

Abstract
Type 1 diabetes involves the specific destruction of the pancreatic islet β-cells, eventually resulting in a complete dependency of exogenous insulin. The clinical onset of diabetes is preceded by the appearance of autoantibodies against β-cell antigens. The human leukocyte antigen (HLA) region is the single most important genetic determinant of Type 1 diabetes susceptibility, yet variability in the HLA region has been estimated to explain only approximately 60% of the genetic influence of the disease. Over 50 identified non-HLA genetic polymorphisms support the notion that genetics alone cannot explain Type 1 diabetes. Several lines of evidence indicate that environmental triggers may be integral in inducing the onset of islet autoimmunity in genetically susceptible individuals. The association between environmental factors and the clinical onset is complicated by observation that the rate of progression to clinical onset may be affected by environmental determinants. Hence, the environment may be aetiological as well as pathogenic. Putative inductive mechanisms include viral, microbial, diet-related, anthropometric and psychosocial factors. Ongoing observational cohort studies such as The Environmental Determinants of Diabetes in the Young (TEDDY) study aim to ascertain environmental determinants that may trigger islet autoimmunity and either speed up or slow down the progression to clinical onset in subjects with persistent islet autoimmunity.
Introduction
The notion that environmental determinants contribute to Type 1 diabetes risk has been considered since the late 1800s when a mumps epidemic in a small Norwegian village was associated with an outbreak of childhood diabetes [1]. The search for an infectious agent such as a virus that would trigger both islet autoimmunity and clinical onset has continued ever since. Numerous viral and also bacterial agents, food items and a combination between food items and viruses have been reported and proposed [1,2]. The reader is referred to other reviews that have examined the subject in the past and more recently [2,3]. Studies of children at increased genetic risk for Type 1 diabetes who have been followed from birth for the development of islet autoantibodies and progression to clinical onset support the notion that Type 1 diabetes is a two-step disease.
The first step would be an environmental determinant that causes islet autoimmunity (Fig. 1). Markers of islet autoimmunity are autoantibodies directed against insulin (IA), glutamic acid decarboxylase 65 (GAD), insulinoma antigen-2 (IA-2) or ZnT8 transporter (ZnT8). These autoantibody tests are robust and standardized [4] and may in part be used to predict clinical onset of diabetes [5]. The larger the number of islet autoantibodies, the higher the risk of progression to clinical onset. The autoantibodies do not provide much insight into the mechanisms by which the autoimmunity is triggered. There is a lack of reliable, standardized and reproducible T- and β-cell tests, along with an ability to identify antigen-presenting cells that would contribute to the actual induction or generation of islet autoimmunity. If we are to understand the mechanism by which islet autoimmunity is triggered, resulting in persistent islet autoantibodies, the period of time leading up to the first appearance of islet autoantibodies needs to be dissected using cellular tests. At present there is no obvious candidate trigger of islet autoimmunity, although recent investigations provide some evidence that enteroviruses may contribute to the development of islet autoimmunity measured as persistent islet autoantibodies.
Figure 1.
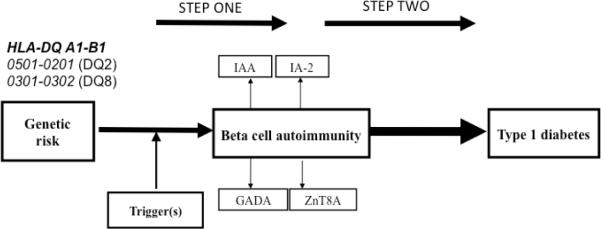
Type 1 diabetes is viewed as a two-step disease. Children may be born with increased genetic risk for Type 1 diabetes. Environmental factors during pregnancy or during neonatal and infancy are thought to induce step one: islet or β-cell autoimmunity marked by the appearance of autoantibodies against insulin (IAA), glutamic acid decarboxylase 65 (GADA), insulinoma antigen-2 (IA-2A), ZnT8 transporter (ZnT8A), alone or in combination. Persistent islet autoantibodies also mark step two, progression to clinical onset of Type 1 diabetes.
The second step of Type 1 diabetes development is progression from persistent islet autoimmunity to clinical onset of diabetes (Fig. 1). It is already well known that an individual may be islet autoantibody positive for months to years before the clinical onset of diabetes. The question must therefore be asked: what environmental factors may affect the progression to clinical onset? Are there environmental factors that are able to slow down progression? Also, are there factors that speed up the progression?
In this brief review, we will first discuss the possible role of environmental factors for step one, i.e. induction of islet autoimmunity. Next, we will examine evidence that environmental factors may affect step two of the disease process, i.e. the progression from islet autoimmunity to clinical onset.
Genetics and the environment
The proband-wise concordance of Type 1 diabetes is approximately 30–50% for monozygotic twins, and approximately 8% in dizygotic twins. The importance of a genetic predisposition for the development of islet autoimmunity against β-cell autoantigen is clear not only in humans, but also in a number of laboratory rodents spontaneously developing autoimmune diabetes. Genetic variability in the human leukocyte antigen (HLA) region has been estimated to explain more than half of the genetic influence in Type 1 diabetes, while over 50 identified non-HLA polymorphisms account for the other half [6]. The reason to briefly review the association between HLA and islet autoimmunity, Type 1 diabetes, or both, is the well-known association between HLA and immune responses to infectious agents. It is as important to ask the question, which virus or bacterial peptides are able to bind to Type 1 diabetes-associated HLA-DQ, as it is to find out which autoantigen peptides bind or do not bind to the trimolecular complex.
Human leukocyte antigen
The HLA system is located on chromosome 6p21 and contains many genes related to the immune system. It is a hugely polymorphic region of the human genome and is divided into three classes, of which class II is the major genetic susceptibility determinant for Type 1 diabetes. HLA class II proteins include the heterodimers coded for at the DP, DQ, and DR loci. Each heterodimer consists of two chains, designated α and β. Specific alleles at the DRB1, DQA1 and DQB1 loci are particularly strongly associated with Type 1 diabetes. The main mechanism of action of the DR and DQ molecules in Type 1 diabetes aetiology is the peptide-binding activity of the HLA class II molecules in antigen-presenting cells (APCs) for T lymphocyte peptide recognition [7]. There are two extended haplotypes that may account for as many as nearly 90% of all children and adolescents who develop Type 1 diabetes [8]. The genotype of highest risk is DRB1*03:01-DQA1*05:01-DQB1*02:01/DRB1*04:xx-DQA1*03:01-DQB1*03:02. This heterozygous genotype is commonly referred to as DR3/DR4 or DQ2/8 [9]. The data in Table 1 illustrate that DQ8 or DQ2 in combination with either DQ6.4, DQ5.1, DQ4 or DQ6.3 may account for as many as 69% of all children and adolescents with Type 1 diabetes diagnosed before 18 years of age. The same genotypes account for 14% of the general population. As the lifetime risk in Sweden is approximately 1.5% only, some subjects are exposed to an environmental exposure that trigger islet autoimmunity (Fig. 1). It is important to note that DQ6.4, DQ5.1, DQ4 or DQ6.3 are only a risk together with DQ8 or DQ2. These haplotypes are not risk factors by themselves. Indeed, some HLA haplotypes have a protective effect—the DRB1*15:01-DQA1*01:02-DQB1*06:02 haplotype, for instance, was found in 20% of a studied population, but only occurred among 1% of patients with Type 1 diabetes and was essentially absent before 10–12 years of age [10].
Table 1.
HLA genotypes among patients with Type 1 diabetes compared with the population genotype frequencies in Sweden
| HLA DQ genotype | % among Type 1 diabetes | % in population | Odds ratio | 95% CI |
|---|---|---|---|---|
| 2/8 | 30 | 3.5 | 11.7 | 9.1–15.1 |
| 8/8 | 11 | 1.7 | 7.5 | 5.2–10.8 |
| 8/6.4 | 5 | 1.2 | 4.3 | 2.6–6.6 |
| 8/5.1 | 10 | 2.7 | 3.8 | 2.8–5.1 |
| 8/4 | 5 | 1.4 | 3.7 | 2.5–5.6 |
| 2/2 | 5 | 1.7 | 3.0 | 2.0–4.4 |
| 8/6.3 | 3 | 2.0 | 1.6 | 1.1–2.4 |
| Total frequency | 69% | 14% |
Data from Delli et al. [43].
HLA class I alleles also contribute to the aetiology of Type 1 diabetes, although less so than class II. The HLA-A and HLA-B genes explain most of the residual influence of the HLA region after taking class II genes into account. HLA-B*39 is of particular significance and is associated with a lower age at diagnosis of Type 1 diabetes [11]. HLA-A*02 increases the risk of Type 1 diabetes in individuals with the high-risk class II DQ2/8 genotype, and HLA-A*02:01 occurs in over 60% of patients with Type 1 diabetes. The class I heterodimers present shorter peptides than class II. The trimolecular complex is particularly important to direct cytotoxic CD8+ T cells towards cells that have been infected with a virus, as virus peptides are presented on class I heterodimers.
Non-HLA genes of importance to the interaction with the environment
A host of genes outside the HLA region have been found to be associated with the risk for Type 1 diabetes [12]. Their individual effects are small compared with HLA-DQ, but would nevertheless contribute either to the risk of islet autoimmunity or to progression to clinical onset in subjects who hade developed islet autoimmunity. Among the strongest non-HLA genes influencing Type 1 diabetes risk is protein tyrosine phosphatase, non-receptor type 22 (PTPN22), located on the short arm of chromosome 1. The protein down-regulates the immune response by inhibiting T-cell antigen receptor (TCR) signalling, and some single-nucleotide polymorphisms (SNPs) are associated with several types of autoimmune diseases. It has been hypothesized that a gain-of-function mutation increasing PTPN22 activity would raise the threshold required for effective T-cell antigen receptor signalling in developing thymocytes, resulting in a lack of negative selection of autoreactive T cells [13]. Hence, it may be speculated that this particular genetic risk factor may be more important to promote progression to diabetes in the islet autoantibody-positive cases than to initiating islet autoimmunity in them.
Insulin is a major β-cell autoantigen in Type 1 diabetes, so it is unsurprising that polymorphisms of the insulin (INS) gene contribute to disease susceptibility. The INS gene is located on chromosome 11 and shows polymorphism in its promoter region. Predisposition to Type 1 diabetes is dependent on a variable number of tandem repeat polymorphisms, with shorter repeats conferring risk and longer repeats providing protection [9]. It is unclear if environmental factors are important to the risk conferred by this genetic factor.
The interleukin-2 receptor-α (IL2RA) that binds interleukin-2 (IL-2) is encoded on chromosome 10 and is expressed on T cells including regulatory T cells. These cells require IL-2 for their growth and survival. Risk variants of IL2RA may impair the function of regulatory T cells, although several other mechanisms have been proposed for the role of IL2RA in autoimmunity [12]. The influence of environmental factors such as virus infections on the function and expression of IL2RA remains to be determined.
Of particular importance to the present analysis of possible effects of environmental factors is the interferon-induced with helicase C domain 1 (IFIH1). IFIH1 is an intracellular detector of viral RNA that mediates the immune response [14]. Reduced IFIH1 expression and protein function protects from Type 1 diabetes, possibly because IFIH1 may be able to stimulate autoreactive T cells during viral infection. The common variant of IFIH1 provides risk, whereas the rare variants confer protection. Some rare variants in the IFIH1 gene are more strongly associated with disease risk than common polymorphisms [14]. The IFIH1 is of particular interest as it may be associated with the risk for islet autoimmunity rather than with risk of diabetes in islet autoantibody-positive individuals.
The environment and the risk for islet autoimmunity—step one
Autoantibodies against GAD65, insulinoma-associated antigen-2, insulin and zinc T8 transporter precede the clinical onset of Type 1 diabetes, with over 94% of patients having one or several of these autoantibodies at the time of diagnosis [15,16]. Subjects may lose islet autoantibodies prior to the clinical onset. A study on twins discordant for Type 1 diabetes concluded that non-shared environmental factors strongly influence the appearance of autoantibodies [17], but the nature of these environmental exposures remains poorly understood. Current data, mainly in children born to mothers with diabetes or having a father with the disease suggest that enterovirus infections alone [17] or enterovirus combined with exposure to gluten [18] may induce islet autoantibodies. Further studies of sufficiently large cohorts such as The Environmental Determinants of Diabetes in the Young (TEDDY) will be needed to identify the possible relation between infections, diet exposures, or both, and appearance of islet autoantibodies. Identifying the environmental triggers of islet autoimmunity thus holds enormous primary prevention therapeutic potential.
The environment and the risk for diabetes in the islet autoimmunity positive—step two
Because of the seasonal variation of Type 1 diabetes onset, a viral contribution to the aetiology of the disease has long been considered. It has been hypothesized that certain viruses may possess a pancreatic tropism, leading to either direct lysis or immune-mediated destruction of β-cells. Several types of viruses have been implicated, yet studies on their relation to Type 1 diabetes have generally yielded weak and non-reproducible results [19]. These results from early studies are not surprising as the prodrome of islet autoimmunity was neither fully understood nor appreciated. Any infection close to the clinical onset of Type 1 diabetes therefore has to be questioned whether it was a trigger or merely an accelerator of an ongoing islet autoimmunity (Table 2). Numerous publications on the relationship between viruses and Type 1 diabetes therefore have to be reconsidered. Were they a trigger of islet autoimmunity or an accelerator?
Table 2.
Possible differences of genetic and environmental factors that may affect step one islet autoimmunity and step two progression to Type 1 diabetes
| Environmental factor | Step one | Step two |
|---|---|---|
| Gestational events | +++ | ++ |
| HLA-DQ genotype | ++++ | ++ |
| Non-HLA genes | ++ | ++++ |
| Virus infection (1–3 years of age) | ++++ | + |
| Virus infection (> 3 years of age) | ++ | ++++ |
| Microbiome | ? | ? |
| Vitamin A and vitamin D deficiencies | ++ | ++++ |
| Overweight or obese (teenagers) | +? | +++ |
A recent report in the Diabetes and Autoimmunity Study in the Young (DAISY) study of islet autoantibody positive indicates that enterovirus infection of children with persistent islet autoantibodies accelerated the disease progression, shortening the time to clinical onset of Type 1 diabetes [20]. It thus appears that further knowledge about the precise virus serotypes related to islet autoimmunity, Type 1 diabetes, or both, is needed before vaccines or other therapies directed against enteroviruses can be developed.
In comparison with viruses, the relation of bacteria to Type 1 diabetes has so far been given little attention. Yet, some studies of recent years suggest that intestinal microbiota may affect the development of islet autoimmunity, Type 1 diabetes, or both. For instance, spontaneous diabetes in non-obese diabetic (NOD) mice may be affected by the microbial environment in the animal housing facility, or by exposure to microbial stimuli such as injection with mycobacteria or various microbial products [21]. NOD mice lacking MyD88 protein (an adaptor for multiple innate immune receptors that recognize microbial stimuli) develop Type 1 diabetes if their gut is free of germs, yet avoid the disease if their microbiota represents that normally found in humans [22]. It has been proposed that modern standards of hygiene have reduced exposure to microbes, which could diminish protective immunity throughout childhood and increase susceptibility to autoimmune diseases (the `hygiene hypothesis') [2].
Diet—vitamins, breastfeeding, cow's milk, gluten and additional factors
Vitamin A and vitamin D both influence the immune system, and so it has long been thought that they may affect autoimmunity [23]. Studies on vitamin A in humans with Type 1 diabetes are limited, but the literature on vitamin D is extensive. In a large-scale prospective study of a Finnish population, high-dose vitamin D supplementation significantly reduced the risk of developing Type 1 diabetes during the 30-year follow-up period [24]. A meta-analysis of five observational studies found that the risk of Type 1 diabetes was significantly reduced in infants who were supplemented with vitamin D compared with those who were not, with some evidence indicating that a high dosage was more beneficial than a low one [25]. Although some aspects of vitamin D in relation to islet autoimmunity, Type 1 diabetes, or both, remain controversial, there seems to be a beneficial effect, possibly by suppressing immune responses (Table 2). Studies on additional vitamins and minerals are limited [26].
Cow's milk has been suggested as a trigger of Type 1 diabetes, especially since cross-reactivity was found between bovine and human insulin [27]. Studies of cow's milk in relation to Type 1 diabetes have, however, led to contradictory results, with some reporting early exposure to cow's milk as a predisposing factor, while others found no causality [28]. Recent findings that polymorphisms in the PTPN22 gene are associated with Type 1 diabetes among children exposed to cow's milk before 6 months, but not after, suggest that an interplay between genetic and environmental factors could explain the contradictory findings on the significance of exposure to cow's milk in Type 1 diabetes [29]. Again, it is not clear whether the most important factor is the trigger of islet autoimmunity or progression to Type 1 diabetes once islet autoantibodies are persistent.
The duration of breastfeeding, related to the age of introduction to cow's milk, has been suggested to defend against autoimmunity. Putative protective mechanisms of breast milk include secretory antibodies, enhancement of the infant's own immune responses, increased β-cell proliferation, or delayed exposure to foreign food antigens. However, the literature is once again conflicting, with studies showing a protective effect, no effect, or even a predisposing effect [30,31].
In addition to the substances mentioned above, there is limited evidence that numerous other nutriments and chemicals may affect the risk for islet autoimmunity, Type 1 diabetes, or both. Omega-3 fatty acids [32], fruit and berry juices [33], and N-nitroso compounds needs further consideration [34].
Anthropometric and psychosocial factors
Increased birthweight and early childhood weight gain are risk factors for subsequent development of Type 1 diabetes, according to a meta-analysis of the subject [35]. The authors propose that maternal gestational diabetes and overweight lead to antenatal overstimulation of fetal β-cells, or an association between genetic factors increasing body weight and the risk of diabetes. The weakness of the analysis is that islet autoimmunity has not been distinguished from clinical onset of Type 1 diabetes. Surrogate markers of insulin resistance and BMI predict progression to Type 1 diabetes in subjects with islet autoimmunity, and weight gain in early life may predict risk of islet autoimmunity in children with a first-degree relative with Type 1 diabetes [36], although other studies have failed to show that excess body weight and insulin resistance influence autoantibody frequency [37]. Certain HLA genotypes linked to diabetes are associated with increased BMI, suggesting that an interplay of genetic and environmental factors affects the role of body weight and childhood obesity in Type 1 diabetes [38]. HLA genotypes conferring risk of Type 1 diabetes have been associated with increased relative birthweight, indicating that these HLA genotypes moderate intrauterine growth [39]. Hence, the epidemiological observation that high birthweight is a risk factor for Type 1 diabetes could result from the effect of HLA variations on antenatal growth [40]. Gestational factors resulting in increased birthweight may be related to islet autoimmunity rather than clinical onset of Type 1 diabetes.
Children with Type 1 diabetes may show accelerated linear growth compared with children without diabetes [41]. This study demonstrated that, when corrected for mid-parental height, children developing diabetes were taller at birth than non-HLA- but not taller than HLA-matched control subjects. Children with diabetes had increased mid-parental height-corrected height up to 18 months of age compared with both HLA- and non-HLA-matched control subjects. These results show that high-risk HLA affects antenatal growth, but that other factors may explain the increased post-natal linear growth in children developing Type 1 diabetes [41].
Psychological mechanisms are linked to hormonal and neuronal signals, and psychological stress may increase insulin resistance, thus burdening the β-cells. Low socio-economic status, high maternal age and psychosocial strain in families, for example, divorce, serious illness or parenting stress, have been identified as risk factors for both persistent islet autoantibodies, Type 1 diabetes, or both [42].
Conclusions
There is great promise to begin to understand the possible role of the environment and the origins of Type 1 diabetes. Most importantly is the recognition and growing understanding that the clinical onset of Type 1 diabetes most often will be preceded by islet autoimmunity. The islet (or β-cell) autoimmunity is defined by islet autoantibodies against one or, more often, several islet autoantigens, such as insulin, GAD65, IA-2 and ZnT8. Current islet autoantibody assays appear robust and the inter-laboratory variation is diminished through international serum exchange exercises to standardize the analyses. The possible role of the environment and the origins of Type 1 diabetes therefore need to be revised. Do environmental factors trigger islet autoimmunity, i.e. islet autoantibodies in subjects at genetic risk, accelerate the disease progression in islet autoantibody positive subjects, or both? It is unproblematic to appreciate that many prior studies on environmental factors at the time of clinical onset of Type 1 diabetes need to be reinterpreted. Was it a trigger or an accelerator of an already established, subclinical disorder of islet autoimmunity that was present at the time of clinical diagnosis? The concept of a two-step disease—islet autoimmunity and clinical onset of Type 1 diabetes in the islet autoantibody positive—should help to eventually understand the role of the environment in the aetiology of islet autoimmunity and Type 1 diabetes.
Acknowledgments
Funding sources The research in the authors' laboratory is funded in part by the National Institutes of Health (DK063861), Juvenile Diabetes Research Foundation, Swedish Research Council, Skåne County Council for Research and Development, Swedish Diabetes Foundation, and the Lund University Medical Faculty for a fellowship (SER).
Footnotes
Competing interests ÅL is a member of the scientific advisory boards of Diamyd Medicals, Stockholm, Sweden, Zealand Pharma A/S, Copenhagen, Denmark and Probi AB, Lund, Sweden.
References
- 1.Yoon JW. The role of viruses and environmental factors in the induction of diabetes. Curr Top Microbiol Immunol. 1990;164:95–123. doi: 10.1007/978-3-642-75741-9_6. [DOI] [PubMed] [Google Scholar]
- 2.Bach JF, Chatenoud L. The hygiene hypothesis: an explanation for the increased frequency of insulin-dependent diabetes. Cold Spring Harb Perspect Med. 2012;2:a007799. doi: 10.1101/cshperspect.a007799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boettler T, von Herrath M. Protection against or triggering of Type 1 diabetes? Different roles for viral infections. Expert Rev Clin Immunol. 2011;7:45–53. doi: 10.1586/eci.10.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torn C, Mueller PW, Schlosser M, Bonifacio E, Bingley PJ. Diabetes Antibody Standardization Program: evaluation of assays for autoantibodies to glutamic acid decarboxylase and islet antigen-2. Diabetologia. 2008;51:846–852. doi: 10.1007/s00125-008-0967-2. [DOI] [PubMed] [Google Scholar]
- 5.Sosenko JM, Krischer JP, Palmer JP, Mahon J, Cowie C, Greenbaum CJ, et al. A risk score for type 1 diabetes derived from autoantibody-positive participants in the Diabetes Prevention Trial—Type 1. Diabetes Care. 2008;31:528–533. doi: 10.2337/dc07-1459. [DOI] [PubMed] [Google Scholar]
- 6.Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of type 1 diabetes: what's next? Diabetes. 2010;59:1561–1571. doi: 10.2337/db10-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32:457–467. doi: 10.1016/j.immuni.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Sanjeevi CB, Falorni A, Kockum I, Hagopian WA, Lernmark A. HLA and glutamic acid decarboxylase in human insulin-dependent diabetes mellitus. Diabet Med. 1996;13:209–217. doi: 10.1002/(SICI)1096-9136(199603)13:3<209::AID-DIA39>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Kockum I, Lernmark A, Dahlquist G, Falorni A, Hagopian WA, Landin-Olsson M, et al. Genetic and immunological findings in patients with newly diagnosed insulin-dependent diabetes mellitus. The Swedish Childhood Diabetes Study Group and The Diabetes Incidence in Sweden Study (DISS) Group. Horm Metab Res. 1996;28:344–347. doi: 10.1055/s-2007-979811. [DOI] [PubMed] [Google Scholar]
- 10.Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57:1084–1092. doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887–892. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med. 2009;360:1646–1654. doi: 10.1056/NEJMra0808284. [DOI] [PubMed] [Google Scholar]
- 13.Gregersen PK. Gaining insight into PTPN22 and autoimmunity. Nat Genet. 2005;37:1300–1302. doi: 10.1038/ng1205-1300. [DOI] [PubMed] [Google Scholar]
- 14.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in diabetes. Diabetes. 2005;54:S52–61. doi: 10.2337/diabetes.54.suppl_2.s52. [DOI] [PubMed] [Google Scholar]
- 16.Wenzlau JM, Liu Y, Yu L, Moua O, Fowler KT, Rangasamy S, et al. A common nonsynonymous single nucleotide polymorphism in the SLC30A8 gene determines ZnT8 autoantibody specificity in type 1 diabetes. Diabetes. 2008;57:2693–2697. doi: 10.2337/db08-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyoty H, Hiltunen M, Knip M, Laakkonen M, Vahasalo P, Karjalainen J, et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes. 1995;44:652–657. doi: 10.2337/diab.44.6.652. [DOI] [PubMed] [Google Scholar]
- 18.Steck AK, Johnson K, Barriga KJ, Miao D, Yu L, Hutton JC, et al. Age of islet autoantibody appearance and mean levels of insulin, but not GAD or IA-2 autoantibodies, predict age of diagnosis of type 1 diabetes: diabetes autoimmunity study in the young. Diabetes Care. 2011;34:1397–1399. doi: 10.2337/dc10-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppieters KT, von Herrath MG. Viruses and cytotoxic T lymphocytes in type 1 diabetes. Clin Rev Allergy Immunol. 2011;41:169–178. doi: 10.1007/s12016-010-8220-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stene LC, Oikarinen S, Hyoty H, Barriga KJ, Norris JM, Klingensmith G, et al. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY) Diabetes. 2010;59:3174–3180. doi: 10.2337/db10-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okada H, Kuhn C, Feillet H, Bach JF. The `hygiene hypothesis' for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyppönen E, Läärä E, Reunanen A, Järvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. 2001;358:1500–1503. doi: 10.1016/S0140-6736(01)06580-1. [DOI] [PubMed] [Google Scholar]
- 25.Zipitis CS, Akobeng AK. Vitamin D supplementation in early childhood and risk of type 1 diabetes: a systematic review and meta-analysis. Arch Dis Child. 2008;93:512–527. doi: 10.1136/adc.2007.128579. [DOI] [PubMed] [Google Scholar]
- 26.Virtanen SM, Knip M. Nutritional risk predictors of beta-cell autoimmunity and type 1 diabetes at a young age. Am J Clin Nutr. 2003;78:1053–1067. doi: 10.1093/ajcn/78.6.1053. [DOI] [PubMed] [Google Scholar]
- 27.Virtanen SM, Laara E, Hypponen E, Reijonen H, Rasanen L, Aro A, et al. Cow's milk consumption, HLA-DQB1 genotype, and type 1 diabetes: a nested case—control study of siblings of children with diabetes. Childhood diabetes in Finland study group. Diabetes. 2000;49:912–917. doi: 10.2337/diabetes.49.6.912. [DOI] [PubMed] [Google Scholar]
- 28.Norris JM, Beaty B, Klingensmith G, Yu L, Hoffman M, Chase HP, et al. Lack of association between early exposure to cow's milk protein and beta-cell autoimmunity. Diabetes Autoimmunity Study in the Young (DAISY) J Am Med Assoc. 1996;276:609–614. doi: 10.1001/jama.1996.03540080031025. 28. [DOI] [PubMed] [Google Scholar]
- 29.Lempainen J, Vaarala O, Mäkelä M, Veijola R, Simell O, Knip M, et al. Interplay between PTPN22 C1858T polymorphism and cow's milk formula exposure in type 1 diabetes. J Autoimmun. 2009;33:155–164. doi: 10.1016/j.jaut.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Kimpimaki T, Erkkola M, Korhonen S, Kupila A, Virtanen SM, Ilonen J, et al. Short-term exclusive breastfeeding predisposes young children with increased genetic risk of Type I diabetes to progressive beta-cell autoimmunity. Diabetologia. 2001;44:63–69. doi: 10.1007/s001250051581. [DOI] [PubMed] [Google Scholar]
- 31.Couper JJ. Environmental triggers of type 1 diabetes. J Paediatr Child Health. 2001;37:218–220. doi: 10.1046/j.1440-1754.2001.00658.x. [DOI] [PubMed] [Google Scholar]
- 32.Norris JM, Yin X, Lamb MM, Barriga K, Seifert J, Hoffman M, et al. Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. J Am Med Assoc. 2007;298:1420–1428. doi: 10.1001/jama.298.12.1420. [DOI] [PubMed] [Google Scholar]
- 33.Virtanen SM, Nevalainen J, Kronberg-Kippila C, Ahonen S, Tapanainen H, Uusitalo L, et al. Food consumption and advanced beta-cell autoimmunity in young children with HLA-conferred susceptibility to type 1 diabetes: a nested case–control design. Am J Clin Nutr. 2012;95:471–478. doi: 10.3945/ajcn.111.018879. [DOI] [PubMed] [Google Scholar]
- 34.Dahlquist GG. Viruses and other perinatal exposures as initiating events for beta-cell destruction. Ann Med. 1997;29:413–417. doi: 10.3109/07853899708999371. [DOI] [PubMed] [Google Scholar]
- 35.Harder T, Roepke K, Diller N, Stechling Y, Dudenhausen JW, Plagemann A. Birth weight, early weight gain, and subsequent risk of type 1 diabetes: systematic review and meta-analysis. Am J Epidemiol. 2009;169:1428–1436. doi: 10.1093/aje/kwp065. [DOI] [PubMed] [Google Scholar]
- 36.Couper JJ, Beresford S, Hirte C, Baghurst PA, Pollard A, Tait BD, et al. Weight gain in early life predicts risk of islet autoimmunity in children with a first-degree relative with type 1 diabetes. Diabetes Care. 2009;32:94–99. doi: 10.2337/dc08-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barker JM, Goehrig SH, Barriga K, Hoffman M, Slover R, Eisenbarth GS, et al. Clinical characteristics of children diagnosed with type 1 diabetes through intensive screening and follow-up. Diabetes Care. 2004;27:1399–1404. doi: 10.2337/diacare.27.6.1399. [DOI] [PubMed] [Google Scholar]
- 38.Carlsson A, Kockum I, Lindblad B, Engleson L, Nilsson A, Forsander G, et al. Low risk HLA-DQ and increased body mass index in newly diagnosed type 1 diabetes children in the Better Diabetes Diagnosis study in Sweden. Int J Obes (Lond) 2011;XX:XXX–XXX. doi: 10.1038/ijo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larsson HE, Lynch K, Lernmark B, Nilsson A, Hansson G, Almgren P, et al. Diabetes-associated HLA genotypes affect birthweight in the general population. Diabetologia. 2005;48:1484–1491. doi: 10.1007/s00125-005-1813-4. [DOI] [PubMed] [Google Scholar]
- 40.Larsson HE, Lynch K, Lernmark B, Hansson G, Lernmark A, Ivarsson SA. Relationship between increased relative birthweight and infections during pregnancy in children with a high-risk diabetes HLA genotype. Diabetologia. 2007;50:1161–1169. doi: 10.1007/s00125-007-0648-6. [DOI] [PubMed] [Google Scholar]
- 41.Larsson HE, Hansson G, Carlsson A, Cederwall E, Jonsson B, Larsson K, et al. Children developing type 1 diabetes before 6 years of age have increased linear growth independent of HLA genotypes. Diabetologia. 2008;51:1623–1630. doi: 10.1007/s00125-008-1074-0. [DOI] [PubMed] [Google Scholar]
- 42.Sepa A, Wahlberg J, Vaarala O, Frodi A, Ludvigsson J. Psychological stress may induce diabetes-related autoimmunity in infancy. Diabetes Care. 2005;28:290–295. doi: 10.2337/diacare.28.2.290. [DOI] [PubMed] [Google Scholar]
- 43.Delli AJ, Lindblad B, Carlsson A, Forsander G, Ivarsson SA, Ludvigsson J, et al. Type 1 diabetes patients born to immigrants to Sweden increase their native diabetes risk and differ from Swedish patients in HLA types and islet autoantibodies. Pediatr Diabetes. 2011;11:513–520. doi: 10.1111/j.1399-5448.2010.00637.x. [DOI] [PubMed] [Google Scholar]