biol Chem. 1997;272:17887–17893. doi: 10.1074/jbc.272.28.17887. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Flynn A, Waskiewicz AJ, Webb BL, Vries RG, Baines IA, Cooper JA, Proud CG. The phosphorylation of eukaryotic initiation factor eIF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J Biol Chem. 1998;273:9373–9377. doi: 10.1074/jbc.273.16.9373. [DOI] [PubMed] [Google Scholar]
- 17.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol. 2004;24:6539–6549. doi: 10.1128/MCB.24.15.6539-6549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 20.Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC, Jr., Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5'-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 21.Merrick WC. Cap-dependent and cap-independent translation in eukaryotic systems. Gene. 2004;332:1–11. doi: 10.1016/j.gene.2004.02.051. [DOI] [PubMed] [Google Scholar]
- 22.Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 1999;18:270–279. doi: 10.1093/emboj/18.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arquier N, Bourouis M, Colombani J, Leopold P. Drosophila Lk6 kinase controls phosphorylation of eukaryotic translation initiation factor 4E and promotes normal growth and development. Curr Biol. 2005;15:19–23. doi: 10.1016/j.cub.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 24.Ueda T, Sasaki M, Elia AJ, Chio, Hamada K, Fukunaga R, Mak TW. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc Natl Acad Sci USA. 2010;107:13984–13990. doi: 10.1073/pnas.1008136107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mavrakis KJ, Zhu H, Silva RL, Mills JR, Teruya-Feldstein J, Lowe SW, Tam W, Pelletier J, Wendel HG. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008;22:2178–2188. doi: 10.1101/gad.1690808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noubade R, Krementsov DN, Del Rio R, Thornton T, Nagaleekar V, Saligrama N, Spitzack A, Spach K, Sabio G, Davis RJ, Rincon M, Teuscher C. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood. 2011;118:3290–3300. doi: 10.1182/blood-2011-02-336552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagaleekar VK, Sabio G, Aktan I, Chant A, Howe IW, Thornton TM, Benoit PJ, Davis RJ, Rincon M, Boyson JE. Translational control of NKT cell cytokine production by p38 MAPK. J Immunol. 2011;186:4140–4146. doi: 10.4049/jimmunol.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rowlett RM, Chrestensen CA, Nyce M, Harp MG, Pelo JW, Cominelli F, Ernst PB, Pizarro TT, Sturgill TW, Worthington MT. MNK kinases regulate multiple TLR pathways and innate proinflammatory cytokines in macrophages. Am J Physiol Gastrointest Liver Physiol. 2008;294:G452–459. doi: 10.1152/ajpgi.00077.2007. [DOI] [PubMed] [Google Scholar]
- 29.Zhong XP, Guo R, Zhou H, Liu C, Wan CK. Diacylglycerol kinases in immune cell function and self-tolerance. Immunol Rev. 2008;224:249–264. doi: 10.1111/j.1600-065X.2008.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases. Blood. 2011;117:4022–4031. doi: 10.1182/blood-2010-08-300731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen S, Wu J, Srivatsan S, Gorentla BK, Shin J, Xu L, Zhong XP. Tight regulation of diacylglycerol-mediated signaling is critical for proper invariant NKT cell development. J Immunol. 2011;187:2122–2129. doi: 10.4049/jimmunol.1100495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong XP, Hainey EA, Olenchock BA, Jordan MS, Maltzman JS, Nichols KE, Shen H, Koretzky GA. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat Immunol. 2003;4:882–890. doi: 10.1038/ni958. [DOI] [PubMed] [Google Scholar]
- 34.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, Zhong XP. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7:1174–1181. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 35.Shen S, Chen Y, Gorentla BK, Lu J, Stone JC, Zhong XP. Critical Roles of RasGRP1 for Invariant NKT Cell Development. J Immunol. 2011;187:4467–4473. doi: 10.4049/jimmunol.1003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Brien TF, Gorentla BK, Xie D, Srivatsan S, McLeod IX, He YW, Zhong XP. Regulation of T-cell survival and mitochondrial homeostasis by TSC1. Eur J Immunol. 2011;41:3361–3370. doi: 10.1002/eji.201141411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudani R, Chapdelaine Y, Faassen Hv H, Smith DK, Shen H, Krishnan L, Sad S. Multiple mechanisms compensate to enhance tumor-protective CD8(+) T cell priming initially: comparison between an acute versus a chronic intracellular bacterium expressing a model antigen. J Immunol. 2002;168:5737–5745. doi: 10.4049/jimmunol.168.11.5737. [DOI] [PubMed] [Google Scholar]
- 38.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shin J, O'Brien TF, Grayson JM, Zhong XP. Differential regulation of primary and memory CD8 T cell immune responses by diacylglycerol kinases. J Immunol. 2012;188:2111–2117. doi: 10.4049/jimmunol.1102265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo R, Wan CK, Carpenter JH, Mousallem T, Boustany RM, Kuan CT, Burks AW, Zhong XP. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc Natl Acad Sci. 2008;105:11909–11914. doi: 10.1073/pnas.0711856105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong XP, Hainey EA, Olenchock BA, Zhao H, Topham MK, Koretzky GA. Regulation of T cell receptor-induced activation of the Ras-ERK pathway by diacylglycerol kinase zeta. J Biol Chem. 2002;277:31089–31098. doi: 10.1074/jbc.M203818200. [DOI] [PubMed] [Google Scholar]
- 42.Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 43.Xie DL, Wu J, Lou YL, Zhong XP. Tumor suppressor TSC1 is critical for T-cell anergy. Proc Natl Acad Sci. 2012;109:14152–14157. doi: 10.1073/pnas.1119744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong XP. An expanded role of the tumor suppressor TSC1 in T cell tolerance. Cell Cycle. 2012;11:3909–3910. doi: 10.4161/cc.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, Powell JD. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol. 2007;178:2163–2170. doi: 10.4049/jimmunol.178.4.2163. [DOI] [PubMed] [Google Scholar]
- 46.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248:205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kjellerup RB, Kragballe K, Iversen L, Johansen C. Pro-inflammatory cytokine release in keratinocytes is mediated through the MAPK signal-integrating kinases. Exp Dermatol. 2008;17:498–504. doi: 10.1111/j.1600-0625.2007.00672.x. [DOI] [PubMed] [Google Scholar]
- 49.Mikkelsen SS, Jensen SB, Chiliveru S, Melchjorsen J, Julkunen I, Gaestel M, Arthur JS, Flavell RA, Ghosh S, Paludan SR. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: dependence on TRAF2 and TAK1. J Biol Chem. 2009;284:10774–10782. doi: 10.1074/jbc.M807272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herdy B, Jaramillo M, Svitkin YV, Rosenfeld AB, Kobayashi M, Walsh D, Alain T, Sean P, Robichaud N, Topisirovic I, Furic L, Dowling RJ, Sylvestre A, Rong L, Colina R, Costa-Mattioli M, Fritz JH, Olivier M, Brown E, Mohr I, Sonenberg N. Translational control of the activation of transcription factor NF-kappaB and production of type I interferon by phosphorylation of the translation factor eIF4E. Nat Immunol. 2012;13:543–550. doi: 10.1038/ni.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cooper HL. Ribsomal ribonucleic acid production and growth regulation in human lymphocytes. J Biol Chem. 1969;244:1946–1952. [PubMed] [Google Scholar]
- 52.Crabtree GR, Clipstone NA. Signal transmission between the plasma membrane and nucleus of T lymphocytes. Annu Rev Biochem. 1994;63:1045–1083. doi: 10.1146/annurev.bi.63.070194.005145. [DOI] [PubMed] [Google Scholar]
- 53.Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–835. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 56.Panja D, Dagyte G, Bidinosti M, Wibrand K, Kristiansen AM, Sonenberg N, Bramham CR. Novel translational control in Arc-dependent long term potentiation consolidation in vivo. J Biol Chem. 2009;284:31498–31511. doi: 10.1074/jbc.M109.056077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen YJ, Tan BC, Cheng YY, Chen JS, Lee SC. Differential regulation of CHOP translation by phosphorylated eIF4E under stress conditions. Nucleic Acids Res. 2010;38:764–777. doi: 10.1093/nar/gkp1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pyronnet S. Phosphorylation of the cap-binding protein eIF4E by the MAPK-activated protein kinase Mnk1. Biochem Pharmacol. 2000;60:1237–1243. doi: 10.1016/s0006-2952(00)00429-9. [DOI] [PubMed] [Google Scholar]
- 59.Scheper GC, Proud CG. Does phosphorylation of the cap-binding protein eIF4E play a role in translation initiation? Eur J Biochem. 2002;269:5350–5359. doi: 10.1046/j.1432-1033.2002.03291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKendrick L, Morley SJ, Pain VM, Jagus R, Joshi B. Phosphorylation of eukaryotic initiation factor 4E (eIF4E) at Ser209 is not required for protein synthesis in vitro and in vivo. Eur J Biochem. 2001;268:5375–5385. doi: 10.1046/j.0014-2956.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 61.Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, Pandolfi PP, Saad F, Sonenberg N. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci. 2010;107:14134–14139. doi: 10.1073/pnas.1005320107. [DOI] [PMC free article] [PubMed] [Google Scholar]