

SUMMARY
“Triple-defective” (3d) mice carrying a mutation in UNC93B1, a chaperone for the endosomal nucleic-acid sensing (NAS) Toll-Like Receptors TLR3, TLR7 and TLR9, are highly susceptible to Toxoplasma gondii infection. However, none of the single or even the triple NAS-TLR deficient animals recapitulated the 3d susceptible phenotype to Toxoplasma infection. Investigating this further, we find that while parasite RNA and DNA activate innate immune responses via the NAS-TLRs 7 and 9, TLR11 and TLR12 working as heterodimers are required for sensing and responding to Toxoplasma profilin. Consequently, the triple TLR7/TLR9/TLR11 deficient mice are highly susceptible to T. gondii infection, recapitulating the phenotype of 3d mice. Humans lack functional TLR11 and TLR12 genes. Consistently, human cells produce high levels of pro-inflammatory cytokines in response to parasite derived RNA and DNA, but not to Toxoplasma profilin supporting a more critical role for NAS-TLRs in human toxoplasmosis.
Keywords: UNC93B1, TLR, Toxoplasma gondii, IL-12, innate immunity
INTRODUCTION
Natural infection with Toxoplasma gondii has been described in more than 300 mammal and 30 avian species. While felines are the definitive hosts, mice – the cats’ prey - are the natural intermediate hosts and main reservoirs of this coccidian parasite. Even though humans are considered “accidental” intermediate hosts, one third of the world population carry a chronic and asymptomatic infection with T. gondii (Robert-Gangneux and Darde, 2012). However, in immune-compromised individuals, the dormant parasite becomes highly virulent, leading to reactivation of the chronic infection, and causing severe disease and lethality (Weiss and Dubey, 2009).
Host resistance to T. gondii infection is primarily dependent on T cell-mediated immunity, and most attention has been focused on IFNγ-producing CD4+ T helper type 1 (Th1) and CD8+ T effector lymphocytes that are critical for the resolution of acute illness and to prevent reactivation of latent infection (Denkers and Gazzinelli, 1998). In addition, activation of MyD88, an universal adaptor for all Toll-Like Receptors (TLRs) (except TLR3) (Gazzinelli and Denkers, 2006; Takeuchi and Akira, 2010), is essential for the optimal production of IL-10, IL-12, TNF-α:, and IFNγ, all of which are important mediators of host survival during primary infection with T. gondii (Scanga et al., 2002; Sukhumavasi et al., 2008). While the three latter cytokines (Gazzinelli et al., 1994; Suzuki, 1999) are critical to control parasite growth through activation of effector mechanisms such as inducible GTPases (Howard et al., 2011), IL-10 prevents an excessive inflammatory response that is lethal to the host (Gazzinelli et al., 1996).
As for the Pathogen Associated Molecular Patterns (PAMPs) that activate TLRs during T. gondii infection, important pieces of the puzzle are still missing. Several parasite products, including glycosylphosphoinositol (GPI) anchors, and heat-shock protein were shown to activate TLR2 and TLR4. Yet, mice lacking such TLRs have a rather mild or no phenotype upon T. gondii infection (Aosai et al., 2006; Debierre-Grockiego et al., 2007). Importantly, the T. gondii profilin-like protein (TgPRF) was shown to activate TLR11, and gene-target disruption of TLR11 results in a partial defect of IL-12 production and increased number of cysts in the brain from mice infected with T. gondii (Plattner et al., 2008; Yarovinsky et al., 2005). However, none of these mice recapitulate the profound phenotype observed in MyD88 Knockout (KO) mice infected with T. gondii (Melo et al., 2010; Scanga et al., 2002; Sukhumavasi et al., 2008) suggesting that other members of the TLR family are involved.
The “triple D” (3d) mouse express an UNC93B1 missense mutant that is incapable to bind the nucleic-acid sensing (NAS) -TLRs (i.e., TLR3, TLR7 and TLR9) (Brinkmann et al., 2007; Tabeta et al., 2006), and therefore, to mediate their translocation from the endoplasmic reticulum (ER) and consequent activation into the endolysosomes (Kim et al., 2008). We have shown that 3d mice are highly susceptible to infection with T. gondii, presenting a profound impairment of IL-12 and consequent delay in IFNγ production (Melo et al., 2010). In the current study, we further defined the role of endosomal TLRs during infection with T. gondii. Our data indicate that TLR7 and TLR9 recognize Toxoplasma DNA and RNA, respectively. On the other hand, we found that both TLR11 and TLR12 are required for UNC93B1-dependent cellular responses to TgPRF. We also report that the triple TLR7/TLR9/TLR11 deficient mice are highly susceptible to T. gondii infection, recapitulating the phenotype of 3d mice. It is noteworthy that while the mouse genome encodes 13 TLRs, the human genome lacks functional TLR11, TLR12 and TLR13 (Roach et al., 2005). Consistently, human cells produce high levels of pro-inflammatory cytokines, including IL-12 and TNF-α in response to parasite derived RNA and DNA, but not to TgPRF. Hence, our results support the hypothesis that NAS-TLRs play an important role in human toxoplasmosis.
RESULTS
RNA and DNA derived from T. gondii tachyzoites activate host cells via TLRs
NAS-TLRs are important cognate receptors for viruses, bacteria, and different protozoan parasites (Alexopoulou et al., 2001; Bartholomeu et al., 2008; Benson et al., 2009; Caetano et al., 2011; Heil et al., 2004; Hemmi et al., 2000; Parroche et al., 2007). Here, we exposed immortalized macrophages expressing transgenic TLR9-GFP with Red CMTPX-labeled tachyzoites. TLR9-GFP co-localized with intracellular parasites in the endolysosomal compartment, but not in the parasitophorous vacuole (PV), which is lysotracker negative (Figure 1A). We demonstate that RNA (Figure 1B) and DNA (Figure 1C) extracted from highly purified tachyzoites stimulate immortalized macrophages or DCs, in a TLR7- or TLR9-dependent manner, respectively. Supplementary Table 1 (available online) list a series of (total number of 92) immunostimulatory mouse B-class-like CpG motifs encoded in the Toxoplasma genome. They are potent activators of DCs via TLR9 (Figure 1D).
Figure 1. T. gondii RNA and DNA activate host cells via TLRs.

(A) Confocal microscopy of immortalized TLR9−/− macrophages stably expressing TLR9-GFP and infected with CMTPX-stained T. gondii. Acidic compartments were stained with LysoTracker White-Blue. Arrows indicate internalized parasites. (B) Immortalized WT, TLR7 KO and TLR9 KO macrophages were stimulated with T.gondii RNA at 2 μg/ml complexed with DOTAP (Roche). (C) DCs were stimulated with T. gondii DNA complexed with DOTAP at 10, 5, 1 and 0.1 μg/ml. (D) DCs were stimulated with T. gondii derived oligonucleotides containing B-class mouse-like stimulatory CpG motifs at 3 mM (black circles), 1 mM (dark grey), 0.3 mM (light grey) and 0.1 mM (white circles). Cytokine levels were measured in the tissue culture supernatants at 24hrs after stimulation. (B-D) Data are represented as mean ± SD of three independent experiments. (*0.01 < p < 0.05, **0.001 < p < 0.01, ***p < 0.001). See also Supplementary Table1.
Combined deficiency of NAS-TLRs does not recapitulate the high susceptibility of 3d mice infected with T. gondii.
We found that none of the single TLR3, TLR7 or TLR9 KO was more susceptible to infection with T. gondii (Melo et al., 2010). Thus, we hypothesize that a combined deficiency of NAS-TLRs would explain the dramatic phenotype observed in the 3d mice. However, none of the double TLR3/TLR7, TLR7/TLR9, TLR7/TLR8, or even the triple TLR3/TLR7/TLR9-deficient mice had an impaired IL-12 and IFNγ production (Figure 2A and data not shown), or was highly susceptible to T. gondii infection. Despite this apparently normal IL-12/IFN-γ response, few of the triple TLR3/TLR7/TLR9 KO mice succumbed during early stage of infection (Figure 2B), which was associated with an increased parasitism in peritoneal cells and spleens (Figure 2C), as well as in the brain (Figure 2D).
Figure 2. Mice deficient in TLR3/TLR7/TLR9 are only partially susceptible to T. gondii infection.

Mice were infected intraperitoneally with 25 cysts of T. gondii ME49 strain. (A) Levels of IL-12p40 and IFNγ were measured in the peritoneal cavity exudate and sera at different times post-infection. (B) Combined survival data from WT (n=20), 3d (n=16) TLR7/9 (n=20) and TLR3/7/9 (n=20) mice from 4 independent experiments. (C) Quantitative real-time PCR analysis was performed on the indicated tissues collected from animals infected with T. gondii. Data are mean of three independent experiments. (D) Cysts counts in the brain determined at 30 days post-infection are the mean from four experiments. (A–D) Data are represented as mean ± SD. Asterisks indicate that difference is statistically significant, when comparing different mouse lineages infected with T. gondii, (NS: not significant, *0.01 < p < 0.05, **0.001 < p < 0.01, ***p < 0.001).
Expression of endosomal TLRs in DCs from mice infected with T. gondii.
Because resistance to experimental infection with T. gondii was only slightly affected in the TLR3/TLR7/TLR9 KO mice, we investigated the role of other TLRs, whose function is also dependent on UNC93B1. We generated a molecular tree of mouse TLRs (Phylogeny.fr), which indicated that TLR12 is closely associated to TLR11 and TLR13, that are also endosomal TLRs (Shi et al., 2011; Zhang et al., 2004) (Figure S1 available online). Amino acid sequence alignment for TLR11 and TLR12 show high degree of (35%) sequence identity, suggesting the possibility that both of them may recognize TgPRF.
As both macrophages and DCs are important sources of IL-12 during T. gondii infection in mice, we evaluated the expression of the endosomal TLRs mRNAs in CD11b+, CD11c+/CD8−, and CD11c+/CD8+ cells. As shown in Figure 3A, mRNA of TLR3, TLR9, TLR11 and TLR12 were expressed in higher levels in the CD11c+/CD8+ cells, being consistent with the hypothesis that this DC subset is the main source of IL-12 in mice infected with T. gondii (Mashayekhi et al., 2011; Yarovinsky et al., 2005). However, a recent study (Goldszmid et al., 2012) indicate that the main IL-12 source in the peritoneal cavity of infected mice are the CD11c+CD8− DCs, which we found to express high levels of TLR3, TLR7 and TLR9, and minimal levels of TLR11 or TLR12. Thus, we speculate that IL-12 production by CD11c+CD8+ DCs and CD11c+CD8− DCs is triggered by parasite TgPRF and nucleic acids, respectively. Macrophages also expressed mRNA for the various endosomal TLRs, but in a lesser amount than DCs.
Figure 3. Endosomal TLRs are highly expressed in CD8α + DCs and upregulated upon T. gondii infection.

(A) Real-time PCR was performed to determine the relative levels of TLR3, TLR7, TLR9, TLR11 and TLR12 mRNA expressed by CD11b+, CD11c+/CD8α+ and CD11c+/CD8α − cells sorted from splenocytes from uninfected controls as well as infected (5 days post-infection) WT mice. TLR mRNA levels were normalized to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Data are represented as mean ±SD of three experiments. (B) HEK 293T cells were transfected with different pairs of plasmids, total lysates immunoprecipitated (IP) with anti-hemagglutinin (anti-HA, top) or anti-Flag (bottom), and analyzed by immunoblot (IB) with anti-Flag (top and bottom). The top membrane was then stripped and re-probed with anti-HA to ensure expression of hemagglutinin-tagged TLR11 (middle). (C) CD11c+ cells were purified from spleen of WT, 3d, TLR7/TLR9, TLR3/TLR7/TLR9 and TLR11 KO mice and stimulated with LPS (100 ng/ml) ODN CpG 1826 (1 μm), R848 (2 μm) or infected with ME49 tachyzoites (MOI 3:1). (D) CD11c+ cells purified from WT, TLR4, TLR7/TLR9 and TLR12 KO mice were stimulated with LPS (100 ng/ml), ODN CpG 1826 (1 μm), STAg (10 μg/ml), or rTgPRF (10 ng/ml) left untreated, or treated with DNAse (100 U/ml), RNAse (10 μg/ml) or Proteinase K (10 μg/ml). IL-12 levels were measured in the supernatant at 24 h after stimulation. (C–D) Data are represented as mean ± SD of four experiments. Asterisks indicate that difference is statistically significant, when comparing cytokines levels from WT to different KO mice, infected or not infected with T. gondii (*0.01 < p < 0.05, **0.001 < p < 0.01, ***p < 0.001). See also Figure S1.
To evaluate whether TLR11 and TLR12 are associated with other endosomal TLRs, HEK 293T cells were transiently co-transfected with different Flag tagged TLRs and TLR11-HA, and TLR11 immunoprecipitated as bait. As shown (Figure 3B), TLR3, TLR7, TLR11, and TLR12, but not TLR4, co-immunoprecipitated with TLR11. UNC93B1 has been reported to physically interact with TLR3, TLR7, TLR9, TLR11 and TLR13 (Brinkmann et al., 2007; Kim et al., 2008; Pifer et al., 2011; Tabeta et al., 2006). Thus, the interpretation of these results is that when pulling-down TLR11, co-immunoprecipitation of TLR3, TLR7, TLR11, and TLR12 is observed because all the endosomal TLRs are bound to UNC93B1.
Next, we purified CD11c+ cells from the spleens of 3d as well as double TLR7/TLR9, triple TLR3/TLR7/TLR9 and single TLR11 KO mice. DCs from 3d mice were responsive to LPS, but not to R848 (TLR7 agonist), CpG ODN (TLR9 agonist), STAg, ME49, or recombinant TgPRF (rTgPRF). The lack of TLR11, but not TLR3/TLR7/TLR9 had a major impact on IL-12 production by splenic DCs exposed to STAg, ME49 or rTgPRF. Nevertheless, compared to rTgPRF, DCs from TLR11 KO or TLR12 KO mice still produced significant amounts of IL-12 when exposed to either STAg or ME-49 (Figures 3C and 3D). As expected, DCs from TLR3/TLR7/TLR9 KOs did not respond to R848 or CpG ODN, whereas DCs from TLR11 KOs produced high levels of IL-12 in response to these TLR agonists (Figure 3C). Treatment with proteinase K, but not with RNAse or DNAse, distroyed the ability of rTgPRF to activate CD11c+ cells (Figure 3D).
Co-localization and heterodimerization of TLR11 and TLR12
Macrophages were genetically engineered to stably express color-tagged TLR11 or TLR12 and used to analyze their subcellular distribution by confocal microscopy. We found that in macrophages, TLR11 and TLR12 co-localize with ER tracker, but not with cholera toxin (a cell surface membrane marker) (Figure 4A). We also transfected HEK 293T cells with different combinations of plasmids encoding fluorescent protein tagged-UNC93B1, -TLR11 or -TLR12 and they all co-localized in the ER (Figure 4B). As control, we transfected HEK 293T cells with a color tagged-TLR4. The pattern of cellular distribution for TLR4 was distinct from TLR11 and TLR12 (Figure 4B). As expected (Latz et al., 2004), TLR4 was primarily expressed at the surface membrane of the transfected cells.
Figure 4. Co-localization and heterodimerization of TLR11 and TLR12.
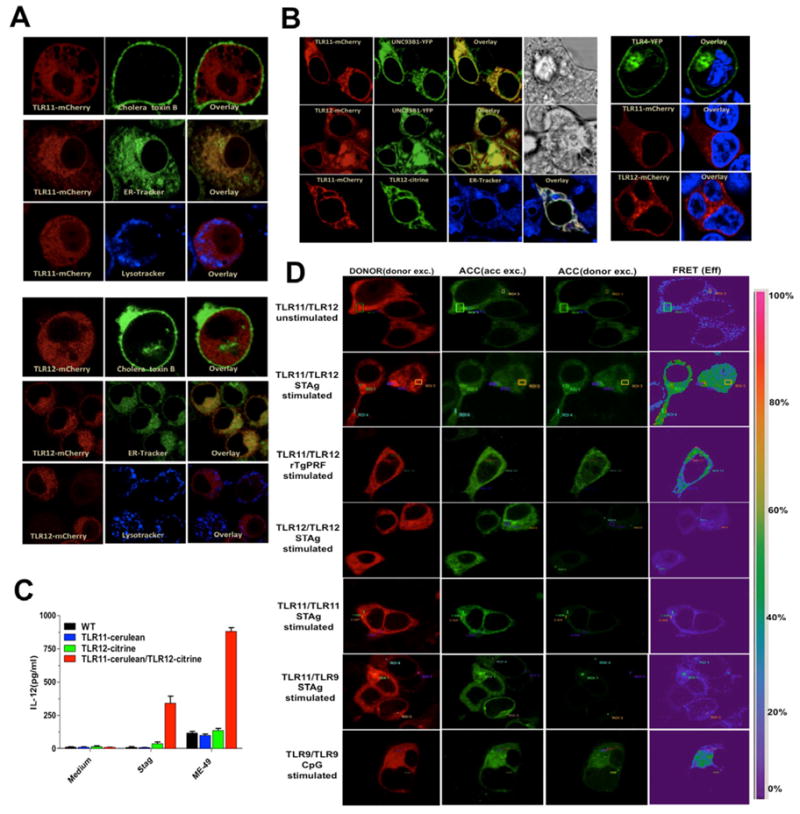
(A) Immortalized macrophages were stably transfected with TLR11- or TLR12-mcherry and imaged post-staining with Cholera toxin B subunit FITC conjugate, LysoTracker White-Blue or ER-Tracker White-Blue. (B) HEK 293T cells transfected with TLR11-mCherry or TLR12-mcherry with UNC93B1-YFP or TLR11-mcherry and TLR12-citrine (left panel) or with TLR4-YFP and stained with Hoechst 33342 as nuclear marker (right panel). (C) Immortalized macrophages stably expressing either TLR11, TLR12 or both were stimulated in vitro with STAg (10 μg/ml) or exposed to live tachyzoites (MOI 3:1) of the ME49 strain and levels of IL-12p40 measured in the supernatants at 24 h post-stimulation. Data are represented as mean ± SD of three experiments. (D) HEK 293T cells were transfected with the plasmids encoding the proteins indicated in the figure; 48 h after transfection, cells were left unstimulated or were stimulated with STAg (10 μg/ml), rTgPRF (100 ng/ml) or CpG 1826 (1 μM) for TLR9/TLR9. FRET between the respective proteins was calculated by measuring sensitized emission (SE) fluorescence using the FRET SE wizard on the Leica SP2 confocal laser-scanning microscope. For each plasmid combination, cerulean (represented in red) was used as donor and citrine (represented in green) as acceptor. Data are from one representative experiment of four.
Importantly, immortalized macrophages produced high levels of IL-12 in response to STAg or ME49 tachyzoites when they were stably transfected with both TLR11 and TLR12, but not with either TLR11 or TLR12 alone (Figure 4C). As dimerization appears to be required for PAMP recognition and activation of TLRs (Latz et al., 2007; Leonard et al., 2008), we used Fluorescence Resonance Energy Transfer (FRET) to evaluate the intermolecular distance between the TIR domains of TLRs. HEK 293T cells were transfected with different combinations of fluorescent TLRs fused to cerulean (Donor) or citrine (Acceptor) and protein-protein interaction evaluated. After STAg or rTgPRF stimulation, a strong FRET signal was observed in cells co-transfected with TLR11 and TLR12, but not with either TLR11/TLR11, TLR12/TLR12 or TLR11/TLR9 stimulated with STAg (Figure 4D) or rTgPRF (not shown). After stimulation with CpG, we observed a strong FRET signal for TLR9/TLR9 homodimers. Based on the results described above, we hypothesized that TLR11/TLR12 work as heterodimers and deficiency of either TLR11 or TLR12 results in impaired IL-12 production during T. gondii infection. Whether formation of TLR11 and TLR12 heterodimers is necessary for recognition of bacterial components (e.g., flagellin) by TLR11 (Mathur et al., 2012; Zhang et al., 2004) remains to be defined.
Quadruple TLR3/TLR7/TLR9/TLR11 KO mice are highly susceptible to T. gondii infection
Despite of a significant impairment on IL-12 response, TLR11 deficient mice still produced higher levels than the 3d mice, being sufficient to induce IFNγ (Figure 5A) and protect mice from death during acute phase of infection (Figure 5B). Nevertheless, TLR11 KOs showed a four-fold increase in cyst numbers (Figure 5). These data left us with the observation that although the NAS-TLRs as well as TLR11 are all required for optimal host responses to T. gondii, neither seemed essential for survival. Hence, we generated a triple TLR7/TLR9/TLR11 and a quadruple TLR3/TLR7/TLR9/TLR11 deficient mice. In contrast to DCs from TLR11 KO, CD11c+ cells purified from spleens of either TLR7/TLR9/TLR11 or TLR3/TLR7/TLR9/TLR11 KO mice did not respond to either STAg or live tachyzoites (ME49) (Figure 6A). As shown in (Figure 6B and S2A), the quadruple KO mice had a major defect in IL-12, IL-6 and MCP1 production, at similar levels to 3d mice. MCP1 is an important chemokyne for the recruitment of inflammatory monocytes and host resistance to T. gondii (Dunay et al., 2008). As presented in (Figure 2SB), by day 3 after infection, WT mice have an increased number of inflammatory monocytes, which was not observed in 3d or quadruple KO mice. The influx of inflammatory monocytes at 3 days post-challenge was partially reduced in TLR3/TLR7/TLR9 KO mice, and may explain the slight enhancement on susceptibility to infection (Figure 2). An impairment in IL-12 production by DCs and inflammatory monocytes was observed both in 3d and quadruple TLR3/TLR7/TLR9/TLR11 KO mice (Figure 6C), causing a delay in IFNγ production (Figure 6D). After challenge with ME49 strain, quadruple KOs were as susceptible as 3d mice, as indicated by the survival curve and increased parasite burden in peritoneal cells, spleen and liver (Figure 6E and 6F). We assume that the influx of neutrophils, inflammatory monocytes and DCs in the highly susceptible mutant/KO mice was too late, and thus, not able to control parasite replication (Figure S2B). Importantly, as shown in (Figure 6G), all 3d and TLR3/TLR7/TLR9/TLR11 KO mice treated with recombinant IL-12 (rIL-12) survived the experimental infection with T. gondii.
Figure 5. TLR11 mice are resistant to T. gondii infection.

Mice were infected intraperitoneally with 25 cysts ME49 strain of T. gondii. (A) Levels of IL-12p40 and IFNγ were measured in the peritoneal cavity exudate and sera from uninfected as well as infected mice. (B) Combined survival data from WT (n=15), 3d (n=12) and TLR11 (n=15) mice from three independent experiments. (C) The cyst numbers in the brain were counted at 30 days post-infection and presented as mean from the three experiments. (A–C) Data are represented as mean ± SD. Asterisks indicate that difference is statistically significant, when comparing to unstimulated controls (NS: not significant, *0.01 < p < 0.05, **0.001 < p < 0.01, and ***p < 0.001).
Figure 6. Quadruple deficient mice have an impaired IL-12 and early IFNγ production and are highly susceptible to T. gondii infection.

(A) CD11c+ cells were purified from spleen of WT, TLR7/TLR9/TLR11, TLR3/TLR7/TLR9 and TLR11 KO mice and stimulated with LPS (100 ng/ml), ODN CpG 1826 (1 μm), R848 (2 μm), STAg (10 μg/ml) or infected with ME49 tachyzoites (MOI 3:1). Data are represented as mean ± SD of two experiments. (B) Levels of IL-12p40 were measured in the peritoneal cavity exudate and sera from uninfected controls and infected mice. Data represent as mean ± SD of four experiments. (C) Mice infected with T. gondii were sacrificed at 5 days post-infection and peritoneal cells analyzed for IL-12 cellular source by intracellular cytokine staining. Dendritic cells (top) were gated for CD11c+/MHC-II+. Inflammatory monocytes (bottom) were gated first for GR1+ and then for CD11b+/F4/80+. Both populations were stained for IL-12p70. Data are from one representative experiment of three that yielded similar results. (D) Levels of IFNγ present in the peritoneal cavity exudate and sera from uninfected controls and infected mice. Data are represented as mean ± SD of four experiments. (E) Combined survival data from WT (n=15), 3d (n=10), TLR7/9/11 KO (n=10) and TLR3/7/9/11 KO (n=15) mice from at least two independent experiments. (F) Quantitative real-time PCR analysis was performed on the indicated tissues collected from animals infected with T. gondii. Data are represented as mean ± SD of three independent experiments. (G) WT, 3d and TLR3/TLR7/TLR9/TLR11 KO were infected i.p. with the ME49 strain of T. gondii (n=5 per group). Mice were treated with 100 ng of recombinant IL-12p70 or vehicle for 6 consecutive days, and mortality was evaluated. Data are from one representative experiment of two that yielded identical results. Asterisks indicate that difference is statistically significant, when comparing different mouse lineages infected with T. gondii. (**0.001 < p < 0.01, and ***p < 0.001). See also Figure S2.
We also generated the triple TLR7/TLR9/TLR11 deficient mice, which was as susceptible as the 3d and quadruple KO mice infected with T. gondii (Figure 6D). As TRIF and Type I IFN are, respectively, the main adaptor molecule and outcome of TLR3 activation we performed experimental infections in TLR3 KO, TRIF KO and Type I IFN Receptor KO mice. Our results show that when compared to WT mice, none of these mice displayed enhanced susceptibility to T. gondii infection (Figures 2SC and 2SD). Consistently, we were unable to detect any increase in the production of IFN-α protein in peritoneal fluids, splenocyte cultures or sera from WT mice infected with T. gondii (data not shown).
Human peripheral blood mononuclear cells (PBMCs) produce high levels of IL-12 in response to T. gondii DNA and RNA, but not rTgPRF
TLR11 and TLR12 are not expressed in human cells (Roach et al., 2005; Zhang et al., 2004). This raised the hypothesis that NAS-TLRs are the key TLRs sensing T. gondii parasites in human cells. The results presented in Figure 7A indicate that when compared to R848 (agonist for human TLR8) and LPS (agonist for TLR4), STAg is a poor stimulator of IL-12p70, TNF-α and IL-1β production by human PBMCs. Furthermore, we were unable to detect production of any cytokine (including IL-12p70) in response to rTgPRF (Figure 7B). Nevertheless, we found that parasite RNA and DNA elicited the production of pro-inflammatory cytokines, including TNF-α, IL-12p40 and IL-12p70, which was more pronounced when PBMCs were primed with IFNγ (Figure 7B). We also searched for immunostimulatory CpG motifs (Supplementary Table 2) that activate human TLR9. In the whole genome, we found 363 of human B-class CpG motifs and 67 of human C-class motifs. Synthetic B and C class-like immunostimulatory oligonucleotides derived from T. gondii genome induced NF-κB activation in HEK cells (Figure 7C), as well as pro-inflammatory cytokines, and in special IL-12p40/IL-12p70, when human PBMCs were primed with IFNγ (Figure 7D).
Figure 7. DNA and RNA from T. gondii activate Human Peripheral Blood Mononuclear Cells (PBMCs).

(A) PBMCs purified from blood of clinically healthy donors were stimulated in vitro with STAg (10 μg/ml), CpG ODN 2007 (1 μM), R848 (2 μM) or LPS (100 ng/ml). Data are represented as mean ± SD. (B) PBMCs primed or not with recombinant human IFNγ (200 U/ml) were stimulated with LPS (100 ng/ml), STAg (10 μg/ml), ME49 RNA (2 μg/ml), ME49 DNA (5 μg/ml), or rTgPRF (1 μg/ml). Parasite RNA and DNA were complexed with DOTAP (Roche). (C) HEK 293T cells were stimulated with T. gondii derived oligonucleotides containing B or C-class human-like stimulatory CpG motifs at 3 μM (black circles), 1 mM (dark grey), 0.3 mM (light grey) and 0.1 mM (white circles). Asterisks indicate that differences are statistically significant when comparing stimulated cells with negative control - ODN 2007GC (top panel) or unstimulated cells (bottom panel). (D) PBMCs primed or not with recombinant human IFNγ (200 U/ml) were stimulated in vitro with T. gondii derived oligonucleotides containing B and C-class human-like stimulatory CpG motifs (5 mM). Cytokine levels were measured in the supernatants at 18 h after stimulation. (B-D) Data are represented as mean ± SD of three independent experiments (*0.01 < p < 0.05, **0.001 < p < 0.01, and ***p < 0.001). See also Supplementary Table 2.
DISCUSSION
We hypothesized that a combined deficiency of NAS-TLRs was responsible for the observed phenotype of 3d mice infected with T. gondii. Our current report indicate that the extreme susceptibility of 3d mice is due to a combined defect on endosomal TLRs, but not simply a deficiency of the NAS-TLRs, also including TLR11 and TLR12. The combined deficiency of endosomal TLRs results in a profound impairment of IL-12 production by both dendritic cells and macrophages, and a subsequent reduction in the levels of IFNγ.
In our previous study (Melo et al., 2010), we suggested, as an alternative mechanism, that UNC93B1 was directly mediating control of tachyzoite replication in the PV. However, induction and translocation of iGTPases to the PV are normal, and IFNγ-activated macrophages from 3d mice effectively controlled parasite replication (Howard et al., 2011). In addition, macrophages lacking functional TLRs, including NAS-TLRs, are not more permissive to parasite growth in vitro. To test this hypothesis in vivo, we generated mixed chimeras and observed that in vivo parasite replication was equal in cells from WT and 3d mice (data not shown). UNC93B1 is also involved in antigen cross presentation (Tabeta et al., 2006), but we have no indication that activation of CD8+ T lymphocytes is impaired in 3d mice infected with intracellular protozoan parasites (Caetano et al., 2011; Melo et al., 2010). Hence, we propose that the main cause of the enhanced susceptibility of 3d mice to T. gondii infection is the defective activation of endosomal TLRs, impaired IL-12 production and inadequate development of protective immunity.
As observed for other intracellular protozoan parasites (Bartholomeu et al., 2008; Caetano et al., 2011; Parroche et al., 2007) we report that Toxoplasma derived DNA and RNA induces the production of pro-inflammatory cytokines by macrophages and DCs. However, the triple TLR3/TLR7/TLR9 KO mice are only slightly more susceptible to infection with T. gondii. In addition, mice deficient in TLR11, which is activated by TgPRF in an UNC93B1-dependent manner (Pifer et al., 2011), survive the acute infection with T. gondii (Yarovinsky et al., 2005). This complicated scenario convinced us to generate the quadruple TLR3/TLR7/TLR9/TLR11 and triple TLR7/TLR9/TLR11 KO mice that upon infection with T. gondii recapitulated the 3d phenotype, resulting in a profound impairment of the IL-12/IFN-γ axis, and unhampered parasite growth.
Importantly, two studies reported that TLR9 is partially responsible for the development of an optimal anti-parasite Th1 response and intestinal pathology, when mice are per-orally infected with cysts of the ME49 strain of T. gondii (Benson et al., 2009; Minns et al., 2006). The interpretation of these results is that DNA of the intestinal microflora serves as a natural adjuvant for mucosal immunity (Hall et al., 2008). However, the adjuvant effect of the intestinal microflora is not observed, when mice are infected intraperitoneally (Benson et al., 2009). Since our main interest was to identify the innate immune receptors for T. gondii, we used the intraperitoneal infection to avoid the interference of the gut microbiota. Hence, collectively, our results indicate that in addition to TLR11, TLR7 and TLR9 are also important sensors of Toxoplasma tachyzoites.
The high similarity of TLR11 and TLR12 found in a phylogenetic analysis, lead us to query if they had similar function, as previously observed for TLR1, TLR2 and TLR6 (Roach et al., 2005; Triantafilou et al., 2006). Indeed, TLR11 and TLR12 co-localized with UNC93B1 in transfected HEK 293T cells, and FRET between TLR11/TLR12 was induced upon activation with Toxoplasma extracts or rTgPRF. This result was not observed when the TLRs were expressed alone, suggesting that homodimerization does not occur. Together, our biochemical, cellular and immunological assays strongly suggest that both TLR11 and TLR12 are endosomal TLRs and act as heterodimers in the recognition of Toxoplasma molecules. Thus, we propose that in mice the primary role of UNC93B1 in host resistance to T. gondii infection is to mediate translocation and function of TLR7, TLR9, TLR11 and TLR12.
The endosomal localization of TLR11 and TLR12, in contrast to the surface membrane TLRs that recognize cell wall components of bacteria (i.e., TLR1, TLR2, TLR4, TLR5, TLR6), is an important finding. The current report further emphasizes that despite the presence of ligands for surface membrane TLRs (Gazzinelli and Denkers, 2006), endosomal TLRs are the critical ones for the in vivo sensing of intracellular protozoan parasites. The protozoan ligands that are recognized by endosomal TLRs appear to reside inside the protozoan parasite: DNA, RNA and TgPRF, all of which are released from the parasite when it is killed in the phagolysosome. As shown for TLR9, it is reasonable to speculate that both TLR11 and TLR12 translocate from the ER and recognize the parasite components in the endolysosomes and not in the PV, which avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways (Mordue et al., 1999). Whereas activation of DCs by TgPRF, STAg or live tachyzoites is mediated by UNC93B1, additional experiments are necessary to confirm this hypothesis.
Another intriguing aspect of the TLR11 and TLR12 biology is their specie specificity. TLR11 in humans is a pseudogene (Zhang et al., 2004), whereas TLR12 is not present in the human genome. As mice are the natural intermediate host and highly exposed to T. gondii infection, we speculate that TLR11 was not only maintained as a functional receptor, but also duplicated as an important mechanism of host resistance to infection. Although TLR11 and TLR12 are not involved in recognition of T. gondii by human cells, one can imagine that they may play an indirect role in human disease, because of the role that the mouse has in transmitting the parasite to cats, which can then transmit to humans.
It is worth mentioning that in mice TLR8 has no known ligands, whereas TLR7 is widely expressed and functions as a receptor for ssRNA (Heil et al., 2004). Complicating, human mDCs and monocytes respond to ssRNA via TLR8. Human TLR7 is active, however, in pDCs (which do not express TLR8) response to RNA viruses (Forsbach et al., 2008). Thus, in consideration of the lack of expression of TLRs 11 and 12 in humans, these data led us to speculate that TLR7, TLR8 and TLR9 are the key TLRs in human toxoplasmosis. Importantly, human PBMCs from uninfected healthy donors produce significant levels of IL-1β and TNF-α when stimulated with parasite-derived DNA and RNA, as well as oligonucleotides containing CpG motifs derived from Toxoplasma genome. Furthermore, when primed with IFNγ, human PBMCs produced high levels of IL-12p40/70 upon stimulation with parasite-derived nucleic acids, but not with STAg or rTgPRF. The precedent for the importance of IFNγ priming in the production of IL-12 also exist in the rodent model of toxoplasmosis (Gazzinelli et al., 1994; Goldszmid et al., 2012).
Finally, despite of being considered an “accidental” intermediate host, one third of the human population in the world is chronically infected with T. gondii (Robert-Gangneux and Darde, 2012). Although, one can imagine the evolutionary pressures that gave rise to TLR11 in mice, it remains unclear why it was downgraded to a non-coding gene in humans. Hence, our findings have important implications for human disease. One has to assume that alternative TLRs are responsible for detecting the parasite, triggering innate immunity and initiating acquired immunity during acute toxoplasmosis in humans. The data presented here show that human PBMCs produce high levels of pro-inflammatory cytokines, including IL-12, in response to T. gondii RNA and DNA, but not rTgPRF. Finally, a recent study reports an association of a TLR9 single nucleotide polymorphism and development of ocular toxoplasmosis (Peixoto-Rangel et al., 2009). Hence, we propose that NAS-TLRs as well as the signaling pathways they activate, are important determinants of resistance to infection and the clinical outcome of human toxoplasmosis.
EXPERIMENTAL PROCEDURES
Ethics Statement
All experiments involving animals were performed in accordance with guidelines set forth by the American Association for Laboratory Animal Science (AALAS), and approved by the Institutional Animal Care and Use Committee (IACUC A-1817) at the University of Massachusetts Medical School (UMASSMED). The protocols and consent forms for experiments with human PBMCs were approved by the Institutional Research Board from UMASSMED (IRB-UMMS H-12328) and from Centro de Pesquisas René Rachou – Fundação Oswaldo Cruz (CEP-CPqRR 11/2006), as well as by the National Ethical Committee (CONEP 13.368) from Ministry of Health in Brazil.
Reagents
All cell culture reagents were obtained from Mediatech. LPS derived from Escherichia coli strain 0111:B4 was purchased from Sigma and re-extracted by phenol chloroform to remove lipopeptides as described (Hirschfeld et al., 2000). R848, a synthetic small molecule agonist for TLR7 was provided by 3M Pharmaceuticals. Phosphorothioate-stabilized unmethylated DNA oligonucleotide-bearing CpG ODN 1826, ODN 2007, ODN 2395, as well as ODNs containing CpG motifs identified in T. gondii genome and qPCR primers were obtained from Integrated DNA Technologies. Purified rTgPRF (Skillman et al., 2012) was kindly provided by Dr. David Sibley (Washington University, St. Louis, MI). Mouse and Human Recombinant Interferon-γ and rIL-12p70 were purchased from eBioscience. ER-Tracker Blue-White dpx, Lysotracker Blue-White dpx, and Cell tracker Red CPMTX were obtained from Molecular Probes. Cholera Toxin B Subunit FITC conjugate from Sigma. Hoechst 33342 to stain the nucleus was bought from Thermo Scientific. Proteinase K was obtained from Ambion and DNAse and RNase from Promega.
Mice and Parasites
C57BL/6 mice were obtained from The Jackson Laboratory. UNC93B1 mutant (3d) mice were kindly provided by Bruce Beutler at The Scripps Research Institute, La Jolla, CA (Tabeta et al., 2006). The single TLR3, TLR7, TLR9, and TLR11 KO mice were kindly provided by Shizuo Akira (Osaka University, Japan) and Richard Flavell (Yale University, Yale, CN), respectively. TLR12 KO mice were provided by Sankar Ghosh (Columbia University, New York, NY). Interbreeding single KO animals generated TLR3/7, TLR7/9, TLR7/8, TLR3/7/9, TLR7/9/11 and TLR3/7/9/11 deficient mice. Age matched (6–8 weeks old) and female groups of WT and KO mice were used in all experiments. The ME49 strain was maintained in C57BL/6 mice by serial inoculation of brain homogenate containing cysts. ME49 tachyzoites were maintained in human foreskin fibroblast cells (Hs27) (Lock, 1953) and used to prepare STAg as previously described (Melo et al., 2010). DNA and RNA were extracted by employing DNeasy Blood and Tissue kit and RNesy Mini Kit from Qiagen.
Genome-wide scanning for TLR9 stimulatory sequences
Both mouse B-like class and human-like B and C-class CpG motifs were searched in the DNA strands of the assembled contigs of T. gondii ME49 genome downloaded from the ToxoDB website (http://toxodb.org/common/downloads/), as previously described (Bartholomeu et al., 2008).
Cell purification
CD11c+ cells were purified using the EasySep® mouse CD11c positive selection kit according to manufacture’s protocol. In some experiments, CD11c+ cells, purified in magnetic beads, were stained with anti-CD8APC and sorted in a FACSAria for PE (CD11c+) or PE/APC (CD11c+/CD8+). Cell purity was checked by FACS.
Measurement cytokine levels
The levels of mouse TNF-α, IFN-γ, IL-12, IL-6 and MCP1 were measured by using DuoSet ELISA kits (R&D Systems). IL- 12p70, IL-1β and TNF-α levels were measured in PBMC culture supernatants by using the Cytometric Bead Array (CBA) Human Inflammatory Cytokines Kit (BD Bioscience) or ELISA for IL-12p40 (R&D Systems).
Flow cytometry
Cells were stained with conjugated antibodies against the surface markers CD11b, CD11c, GR1, F4/80 and MHC-II (eBioscience). For intracellular measurement of cytokines, cells cultivated for 8h in presence of GolgiPlug (BD Bioscience), surface stained, fixed with 4% formaldehyde, permeabilized with PBS + Tween®20 0.5% and incubated with Phycoerythrina anti-IL-12p70 (BD Bioscience). Subsequently cells were washed and analyzed by flow cytometry in an LSRII cytometer (BD Bioscience).
Quantitative real-time PCR
Total DNA from peritoneal exudate cells, spleen and liver was used for amplification of T. gondii B1 gene (Melo et al., 2010). Relative quantification was performed using standard curve analysis of purified parasite DNA. For TLRs expression, the following primers were used: TLR3 Forward: 5’-ATAAAATCCTTGCGTTGCGAAGT -3’; TLR3 Reverse: 5’-TGTTCAAGAGGAGGGCGAATAA–3’; TLR7 Forward: 5’-TCTTTGGGTTTCGATGGTTTCC-3’; TLR7 Reverse: 5’-GCAGTCCACGATCACATGGG –3’; TLR9 Forward: 5’-ACGGGAACTGCTACTACAAGA-3’; TLR9 Reverse: 5’-CCCAGCTTGACAATGAGGTTAT–3’; TLR11 Forward:5’-AGAGCTGGCTGGTATGTTCC-3’; TLR11 Reverse: 5’-GTGTTCTTGTCAGGTCCAGAATC-3;’ TLR12 Forward: 5’-CCAGGACTGCACCTTTTGG-3’; TLR12 Reverse: 5’–GTGACACTGGTTGTACGCAAT -3’.
Immunoprecipitation
HEK 293T cells were co-transfected with bait and prey constructs. Forty-eight hours post-transfection, cells were lysed in Ripa lysis buffer (Sigma) with Complete Mini Protease Inhibitors (Roche). Equal amount of cell lysate were used for immunoprecipitation with monoclonal anti-HA (Sigma) bound to Protein A Agarose (Invitrogen). Eluted proteins were electrophoresed by SDS-PAGE, transferred to nitrocellulose (BIO-RAD), and blotted using monoclonal antibody anti-HA or anti-Flag (Sigma).
Plasmid construction and viral transduction
TLR11 and TLR12 from InvivoGen plasmids were cloned into mCherry, Cerulean and Citrine-tagged vectors that were modified from the original pCLXSN backbone from Imgenex. All tags were fused in the C-terminal portion of the TLRs. Recombinant retroviruses were produced as previously described (Mann et al., 1983; Melo et al., 2010).
Confocal microscopy
We used an inverted Leica LSM TSC SP2 AOBS microscopy and a 1.4 NA 63x plan apochromat objective (Zeiss). Cells were cultured on glass-bottom 35-mm tissue-culture dishes (Matek), and dual or triple color images acquired by consecutive scanning. For Fluorescence Energy Resonance Transfer (FRET) experiments, HEK293T cells were transiently transfected with the indicated plasmids and 48 h later stimulated for 4 h with STAg or rTgPRF, or for 1h with CpG 1826. FRET between the respective proteins were calculated by measuring sensitized emission (SE) fluorescence using the FRET SE wizard on the Leica SP2 confocal laser-scanning microscope. In each case, cerulean-tagged protein acted as the donor fluorophore whereas the citrine-tagged protein functioned as the acceptor fluorophore. Excitation wavelengths for the donor and acceptor were 405 nM and 514 nM, respectively. The FRET efficiency is shown as a color-coded scale of values between 0 and 100%.
Experiments with human Peripheral Blood Mononuclear Cells (PBMCs)
PBMCs were enriched on-site by gradient centrifugation over Ficoll-Paque™ plus (GE-Healthcare). Cells were plated at 3 x105 cells/well, and cytokines measured 18 h post-stimulation with STAg, rTgPRF, CpG oligonucleotides, R848, LPS, ME49 DNA or RNA .
Luciferase Assay
HEK293 cells stably expressing human TLR9 and a luciferase gene under the control of the pELAM promoter containing NF-κB sites were used for testing the activity of T. gondii derived ODNs. We also used pRL vector expressing a Renilla luciferase gene (Promega) for constitutive protein expression, and assays revealed by Dual luciferase reporter assay system (Promega) substrate (Bartholomeu et al., 2008; Latz et al., 2007).
Statistical Analysis
All data were analysed using an unpaired, two-tailed S tudent’s t test with a 95% confidence interval (Prism; GraphPad Software, Inc.). All data are represented as means ± SD.
Supplementary Material
T. gondii DNA and RNA activate innate immune cells via Nucleic Acid Sensing TLR7 and 9
TLR11 and TLR12 heterodimers are required for cellular responses to T. gondii profilin
TLR7/TLR9/TLR11 triple deficient mice are highly susceptible to T. gondii infection
Human cells lack TLR11/12 and respond to T. gondii DNA and RNA, but not Tg profilin
Acknowledgments
The authors would like to thank A. Cerny for animal husbandry and genotyping, as well as A. Sher and D. Jankovic for discussions during the development of this study. We are also grateful to D. Sibley for providing rTgPRF. This work was supported by the National Institute of Health (NIAID - R01 AI071319-01), and the National Institute of Science and Technology for Vaccines (INCTV/CNPq/FAPEMIG). WAA and ERM were supported by fellowships from CNPq (Brazil) and DGAPA (Mexico), respectivelly.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Aosai F, Rodriguez Pena MS, Mun HS, Fang H, Mitsunaga T, Norose K, Kang HK, Bae YS, Yano A. Toxoplasma gondii-derived heat shock protein 70 stimulates maturation of murine bone marrow-derived dendritic cells via Toll-like receptor 4. Cell Stress Chaperones. 2006;11:13–22. doi: 10.1379/CSC-138R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeu DC, Ropert C, Melo MB, Parroche P, Junqueira CF, Teixeira SM, Sirois C, Kasperkovitz P, Knetter CF, Lien E, et al. Recruitment and endo-lysosomal activation of TLR9 in dendritic cells infected with Trypanosoma cruzi. J Immunol. 2008;181:1333–1344. doi: 10.4049/jimmunol.181.2.1333. [DOI] [PubMed] [Google Scholar]
- Benson A, Pifer R, Behrendt CL, Hooper LV, Yarovinsky F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe. 2009;6:187–196. doi: 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano BC, Carmo BB, Melo MB, Cerny A, dos Santos SL, Bartholomeu DC, Golenbock DT, Gazzinelli RT. Requirement of UNC93B1 reveals a critical role for TLR7 in host resistance to primary infection with Trypanosoma cruzi. J Immunol. 2011;187:1903–1911. doi: 10.4049/jimmunol.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debierre-Grockiego F, Campos MA, Azzouz N, Schmidt J, Bieker U, Resende MG, Mansur DS, Weingart R, Schmidt RR, Golenbock DT, et al. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J Immunol. 2007;179:1129–1137. doi: 10.4049/jimmunol.179.2.1129. [DOI] [PubMed] [Google Scholar]
- Denkers EY, Gazzinelli RT. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev. 1998;11:569–588. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, Sibley LD. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsbach A, Nemorin JG, Montino C, Muller C, Samulowitz U, Vicari AP, Jurk M, Mutwiri GK, Krieg AM, Lipford GB, et al. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J Immunol. 2008;180:3729–3738. doi: 10.4049/jimmunol.180.6.3729. [DOI] [PubMed] [Google Scholar]
- Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol. 2006;6:895–906. doi: 10.1038/nri1978. [DOI] [PubMed] [Google Scholar]
- Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri G, Sher A. Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J Immunol. 1994;153:2533–2543. [PubMed] [Google Scholar]
- Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, Kelsall B, Trinchieri G, Sher A. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity. 2012;36:1047–1059. doi: 10.1016/j.immuni.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, Zhu Q, Grigg ME, Berzofsky JA, Belkaid Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–649. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- Howard JC, Hunn JP, Steinfeldt T. The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii. Curr Opin Microbiol. 2011;14:414–421. doi: 10.1016/j.mib.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–238. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, et al. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–779. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- Leonard JN, Ghirlando R, Askins J, Bell JK, Margulies DH, Davies DR, Segal DM. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc Natl Acad Sci U S A. 2008;105:258–263. doi: 10.1073/pnas.0710779105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock JA. Cultivation of Toxoplasma gondii in tissue culture in mammalian cells. Lancet. 1953;1:324–325. doi: 10.1016/s0140-6736(53)90996-9. [DOI] [PubMed] [Google Scholar]
- Mann R, Mulligan RC, Baltimore D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell. 1983;33:153–159. doi: 10.1016/0092-8674(83)90344-6. [DOI] [PubMed] [Google Scholar]
- Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, Sher A, Ploegh HL, Murphy TL, Sibley LD, et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. 2011;35:249–259. doi: 10.1016/j.immuni.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur R, Oh H, Zhang D, Park SG, Seo J, Koblansky A, Hayden MS, Ghosh S. A mouse model of salmonella typhi infection. Cell. 2012;151:590–602. doi: 10.1016/j.cell.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo MB, Kasperkovitz P, Cerny A, Konen-Waisman S, Kurt-Jones EA, Lien E, Beutler B, Howard JC, Golenbock DT, Gazzinelli RT. UNC93B1 mediates host resistance to infection with Toxoplasma gondii. PLoS Pathog. 2010;6:e1001071. doi: 10.1371/journal.ppat.1001071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minns LA, Menard LC, Foureau DM, Darche S, Ronet C, Mielcarz DW, Buzoni-Gatel D, Kasper LH. TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J Immunol. 2006;176:7589–7597. doi: 10.4049/jimmunol.176.12.7589. [DOI] [PubMed] [Google Scholar]
- Mordue DG, Hakansson S, Niesman I, Sibley LD. Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways. Exp Parasitol. 1999;92:87–99. doi: 10.1006/expr.1999.4412. [DOI] [PubMed] [Google Scholar]
- Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A. 2007;104:1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto-Rangel AL, Miller EN, Castellucci L, Jamieson SE, Peixe RG, de Elias LS, Correa-Oliveira R, Bahia-Oliveira LM, Blackwell JM. Candidate gene analysis of ocular toxoplasmosis in Brazil: evidence for a role for toll-like receptor 9 (TLR9) Mem Inst Oswaldo Cruz. 2009;104:1187–1190. doi: 10.1590/s0074-02762009000800019. [DOI] [PubMed] [Google Scholar]
- Pifer R, Benson A, Sturge CR, Yarovinsky F. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J Biol Chem. 2011;286:3307–3314. doi: 10.1074/jbc.M110.171025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner F, Yarovinsky F, Romero S, Didry D, Carlier MF, Sher A, Soldati-Favre D. Toxoplasma profilin is essential for host cell invasion and TLR11-dependent induction of an interleukin-12 response. Cell Host Microbe. 2008;3:77–87. doi: 10.1016/j.chom.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A. 2005;102:9577–9582. doi: 10.1073/pnas.0502272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Gangneux F, Darde ML. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin Microbiol Rev. 2012;25:264–296. doi: 10.1128/CMR.05013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, Medzhitov R, Sher A. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- Shi Z, Cai Z, Sanchez A, Zhang T, Wen S, Wang J, Yang J, Fu S, Zhang D. A novel Toll-like receptor that recognizes vesicular stomatitis virus. J Biol Chem. 2011;286:4517–4524. doi: 10.1074/jbc.M110.159590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skillman KM, Daher W, Ma CI, Soldati-Favre D, Sibley LD. Toxoplasma gondii profilin acts primarily to sequester G-actin while formins efficiently nucleate actin filament formation in vitro. Biochemistry. 2012;51:2486–2495. doi: 10.1021/bi201704y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhumavasi W, Egan CE, Warren AL, Taylor GA, Fox BA, Bzik DJ, Denkers EY. TLR adaptor MyD88 is essential for pathogen control during oral toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol. 2008;181:3464–3473. doi: 10.4049/jimmunol.181.5.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y. Genes, cells and cytokines in resistance against development of toxoplasmic encephalitis. Immunobiology. 1999;201:255–271. doi: 10.1016/S0171-2985(99)80066-7. [DOI] [PubMed] [Google Scholar]
- Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, Triantafilou K. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem. 2006;281:31002–31011. doi: 10.1074/jbc.M602794200. [DOI] [PubMed] [Google Scholar]
- Weiss LM, Dubey JP. Toxoplasmosis: A history of clinical observations. Int J Parasitol. 2009;39:895–901. doi: 10.1016/j.ijpara.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.