

Abstract
Linoleic acid (LA) and LA-esters are the precursors of LA hydroperoxides, which are readily converted to 9- and 13-hydroxy-octadecadienoic acid (HODE) and 9- and 13-oxo-octadecadienoic acid (oxo ODE) metabolites in vivo. These four oxidized LA metabolites (OXLAMs) have been implicated in a variety of pathological conditions. Therefore, their accurate measurement may provide mechanistic insights into disease pathogenesis. Here we present a novel quadrupole time-of-flight mass spectrometry (Q-TOFMS) method for quantitation and identification of target OXLAMs in rat plasma. In this method, the esterified OXLAMs were base-hydrolyzed and followed by liquid-liquid extraction. Quantitative analyses were based on one-point standard addition with isotope dilution. The target metabolites were quantified by multiple reaction monitoring (MRM) extracted ion chromatograms generated post-acquisition with 10 ppm extraction window. The limit of quantitation was 9.7–35.9 nmol/L depending on the metabolite. The method was reproducible with coefficient of variation below 18.5%. Mean concentrations of target metabolites were 57.8, 123.2, 218.1, and 57.8 nmol/L for 9-HODE, 13-HODE, 9-oxoODE, and 13-oxoODE, respectively. Plasma levels of total OXLAMs were 456.9 nmol/L, which correlated well with published concentrations obtained by gas chromatography/mass spectrometry (GC/MS). The concentrations were also obtained utilizing a standard addition curve approach. The calibration curves were linear with correlation coefficients > 0.991. Concentrations of 9-HODE, 13-HODE, 9-oxoODE, and 13-oxoODE were 84.0, 138.6, 263.0, and 69.5 nmol/L, respectively, which were consistent with the results obtained from one-point standard addition. Target metabolites were simultaneously characterized based on accurate Q-TOFMS data. This is the first study of secondary LA metabolites using Q-TOFMS.
Keywords: Q-TOFMS, hydroxy-octadecadienoic acid, oxo-octadecadienoic acid, linoleic acid, rat plasma
1. Introduction
Linoleic acid (LA, 18:2n-6) is the most abundant polyunsaturated fatty acid (PUFA) in human diets (U.S. Department of Agriculture, 2010, 2007–2008) and a major component of mammalian tissues. LA can be oxidized to lipid hydroperoxides either enzymatically by 12/15-lipoxygenase (Kuhn et al., 1990; Navab et al., 2000; Shureiqi and Lippman, 2001), cyclooxygenase (Hampton et al., 1998), and/or cytochrome P450 (Oliw et al., 1993), or non-enzymatically by the action of reactive oxygen species (Niki, 2009) (Figure 1). The two major biochemical pathways for conversion of LA to oxidized LA metabolites (OXLAMs) begin with formation of the conjugated hydroperoxydienes 9- and 13-hydroperoxy-octadecadienoic acid (9-HPODE and 13-HPODE), which are rapidly reduced by glutathione-dependent peroxidase to the alcohols 9- and 13-hydroxy-octadecadienoic acid (9- and 13-HODE), respectively (Imaizumi et al., 2010). Both 9- and 13-HODE can be converted to the ketones 9- and 13-oxo-octadecadienoic acid (9- and 13-oxoODE) via hydroxy-fatty acid dehydrogenase.
Figure 1. Formation of oxidized LA metabolites.

LA can be enzymatically or non-enzymatically converted to the LA hydroperoxides (9- and 13-HPODE), which are readily converted to hydroxyl (9- and 13-HODE) and ketone (9- and 13-oxoODE) derivatives in vivo. The initial step can be catalyzed by 12/15-lipoxygenase, cyclooxygenase, or cytochrome P450. Abbreviations: LA: linoleic acid; HPODE; hydroperoxy-octadecadienoic acid; HODE: hydroxy-octadecadienoic acid; oxoODE: oxo-octadecadienoic acid.
These four OXLAMs are bioactive in several organ systems (Engels et al., 1995; Haas et al., 1990; Ku et al., 1992; Patwardhan et al., 2009), and have been mechanistically linked to the etiology of pathological conditions ranging from chronic pain (Patwardhan et al., 2010; Patwardhan et al., 2009) to atherosclerosis (Brooks et al., 1970; Jira et al., 1998; Spiteller, 1998). OXLAMs, which are elevated in blood in Alzheimer’s disease (Yoshida et al., 2009) and non-alcoholic steatohepatitis (NASH) (Feldstein et al., 2010), have been proposed as circulating biomarkers for both conditions.
OXLAM analyses have been carried out by many methods including high performance liquid chromatography (HPLC) (Browne and Armstrong, 2000), gas chromatography-mass spectrometry (GC-MS) (Johnson et al., 1997; Yoshida et al., 2006; Yoshida et al., 2005; Yoshida et al., 2009), enzyme linked immunosorbent assay (ELISA) (Spindler et al., 1997), and tandem mass spectrometry (LC-MS/MS). HPLC methods are not specific and only allow for comparison with synthetic standards. The GC-MS technique is limited to the volatile compounds and thermolabile compounds. Additionally, it requires chemical derivatization and is unable to provide detailed information on the molecular species. The main drawback of ELISA is the lack of specificity and inability to distinguish and quantify multiple isoforms of compounds. Lately, LC-MS/MS has become more important in this field, owing to better sensitivity and specificity, yielding lower limits of detection and quantitation. Triple quadrupole LC-MS/MS and Q-trap measurements have been used to quantify OXLAMs in human body fluids or tissues (Feldstein et al., 2010; Shishehbor et al., 2006) and in rodent plasma and tissues (Imaizumi et al., 2010; Kubala et al., 2010; Liu et al., 2010). Many studies have demonstrated the qualitative and quantitative usefulness of Q-TOFMS based techniques in various analytic fields, including toxicology and therapeutic drug monitoring (Decaestecker et al., 2000), illicit drugs and metabolites in urban sewage (Gonzalez-Marino et al., 2012), pesticides (Hernandez et al., 2005), and sterol lipid quantitation (Wewer et al., 2011). So far, Q-TOFMS technology has not been explored to measure secondary metabolites of LA.
In this study, we developed a sensitive and specific Q-TOFMS method for the quantification of targeted OXLAMs in rat plasma by standard addition with isotope dilution and simultaneous identification of the target metabolites. The method was evaluated by using a standard addition curve method, and comparing results with published results obtained by GC/MS.
2. Materials and Methods
2.1. Materials
(±)9-hydroxy-10E,12Z-octadecadienoic acid (9-HODE), (±)13-hydroxy-10E,12Z-octadecadienoic acid (13-HODE), 9-oxo-10E,12Z-octadecadienoic acid (9-oxoODE), 13-oxo-9Z,11E-octadecadienoic acid (13-oxoODE), 13S-hydroxy-9Z,11E-octadecadienoic-9,10,12,13-d4 acid (13-HODE-d4), and linoleic acid-9,10,12,13-d4 (LA-d4) were purchased from Cayman Chemical Co. (Ann Arbor, MI). Diethylenetriamine pentaacetic acid (DTPA), butylated hydroxytoluene (BHT), 1N hydrochloric acid (37%), acetic acid, and potassium hydroxide were obtained from Sigma-Aldrich (St. Louis, MO). HPLC grade hexane, methanol, water, and LC-MS grade acetonitrile, water and isopropanol were purchased from Fisher Scientific (Fair Lawn, NJ).
2.2. Animals and diets
The experimental protocol for the animal studies was approved by the Animal Care and Use Committee of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23). Male F344 (CDF) rat pups (18–21 days old) and their surrogate mothers, purchased from Taconic (Rockville, MD, USA), were housed in an animal facility with regulated temperature, humidity, and a 12 h light/12 h dark cycle. The pups were allowed to nurse until 21 days old. Lactating rats were allowed free access to water and rodent chow formulation NIH-31 18-4, which contained 4% (wt/wt) crude fat (Zeigler Bros., Gardners, PA) and whose fatty acid composition has been reported (Igarashi et al., 2009). After 15 weeks on the diet, the rats were implanted with an osmotic mini-pump (Alzet®, Model 2004; 0.25 μL/h, Cupertino, CA, USA) used to deliver artificial cerebrospinal fluid (140 mmol/L NaCl, 3.0 mmol/L KCl, 2.5 mmol/L CaCl2, 1.0 mmol/L MgCl2, and 1.2 mmol/L NaPO4, pH 7.4) at a rate of 0.5 μg/hr to the fourth ventricle for 2 days. The rats were euthanized with CO2 and decapitated. Trunkal blood was collected into ethylenediaminetetraacetic acid-containing centrifuge tubes, centrifuged, and plasma was removed and kept frozen at −80 °C until analysis.
2.3. Instrumentation
2.3.1. Liquid Chromatography
Analyses of target oxidized LA metabolites were conducted by Q-TOFMS. The liquid chromatography system used was an Agilent 1200 SL liquid chromatography series (Agilent Corporation, Palo Alto, CA). Reversed phase HPLC was performed on a C18 column (Luna C18, 3 μ, 2 mm × 150 mm, Phenomenex, CA). The optimized mobile phase consisted of water : acetonitrile : acetic acid (90:10:0.04, v/v/v) (mobile phase A) and acetonitrile: isopropanol (80:20, v/v) (mobile phase B). The column was eluted with the following gradient: 30% B at 0 min, 58% B at 1 min, 68% B at 20 min, 70% B at 23 min, 100% B from 24 to 27 min, 30% B from 27.1 to 30 min. The flow rate was 0.2 mL/min. Aliquots of 50 uL of the extracts of LA metabolites from rat plasma were injected in Q-TOFMS.
2.3.2. Mass spectrometry (MS)
Q-TOFMS analyses were performed on an Agilent Accurate-Mass quadrupole time-of-flight 6540 mass spectrometer. The main parameters were optimized by making multiple injections of solvent standard solutions containing the target analytes while making incremental changes in instrument values. The final parameters chosen were based on the best response for most compounds: fragmentor (150 V), skimmer (40 V), nozzle voltage (1500 V), Vcap (2500 V), nebulizer (40 psig), drying gas (8 L/min, 350 °C), and sheath gas (10 L/min, 350 °C).
Data acquisition was performed in target MS/MS mode so that MS spectra were recorded simultaneously with MS/MS spectra. The mass range for MS full scan was acquired from 100 to 1100 m/z at a rate of 0.6 spectra/s. For tandem MS/MS analysis, the spectra were collected over the m/z 100–400 range at a scan rate 0.6 spectra/s. Negative ion mode was used for the characterization and detection of OXLAM products. To maintain mass accuracy during the run time, a reference mass solution was continuously introduced along with the LC stream for real-time mass calibration. Ions at m/z 112.985587 and 980.016375 were used as references. Spectral data were acquired at 2 GHz (extended dynamic range mode) when used for quantification measurements. A target list of the analytes of interest was generated with the precursor ion (deprotonated molecule): 295.2278, 293.2122, and 299.2530 for HODEs, oxoODEs, and internal standard, respectively.
Quantitative determination of targeted OXLAMs was carried out using multiple reaction monitoring (MRM) channels reconstructed from MS/MS spectra. Collision energies were optimized. Table 2 depicts the multiple reaction monitoring (precursor ion → fragment ion) used for the analyses and their corresponding collision induced dissociation energy. The MRM extracted ion chromatograms (EICs) of individual compounds were extracted with the exact mass of each compound using a 10 ppm mass extraction window. Metabolites were identified by comparing MS/MS spectra of target metabolites in plasma with spectra of authentic standards.
Table 2.
Multiple reaction monitoring (MRM) transitions and their corresponding collision energies of oxidized LA metabolites
| Compound | Quantifier MRM | Collision Energy (eV) |
|---|---|---|
| 9-HODE | 295.2278/171.1026 | 15 |
| 13-HODE | 295.2278/195.1390 | 15 |
| 9-oxoODE | 293.2122/185.1183 | 13 |
| 13-oxoODE | 293.2122/113.0968 | 13 |
| 13-HODE-d4 | 299.2530/198.1579 | 15 |
MassHunter workstation software in version B.04.00 was used for the control of the system, data acquisition, qualitative and quantitative analysis.
2.4. Preparation of standard solutions
Primary stock solutions (100 ng/μL) of 9-HODE, 13-HODE, 9-oxoODE, 13-oxoODE, and 13-HODE-d4 obtained from Cayman Chemical were prepared in ethanol and stored at −80 °C until use. Working standard mixtures of the test compounds were prepared in methanol by mixing appropriate volumes of individual analyte stock and diluting with methanol. The IS was prepared in the same manner. The final concentrations of working standard solutions were 1, 2, 5, 10, 20, 50, 100, and 200 ng/mL corresponding to a range from 6.8 to 1351.4 and 6.8 to 1360.5 nmol/L for HODEs and oxoODEs, respectively. The final concentration of the working solution for deuterated internal standard was 668.9 nmol/L.
2.5. Alkaline hydrolysis of the esters
In order to have a better assessment of lipid peroxidation, it is necessary to analyze target OXLAMs in plasma from both unesterified and esterified lipids. Therefore, esterified OXLAMs must first to be hydrolyzed. The alkaline hydrolysis was based on the method described by Feldstein et al (2010) with slight modifications, as follows. Frozen plasma samples (stored at −80 °C) were thawed on ice. All Kimble glass culture tubes (8 mL screw-capped) used for the experiment were placed in the ice bath. A 50 μL plasma aliquot was transferred into a glass culture tube containing 10 μL of mixture of antioxidant solution containing 50 mM BHT and 200 mM DTPA. 100 μL of internal standards 13-HODE-d4 working solution (5 ng) was added to the plasma. The samples were treated with 0.2 M KOH (final concentration) in methanol. The reaction mixtures in a total volume of 0.5 mL were vortexed, perfused with nitrogen, and sealed with cap wrapping using Parafilm M (Fisher Scientific, Pittsburgh, PA). The samples were then heated at 60 °C for 30 min.
To determine the time needed until the hydrolysis reaction was complete while minimizing degradation, a short time-course for alkaline hydrolysis was conducted following the procedure described above. Hydrolysis reactions were allowed to proceed for 20, 30, and 40 min. The optimal incubation time was selected as 30 min for all subsequent determinations.
We also examined the potential of autoxidation during hydrolysis by employing deuterated LA-d4. We added 10 μL deuterated LA-d4 (1 ng) in ethanol to 50 μL plasma. Alkaline hydrolysis was conducted following the procedure described above without addition of OXLAM internal standard. One sample was hydrolyzed for 30 min and the other was conducted in parallel without incubation.
2.6. Extraction of LA metabolites
After alkaline hydrolysis, the samples were allowed to cool from 60 °C to room temperature and then placed on the ice bath. All procedures were conducted on ice to minimize autoxidation during sample preparation. The reaction mixture was acidified to pH 3 with 0.5 N HCl. The samples were extracted by adding 3 mL hexane, perfused with nitrogen, capped, vortex mixed for 1 min, and then centrifuged at 3000 rpm for 10 min at 4 °C. The upper organic phase was transferred into a new glass centrifuge tube. The extraction with hexane was repeated one more time. The organic layers were combined and evaporated to dryness under nitrogen. The residue was reconstituted in 200 uL of 8:2 methanol-water containing 0.04% acetic acid. The solution was transferred to an amber injection vial, flushed with nitrogen and analyzed by Q-TOFMS.
2.7. Preparation of samples
For the one-point standard addition method, each pooled plasma sample was divided into two aliquots (50 μL): the first was blank plasma and the second was spiked with a mixture of the target metabolite standards (1 ng each). Samples were then hydrolyzed and extracted following the same procedure as described above.
For the standard addition curve method, the pooled plasma aliquot (50 μL) was used for preparation of calibration samples. The calibration curves were constructed by spiking varying amounts (0.1, 0.2, 0.5, 1, 2, 5, 10, and 20 ng) of a mixture of working standard solutions. The measurement range in quantitation was 6.8–1360.5 nmol/L plasma. Samples were then subjected to alkaline hydrolysis and solvent extraction as stated above.
2.8. Sample preparation for reproducibility testing
Sample preparation procedure was same as those of the one-point standard addition described above. Experiments were carried out at two concentrations (pooled blank plasma and pooled plasma spiked with 1 ng standard mixture solution in 50 μL plasma) in replicate (n = 3 for blank plasma, n = 4 for plasma fortified with standards). Reproducibility was expressed as the coefficient of variation (CV) of the peak area ratio of the target analyte to that of the internal standard and calculated using the equation CV% = SD/Mean × 100%.
2.9. Measurement
The endogenous concentrations of the target OXLAMs in the pooled plasma were measured by the method of one-point standard addition (Eq. 1), utilizing known amounts of standard mixture spiked in 50 μL pooled plasma for the nominal value of each standard sample.
| (Eq. 1) |
where Cen is the endogenous concentration of individual metabolite in the sample; R1 is the mean area ratio of the target metabolite and internal standard measured from pooled blank plasma; R2 is the mean area ratio of the target metabolite and internal standard measured from pooled plasma fortified with standard solution (1 ng/50 uL plasma).
To evaluate the one-point standard addition method, we performed a standard addition curve method. The calibration curves were constructed by plotting the peak area ratio of the target metabolite to that of the internal standard versus theoretical concentrations. A linear equation (y = ax + b) was derived from the calibration curve. By extrapolation to y = 0, unknown endogenous concentrations of target metabolites were calculated by the equation x = − (−b/a).
3. Results and Discussion
3.1. Optimization of analytical procedure
3.1.1. Optimization of incubation time for alkaline hydrolysis
In order to evaluate completion of hydrolysis of esterified lipids with minimal potential for autoxidation and degradation of target metabolites, a short time course was determined by incubating samples for 20, 30, and 40 min at 60 °C. The levels of each free target metabolite were stabilized at the period of 20–30 min (data not shown). Thus, a suitable reaction time of 30 min was selected for the hydrolysis. Similar incubation conditions using 0.2 M NaOH at 60 °C for 30 min to hydrolyze esterified 9- and 13-HODEs have been described by Shishehbor et al (2006).
The likelihood of autoxidation was examined using a hydrolysis process with deuterated LA-d4, with percent LA-d4 remaining determined by comparison of the peak area of LA-d4 after the 30-min incubation period to the peak area without incubation. The results showed 99.5% remaining at the end of the 30-min hydrolysis, suggesting no autoxidation during this process.
3.1.2. Optimization of LC-TOFMS conditions
In order to develop a sensitive and specific Q-TOFMS method, we optimized the LC, MS and MS/MS conditions with the peak area selected as a criterion for optimization.
3.1.2.1 Optimization of LC conditions
Since OXLAMs contain a carboxylic acid functional group, negative electrospray ionization (ESI) mode by generation of [M-H]− precursor ions was used for LC optimization. LC conditions were optimized to achieve maximal sensitivity of the mass spectrometry detection. The mobile phase optimization was performed by adjusting organic solvent type, the amount of organic modifiers, pH, and buffers. The final optimum mobile phase composition was: water-acetonitrile-acetic acid (90:10:0.04, v/v/v) (mobile phase A) and acetonitrile-isopropanol (80:20, v/v) (mobile phase B). Slightly acidic conditions using acetic acid provided better ionization efficiency, as pointed out by Wu et al (2004). Under these conditions using flow injections, MS and MS/MS parameters were optimized for ionization efficiency of precursor ions and sensitivity of product ions.
3.1.2.2 Optimization of MS and MS/MS detection
Sensitivity of precursor ions depends critically on the tuning of instrumental parameters. Basic source parameters (drying gas and sheath gas) were tuned and listed in the Methods section. The optimization of the key parameters (fragmentor voltage, capillary voltage, nozzle voltage, and skimmer) was done by injection of a mixture of standard solutions using above optimized LC conditions. Figure 2a shows the relation between signal intensity of selected precursor ion masses of individual metabolite and fragmentor voltage in the range of 60–180 V. The highest responses were achieved for fragmentor voltage in the range of 130–160 V. The fragmentor voltage value of 150 V was selected.
Figure 2. Effect of voltage parameters on ionization of target compounds: 9-HODE [triangles], 13-HODE [diamonds], 9-oxoODE [squares], and 13-oxoODE [circles]. (a) fragmentor voltage, (b) capillary voltage, (c) nozzle voltage, and (d) skimmer voltage.

Mobile phase: water-acetonitrile-acetic acid (90:10:0.04) (mobile phase A) and acetonitrile-isopropanol (80:20) (mobile phase B), flow rate = 0.2 mL/min, drying gas temperature 350 °C.
Evaluation of capillary voltage between 1000–4000 V indicated that signal intensity increased with the increment of capillary voltage to 2500 V, without substantial changes above this threshold (Fig. 2b). Therefore, capillary voltage used for MS analyses was set as 2500 V.
Nozzle voltage and skimmer were also optimized for the detection of these target metabolites. The nozzle voltage value of 1500 V was selected as a compromise between monitored compounds and sensitivity (Fig. 2c). The highest value (40 V) was selected for skimmer (Fig. 2d).
Since multiple isomeric and isobaric lipid species differed by positions of fatty acid chain double bonds, and their oxidized metabolites are further complicated with regio-, stereo-, and enantio-isomers, MRM channels were used for detection and quantitation of OXLAMs instead of choosing MS full scan channels. The selection of product ion to reconstruct MRM transition for each target metabolite was based on specificity and sensitivity. Although the targeted metabolites are two pairs of closely related regio-isomers, unique collision-induced decomposition product ions could be found and specificity basing on the MRM transitions can be achieved under the current chromatographic conditions. No cross talk was observed among their MRMs (data not shown) under current chromatography conditions.
The collision energy for each analyte was optimized in the range of 10–30 V in an attempt to obtain maximal signal intensities of the desired fragment ions (Fig. 3). Since 9-HODE was co-eluted with 13-HODE, a compromised setting for collision energy was 13 eV. The optimum collision energy for 9-oxoODE and 13-oxoODE was 15 eV. Table 2 shows specific dissociation reactions used in the MRM mode and the corresponding collision energies.
Figure 3. Effect of collision energy on intensity of fragment ions.
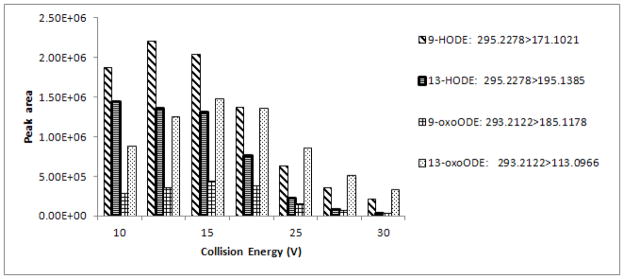
Mobile phase: water-acetonitrile-acetic acid (90:10:0.04) (mobile phase A) and acetonitrile-isopropanol (80:20) (mobile phase B), flow rate = 0.2 mL/min, drying gas temperature 350 °C, fragmentor voltage 150 V, capillary voltage 2500 V, nozzle voltage 1500 V, skimmer 40 V.
3.2. Target metabolite profiling
All four target metabolites were identified in rat plasma. Figure 4 demonstrates representative MRM extracted ion chromatograms (EICs) of the four target metabolites in the presence of internal standard obtained from a non-spiked blank plasma sample (Fig. 4a) and a pooled blank plasma spiked with authentic standards at the level of 337.8 and 340.1 nmol/L for HODEs and oxoODEs (Fig. 4b). The retention times of 9-HODE, 13-HODE, 9-oxoODE, 13-oxoODE, and 13-HODE-d4 (IS) were 17.0, 16.8, 19.8, 18.1 and 16.4 min respectively.
Figure 4. Reconstructed multiple reaction monitoring (MRM) chromatograms from rat plasma extracts after alkaline hydrolysis.

(A) pooled blank plasma sample; (B) pooled blank plasma fortified with 100 ng/mL individual standard.
3.3. MS/MS analysis of target metabolites
MS/MS spectra of representative target metabolites obtained from a pooled blank plasma and solvent standard mixture are shown in Figure 5 along with the interpretation of fragmentation patterns of these metabolites. The MS/MS spectra of authentic 9-HODE and 13-HODE standards showed the same deprotonated molecular ion [M-H]− at m/z 295.2278 (Fig. 5b and 5d). The high abundant fragment ions at m/z 277.2177 resulted from a neutral loss of H2O. Cleavage between the carbon-carbon bond adjacent to the hydroxyl group gave major signature fragment ions at m/z 171.1021 and 195.1385 for 9-HODE and 13-HODE respectively (Fig. 5b and 5d). In comparison, blank plasma sample demonstrated the same pattern of product ions of m/z 171.1021 and 277.2177 for 9-HODE (Fig. 5a) and 195.1385 and 277.2177 for 13-HODE (Fig. 5c), which confirmed the identity of 9-HODE and 13-HODE from rat plasma. The identical fragmentation patterns of 9-HODE and 13-HODE were reported by Wheelan et al (1993).
Figure 5. MS/MS spectra and fragmentation analysis of target oxidized linoleic acid metabolites (OXLAMs).

(a) 9-HODE from pooled blank plasma sample, (b) 9-HODE authentic standard; (c) 13-HODE from pooled blank plasma sample, (d) 13-HODE authentic standard; (e) 9-oxoODE from pooled blank plasma sample, (f) 9-oxoODE authentic standard; (g) 13-oxoODE from pooled blank plasma sample, and (h) 13-oxoODE authentic standard.
The MS/MS spectra of authentic standards 9-oxoODE and 13-oxoODE showed the same deprotonated molecular ion [M-H]− at m/z 293.2122. The predominant characteristic ions at m/z 185.1178 for 9-oxoODE and 113.0966 for 13-oxoODE were formed by cleavage of the double bond adjacent to the carbonyl group (Fig. 5f and 5h); similar fragmentation pathways for 5-oxo-ETE, 12-oxo-ETE and 15-oxo-ETE were described by Murphy et al (2005). The minor fragment ions of 9-oxoODE at m/z 125.0967 resulted from the ion at m/z 185.1178 by loss of CH3COOH (Fig. 5f). The minor fragment ion of 13-oxoODE at m/z 249.2224 resulted from the loss of CO2 (Fig. 5h). The small ion of 13-oxoODE at m/z 195.139 was visible, which was further fragmented to form the ion of m/z 167.1067 by loss of C2H4 (Fig. 5h). The MS/MS spectra of endogenous 9-oxoODE and 13-oxoODE in a rat plasma sample showed the same fragments as the authentic standards, with m/z 185.1178 and 125.0967 for 9-oxoODE (Fig. 5e), and m/z 113.0966, 167.1077 and 249.2224 for 13-oxoODE (Fig. 5g). Similar fragmentation patterns of 9-oxoODE and 13-oxoODE have been observed (Hall and Murphy, 1998).
Based on accurate mass measurements of molecular ions and product ions, fragmentation analysis, and retention time comparison with authentic standards, these target metabolites were identified in rat plasma.
Table 3 lists the elemental compositions, mass accuracy errors for precursors and specific product ions of all four target metabolites in plasma samples at three concentration levels (pooled blank plasma, pooled plasma spiked with standard mixture at 20 and 200 ng/mL) based on n = 8 replicates (combined four replicates on two different days). The mass errors between theoretical and observed values for both precursors and product ions were less than 10 ppm (1 mDa) for all of the target metabolites studied. There have been no universally accepted criteria for mass accuracy. In publications, mass accuracies of 2 or 10 mDa (Hernandez et al., 2004) or 10 ppm (Wang and Leung, 2007) for product ions have been suggested. The wider mass error windows of 20 ppm or 50 mD have been used as acceptable criteria (Badoud et al., 2009; Vonaparti et al., 2010). All of these criteria were fulfilled by our method.
Table 3.
Elemental composition and accurate mass measurement for precursor and product ions obtained in pooled blank plasma or fortified at the level of 20 and 200 ng/mL plasma (n = 8)a
| Compound | Molecular formula | Level of interest (ng/mL) | Precursor ion (m/z)b | Product ions | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| [M-H]− | Average mass error (Δppm)c | Elemental composition | Theoretical massb | Average mass error (Δppm)c | |||
| 9-HODE | C18H32O3 | 0 | 295.2278 | 1.8 ± 0.8 | C9H16O3 | 171.1021 | 3.9 ± 0.5 |
| 20 | 2.2 ± 0.5 | 3.5 ± 1.1 | |||||
| 200 | 2.5 ± 0.3 | 3.8 ± 0.3 | |||||
|
| |||||||
| 13-HODE | C18H32O3 | 0 | 295.2278 | 2.2 ± 0.4 | C12H20O2 | 195.1385 | 3.7 ± 0.7 |
| 20 | 2.2 ± 0.4 | 3.8 ± 0.4 | |||||
| 200 | 2.6 ± 0.3 | 3.8 ± 0.3 | |||||
|
| |||||||
| 9-oxoODE | C18H30O3 | 0 | 293.2122 | −2.9 ± 5.7 | C10H17O3 | 185.1178 | 4.4 ± 1.6 |
| 20 | −2 ± 4.5 | 4.6 ± 0.9 | |||||
| 200 | 0.9 ± 0.9 | 3.4 ± 0.9 | |||||
|
| |||||||
| 13-oxoODE | C18H30O3 | 0 | 293.2122 | 1.7 ± 0.6 | C7H14O | 113.0966 | 4.8 ± 1.2 |
| 20 | 2.0 ± 0.4 | 5.2 ± 1.0 | |||||
| 200 | 2.3 ± 0.2 | 5.6 ± 0.5 | |||||
Data combined four replicates on two different days
Theoretical value rounded to the fourth decimal place
Average and standard deviation of mass error (theoretical value)
3.4. Quantitation
3.4.1. Limit of quantitation
As blank plasma samples with negligible levels of target metabolites are not available, the limit of quantitation (LOQ) of the method was estimated based on the signal to noise ratio of the MRM chromatograms as shown in Figure 4a. LOQ with the signal to noise ratio of 10 was estimated to be 9.7, 18.5, 35.9 and 26.8 nmol/L for 9-HODE, 13-HODE, 9-oxoODE, and 13-oxoODE, respectively.
3.4.2. Reproducibility
The reproducibility of the method for each target metabolite was evaluated through relative standard deviation of the replicate measurements, which reflected errors from three analytical processes including alkaline hydrolysis, liquid-liquid extraction and Q-TOFMS analysis. The results obtained at two concentration levels are summarized in Table 4. The coefficient of variation values ranged from 6.4 to 18.5% at blank plasma level and 6.3 to 14% at the level of plasma spiked with 1 ng standard mixture solution, indicating that the method has an acceptable reproducibility.
Table 4.
Reproducibility data calculated from peak area ratio of individual target metabolite to internal standard in pooled blank plasma (n = 3) and pooled plasma aliquot (50 μL) fortified with 1 ng of standard mixture (n = 4)
| Compound | Pooled blank plasma | Pooled blank plasma fortified with 1 ng of standard mixture | ||
|---|---|---|---|---|
|
| ||||
| Peak area ratio | CV% | Peak area ratio | CV% | |
| Mean ± SD | Mean ± SD | |||
| n = 3 | n = 4 | |||
| 9-HODE | 0.145 ± 0.027 | 18.5 | 0.313 ± 0.035 | 11.3 |
| 13-HODE | 0.293 ± 0.027 | 9.3 | 0.454 ± 0.028 | 6.3 |
| 9-oxoODE | 0.113 ± 0.007 | 6.4 | 0.148 ± 0.021 | 14.0 |
| 13-oxoODE | 0.054 ± 0.009 | 17.2 | 0.119 ± 0.012 | 10.2 |
Coefficient of variation reflected errors from three analytical processes including alkaline hydrolysis, liquid-liquid extraction and LC-MS/MS analysis.
3.4.3. Quantitation of OXLAMs
Quantitation was carried out using reconstructed MRM channels with Q-TOF MS/MS analyses applying narrow extracted ion windows (10 ppm) for each target metabolite.
The plasma concentrations expressed as the mean of three replicates, calculated by Equation 1 (Eq. 1) as stated in the one-point standard addition method, corresponded to mean concentrations of 57.8 ± 18.7, 123.2 ± 31.1, 218.1 ± 53.7, and 57.8 ± 19.2 nmol/L for 9-HODE, 13-HODE, 9-oxoODE, and 13-oxoODE, respectively (Table 5). The plasma level of total OXLAMs was 456.9 ± 76.4 nmol/L. The total 9-position oxidized metabolites (9-oxLA: 9-HODE and 9-oxoODE) accounted for 275.9 nmol/L. In contrast, the total 13- position oxidized metabolites (13-oxLA: 13-HODE and 13-oxoODE) accounted for 181 nmol/L. The ratio of 9-oxLA/13-oxLA was 1.5. The major metabolites were 9-oxoODE and 13-HODE, which accounted for 47.7% and 27% of the total OXLAMs, respectively. The proportions of 9-HODE and 13-oxoODE were less abundant, with each accounting for 13% of total OXLAMs. The levels of OXLAMs were also normalized to the level of precursor unesterified LA in plasma as shown in Table 5.
Table 5.
Endogenous concentration of target oxidized LA metabolites (OXLAMs) measured by method of one-point standard addition and standard addition curve extrapolation and ratio of plasma levels of OXLAMs to precursor LA
| Compound | One-point standard addition | Standard addition curve extrapolation | |
|---|---|---|---|
|
| |||
| Mean ± SD (nmole/L) | Ratio of OXLAMs/unesterified LA* (nmol/μmol) | (nmole/L) | |
| 9-HODE | 57.9 ± 18.7 | 1.0 ± 0.3 | 84.0 |
| 13-HODE | 123.2 ± 31.1 | 2.1 ± 0.5 | 138.6 |
| 9-oxoODE | 218.1 ± 53.7 | 3.8 ± 0.9 | 263.0 |
| 13-oxoODE | 57.8 ± 19.2 | 1.0 ± 0.3 | 69.5 |
|
| |||
| Total OXLAMs | 456.7 ± 76.4 | 7.9 ± 1.3 | 555.1 |
: the plasma concentrations of unesterified linoleic acid, esterified linoleic acid, and total linoleic acid were 59.7, 1514.3 and 1574.0 μM/L, respectively, which were determined by GC.
We also prepared a standard addition curve in order to assess the accuracy of the one-point standard addition method. Excellent linearity (R2 ≥ 0.991) was achieved for all target metabolites (Fig. 6). The calibration was linear in the range of 6.8 to 1360.5 nmol/L for 9-HODE and 6.8 to 1351.4 nmol/L for 13-oxoODE. Linearity ranged from 13.5 to 1351.4, 34.0 to 1360.5 nmol/L for 13-HODE and 9-oxoODE, respectively. The endogenous concentration of these target metabolites in pooled plasma sample was derived by extrapolation of the standard addition curve to the x intercept, where y = 0. Values for the extrapolated concentrations of 9-HODE, 13-HODE, 9-oxo ODE, and 13-oxo ODE were 84.0, 138.6, 263.0 and 69.5 nmol/L, respectively (Table 5). The plasma level of total OXLAMs was 560.2 nmol/L. The ratio of 9-oxLA/13-oxLA was 1.7.
Figure 6. Calibration standard addition curve plots for 9-HODE, 13-HODE, 9-oxoODE, and 13-oxoODE.

Preparation of the standard solutions and samples for standard addition curves are described in the Method section. The standard addition curves were prepared by spiking 50 μL aliquots of plasma with a fixed amount of internal standard and known amounts of standards. Samples were hydrolyzed and extracted, analyzed by multiple reaction monitoring (MRM) employed to monitor the analytes are listed in the figure. Extrapolation of the standard addition curve to y = 0 gave endogenous plasma concentrations of 84.0, 138.6, 263.0, and 69.5 nmol/L for 9-HODE, 13-HODE, 9-oxoODE, and 13-oxoODE, respectively.
These concentrations are comparable to those obtained by the one-point standard addition method, verifying the suitability and equivalence of both methods for analyzing target OXLAMs in plasma samples. Furthermore, our total OXLAM values are comparable to those previously reported by Niki et al (666 ± 240 nM) (2005). In their method, esterified forms of oxoODEs were first reduced to HODEs, and samples were hydrolyzed, then total OXLAMs were measured as total HODEs by GC/MS. Collectively, the consistency of our data with internal and published data indicate that reconstructed MRM channels with Q-TOF MS/MS analysis can be used to reliably quantify secondary LA metabolites.
The reported regio-selectivity of measured OXLAMs in human plasma, represented by the total 9-oxLA/13-oxLA ratio, varied from 1.4 in subjects with NASH (Feldstein et al., 2010) and 0.9 in the serum of subjects with chronic pancreatitis (Stevens et al., 2012) to 0.8 in a chronic pain population (Ramsden et al, unpublished data). Shishehbor et al (2006) reported a 9-HODE/13-HODE ratio of 0.3, without analysis of oxoODEs. The ratio of 9-oxLA/13-oxLA varies by species and organ (Liu et al., 2010). Lipid peroxidation is affected by many factors such as pathological conditions, diets, enzymes, in vivo pro-oxidant/antioxidant ratio, age, species, and organ type. Since secondary LA metabolites play important roles in pathological conditions, in a broader perspective, their accurate measurement may provide mechanistic insights into disease pathogenesis.
Summary
This paper is the first to report a novel Q-TOFMS method for identification and quantitation of the target oxidized LA metabolites in rat plasma. The method was also able to provide sensitive and simultaneous elucidation of target metabolites in a single run. Mass accuracies of better than 10 ppm (1 mDa) were obtained for both precursor and product ions. The limit of quantitation ranged from 9.7 to 35.9 nmol/L depending on the target metabolite. The method was reproducible with CV% ranging from 6.4–18.5%. The mean concentrations of target metabolites were 57.8, 123.2, 218.1, and 57.8 nmol/L for 9-HODE, 13-HODE, 9-oxoODE, and 13-oxoODE, respectively. The mean plasma concentration of total OXLAMs was 456.9 ± 76.4 nmol/L, which is consistent with the results of standard addition curve extrapolation and those obtained by GC/MS. This method can be extended to measure OXLAMs in other body fluids or tissues of interest and by adding further molecular species to the target MS/MS list if needed.
Table 1.
Fatty acid composition of n-6 PUFA adequate diet
| Fatty acid | μmol/g food | % of total fatty acid |
|---|---|---|
| 12:0 | 54.6 ± 3.3 | 29.0 |
| 14:0 | 23.5 ± 1.4 | 12.5 |
| 14:1n-5 | 0.06 ± 0.01 | 0.03 |
| 16:0 | 18.2 ± 1.0 | 9.7 |
| 16:1n-7 | 0.08 ± 0.01 | 0.04 |
| 18:0 | 17.1 ± 1.0 | 9.0 |
| 18:1n-9 | 14.4 ± 0.8 | 7.7 |
| 18:2n-6 | 52.1 ± 7.6 | 27.6 |
| 18:3n-3 | 8.5 ± 0.5 | 4.5 |
|
| ||
| Saturated | 113.5 ± 6.6 | 60.1 |
| Monounsaturated | 14.6 ± 0.8 | 7.7 |
| n-6 PUFA | 52.1 ± 7.6 | 27.6 |
| n-3 PUFA | 8.5 ± 0.5 | 4.5 |
| n-6/n-3 | 6.1 | |
Values are mean ± SD (n = 3).
PUFA: polyunsaturated fatty acid.
Acknowledgments
The research was supported by the intramural programs of the National Institute on Aging, National Institute on Alcohol Abuse and Alcoholism, and Warren Grant Magnuson Clinical Center, National Institutes of Health.
Footnotes
No author has a conflict of interest with regard to this manuscript.
References
- Badoud F, Grata E, Perrenoud L, Avois L, Saugy M, Rudaz S, Veuthey JL. Fast analysis of doping agents in urine by ultra-high-pressure liquid chromatography-quadrupole time-of-flight mass spectrometry i. Screening analysis. Journal of Chromatography A. 2009;1216:4423–4433. doi: 10.1016/j.chroma.2009.03.033. Available at http://www.ncbi.nlm.nih.gov/pubmed/19342059. DOI S0021-9673(09)00431-2 [pii] 10.1016/j.chroma.2009.03.033. [DOI] [PubMed] [Google Scholar]
- Brooks CJ, Harland WA, Steel G, Gilbert JD. Lipids of human atheroma: Isolation of hydroxyoctade cadienoic acids from advanced aortal lesions. Biochimica et Biophysica Acta. 1970;202:563–566. doi: 10.1016/0005-2760(70)90131-1. Available at http://www.ncbi.nlm.nih.gov/pubmed/5445332. [DOI] [PubMed] [Google Scholar]
- Browne RW, Armstrong D. Hplc analysis of lipid-derived polyunsaturated fatty acid peroxidation products in oxidatively modified human plasma. Clinical Chemistry. 2000;46:829–836. Available at http://www.ncbi.nlm.nih.gov/pubmed/10839772. [PubMed] [Google Scholar]
- Decaestecker TN, Clauwaert KM, Van Bocxlaer JF, Lambert WE, Van den Eeckhout EG, Van Peteghem CH, De Leenheer AP. Evaluation of automated single mass spectrometry to tandem mass spectrometry function switching for comprehensive drug profiling analysis using a quadrupole time-of-flight mass spectrometer. Rapid Communications in Mass Spectrometry. 2000;14:1787–1792. doi: 10.1002/1097-0231(20001015)14:19<1787::AID-RCM94>3.0.CO;2-S. Available at http://www.ncbi.nlm.nih.gov/pubmed/11006586. DOI 10.1002/1097-0231(20001015)14:19<1787::AID-RCM94>3.0.CO;2-S [pii] 10.1002/1097-0231(20001015)14:19<1787::AID-RCM94>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Engels F, van Houwelingen AH, Buckley TL, van de Velde MJ, Henricks PA, Nijkamp FP. Airway hyperresponsiveness induced by 13-hydroxyoctadecadienoic acid (13-hode) is mediated by sensory neuropeptides. Advances in Prostaglandin, Thromboxane, and Leukotriene Research. 1995;23:361–363. Available at http://www.ncbi.nlm.nih.gov/pubmed/7732872. [PubMed] [Google Scholar]
- Feldstein AE, Lopez R, Tamimi TA, Yerian L, Chung YM, Berk M, Zhang R, McIntyre TM, Hazen SL. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Journal of Lipid Research. 2010;51:3046–3054. doi: 10.1194/jlr.M007096. Available at http://www.ncbi.nlm.nih.gov/pubmed/20631297. DOI jlr.M007096 [pii] 10.1194/jlr.M007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Marino I, Quintana JB, Rodriguez I, Gonzalez-Diez M, Cela R. Screening and selective quantification of illicit drugs in wastewater by mixed-mode solid-phase extraction and quadrupole-time-of-flight liquid chromatography-mass spectrometry. Analytical Chemistry. 2012;84:1708–1717. doi: 10.1021/ac202989e. Available at http://www.ncbi.nlm.nih.gov/pubmed/22185484. DOI 10.1021/ac202989e. [DOI] [PubMed] [Google Scholar]
- Haas TA, Bertomeu MC, Bastida E, Buchanan MR. Cyclic amp regulation of endothelial cell triacylglycerol turnover, 13-hydroxyoctadecadienoic acid (13-hode) synthesis and endothelial cell thrombogenicity. Biochimica et Biophysica Acta. 1990;1051:174–178. doi: 10.1016/0167-4889(90)90190-o. Available at http://www.ncbi.nlm.nih.gov/pubmed/2155665. [DOI] [PubMed] [Google Scholar]
- Hall LM, Murphy RC. Analysis of stable oxidized molecular species of glycerophospholipids following treatment of red blood cell ghosts with t-butylhydroperoxide. Analytical Biochemistry. 1998;258:184–194. doi: 10.1006/abio.1998.2602. Available at http://www.ncbi.nlm.nih.gov/pubmed/9570828. DOI S0003-2697(98)92602-1 [pii] 10.1006/abio.1998.2602. [DOI] [PubMed] [Google Scholar]
- Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92:3007–3017. Available at http://www.ncbi.nlm.nih.gov/pubmed/9787133. [PubMed] [Google Scholar]
- Hernandez F, Ibanez M, Sancho JV, Pozo OJ. Comparison of different mass spectrometric techniques combined with liquid chromatography for confirmation of pesticides in environmental water based on the use of identification points. Analytical Chemistry. 2004;76:4349–4357. doi: 10.1021/Ac049768i. Available at <Go to ISI>://000223010100028. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Pozo OJ, Sancho JV, Lopez FJ, Marin JM, Ibanez M. Strategies for quantification and confirmation of multi-class polar pesticides and transformation products in water by lc-ms2 using triple quadrupole and hybrid quadrupole time-of-flight analyzers. Trac-Trends in Analytical Chemistry. 2005;24:596–612. doi: 10.1016/j.trac.2005.04.007. Available at <Go to ISI>://000231437100016. [DOI] [Google Scholar]
- Igarashi M, Gao F, Kim HW, Ma KZ, Bell JM, Rapoport SI. Dietary n-6 pufa deprivation for 15 weeks reduces arachidonic acid concentrations while increasing n-3 pufa concentrations in organs of post-weaning male rats. Biochimica Et Biophysica Acta-Molecular and Cell Biology of Lipids. 2009;1791:132–139. doi: 10.1016/j.bbalip.2008.11.002. Available at <Go to ISI>://000263616000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi S, Grijalva V, Navab M, Van Lenten BJ, Wagner AC, Anantharamiah GM, Fogelman AM, Reddy ST. L-4f differentially alters plasma levels of oxidized fatty acids resulting in more anti-inflammatory hdl in mice. Drug Metab Lett. 2010;4:139–148. doi: 10.2174/187231210791698438. Available at http://www.ncbi.nlm.nih.gov/pubmed/20642447. DOI BSP/DML/E-Pub/00042 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jira W, Spiteller G, Carson W, Schramm A. Strong increase in hydroxy fatty acids derived from linoleic acid in human low density lipoproteins of atherosclerotic patients. Chemistry and Physics of Lipids. 1998;91:1–11. doi: 10.1016/s0009-3084(97)00095-9. Available at http://www.ncbi.nlm.nih.gov/pubmed/9488997. DOI S0009-3084(97)00095-9 [pii] [DOI] [PubMed] [Google Scholar]
- Johnson JA, Blackburn ML, Bull AW, Welsch CW, Watson JT. Separation and quantitation of linoleic acid oxidation products in mammary gland tissue from mice fed low- and high-fat diets. Lipids. 1997;32:369–375. doi: 10.1007/s11745-997-0047-7. Available at http://www.ncbi.nlm.nih.gov/pubmed/9113624. [DOI] [PubMed] [Google Scholar]
- Ku G, Thomas CE, Akeson AL, Jackson RL. Induction of interleukin 1 beta expression from human peripheral blood monocyte-derived macrophages by 9-hydroxyoctadecadienoic acid. Journal of Biological Chemistry. 1992;267:14183–14188. Available at http://www.ncbi.nlm.nih.gov/pubmed/1629217. [PubMed] [Google Scholar]
- Kubala L, Schmelzer KR, Klinke A, Kolarova H, Baldus S, Hammock BD, Eiserich JP. Modulation of arachidonic and linoleic acid metabolites in myeloperoxidase-deficient mice during acute inflammation. Free Radical Biology and Medicine. 2010;48:1311–1320. doi: 10.1016/j.freeradbiomed.2010.02.010. Available at http://www.ncbi.nlm.nih.gov/pubmed/20156554. DOI S0891-5849(10)00097-3 [pii] 10.1016/j.freeradbiomed.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Belkner J, Wiesner R, Alder L. Occurrence of 9- and 13-keto-octadecadienoic acid in biological membranes oxygenated by the reticulocyte lipoxygenase. Archives of Biochemistry and Biophysics. 1990;279:218–224. doi: 10.1016/0003-9861(90)90484-g. Available at http://www.ncbi.nlm.nih.gov/pubmed/2112367. [DOI] [PubMed] [Google Scholar]
- Liu W, Yin H, Akazawa YO, Yoshida Y, Niki E, Porter NA. Ex vivo oxidation in tissue and plasma assays of hydroxyoctadecadienoates: Z,e/e,e stereoisomer ratios. Chemical Research in Toxicology. 2010;23:986–995. doi: 10.1021/tx1000943. Available at http://www.ncbi.nlm.nih.gov/pubmed/20423158. DOI 10.1021/tx1000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RC, Barkley RM, Zemski Berry K, Hankin J, Harrison K, Johnson C, Krank J, McAnoy A, Uhlson C, Zarini S. Electrospray ionization and tandem mass spectrometry of eicosanoids. Analytical Biochemistry. 2005;346:1–42. doi: 10.1016/j.ab.2005.04.042. Available at http://www.ncbi.nlm.nih.gov/pubmed/15961057. DOI S0003-2697(05)00338-6 [pii] 10.1016/j.ab.2005.04.042. [DOI] [PubMed] [Google Scholar]
- Navab M, Hama SY, Anantharamaiah GM, Hassan K, Hough GP, Watson AD, Reddy ST, Sevanian A, Fonarow GC, Fogelman AM. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: Steps 2 and 3. Journal of Lipid Research. 2000;41:1495–1508. Available at http://www.ncbi.nlm.nih.gov/pubmed/10974057. [PubMed] [Google Scholar]
- Niki E. Lipid peroxidation: Physiological levels and dual biological effects. Free Radical Biology and Medicine. 2009;47:469–484. doi: 10.1016/j.freeradbiomed.2009.05.032. Available at http://www.ncbi.nlm.nih.gov/pubmed/19500666. DOI S0891-5849(09)00330-X [pii] 10.1016/j.freeradbiomed.2009.05.032. [DOI] [PubMed] [Google Scholar]
- Niki E, Yoshida Y, Saito Y, Noguchi N. Lipid peroxidation: Mechanisms, inhibition, and biological effects. Biochemical and Biophysical Research Communications. 2005;338:668–676. doi: 10.1016/j.bbrc.2005.08.072. Available at http://www.ncbi.nlm.nih.gov/pubmed/16126168. DOI S0006-291X(05)01776-6 [pii] 10.1016/j.bbrc.2005.08.072. [DOI] [PubMed] [Google Scholar]
- Oliw EH, I, Brodowsky D, Hornsten L, Hamberg M. Bis-allylic hydroxylation of polyunsaturated fatty acids by hepatic monooxygenases and its relation to the enzymatic and nonenzymatic formation of conjugated hydroxy fatty acids. Archives of Biochemistry and Biophysics. 1993;300:434–439. doi: 10.1006/abbi.1993.1059. Available at http://www.ncbi.nlm.nih.gov/pubmed/8424677. DOI S0003-9861(83)71059-3 [pii] 10.1006/abbi.1993.1059. [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Akopian AN, Ruparel NB, Diogenes A, Weintraub ST, Uhlson C, Murphy RC, Hargreaves KM. Heat generates oxidized linoleic acid metabolites that activate trpv1 and produce pain in rodents. Journal of Clinical Investigation. 2010;120:1617–1626. doi: 10.1172/JCI41678. Available at http://www.ncbi.nlm.nih.gov/pubmed/20424317. DOI 10.1172/JCI4167841678 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan AM, Scotland PE, Akopian AN, Hargreaves KM. Activation of trpv1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18820–18824. doi: 10.1073/pnas.0905415106. Available at http://www.ncbi.nlm.nih.gov/pubmed/19843694. DOI 0905415106 [pii] 10.1073/pnas.0905415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishehbor MH, Zhang R, Medina H, Brennan ML, Brennan DM, Ellis SG, Topol EJ, Hazen SL. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radical Biology and Medicine. 2006;41:1678–1683. doi: 10.1016/j.freeradbiomed.2006.09.001. Available at http://www.ncbi.nlm.nih.gov/pubmed/17145556. DOI S0891-5849(06)00562-4 [pii] 10.1016/j.freeradbiomed.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shureiqi I, Lippman SM. Lipoxygenase modulation to reverse carcinogenesis. Cancer Research. 2001;61:6307–6312. Available at http://www.ncbi.nlm.nih.gov/pubmed/11522616. [PubMed] [Google Scholar]
- Spindler SA, Clark KS, Blackburn ML, Bull AW, Reddy RG. Occurrence of 13(s)-hydroxyoctadecadienoic acid in biological samples. Prostaglandins. 1997;54:875–880. doi: 10.1016/s0090-6980(97)00185-8. Available at http://www.ncbi.nlm.nih.gov/pubmed/9533182. DOI S0090698097001858 [pii] [DOI] [PubMed] [Google Scholar]
- Spiteller G. Linoleic acid peroxidation--the dominant lipid peroxidation process in low density lipoprotein--and its relationship to chronic diseases. Chemistry and Physics of Lipids. 1998;95:105–162. doi: 10.1016/s0009-3084(98)00091-7. Available at http://www.ncbi.nlm.nih.gov/pubmed/9853364. DOI S0009308498000917 [pii] [DOI] [PubMed] [Google Scholar]
- Stevens T, Berk MP, Lopez R, Chung YM, Zhang R, Parsi MA, Bronner MP, Feldstein AE. Lipidomic profiling of serum and pancreatic fluid in chronic pancreatitis. Pancreas. 2012;41:518–522. doi: 10.1097/MPA.0b013e31823ca306. Available at http://www.ncbi.nlm.nih.gov/pubmed/22504378. DOI 10.1097/MPA.0b013e31823ca30600006676-201205000-00029 [pii] [DOI] [PubMed] [Google Scholar]
- U.S. Department of Agriculture, A.R.S, 2010. What we eat in america. NHANES; 2007–2008. [Google Scholar]
- Vonaparti A, Lyris E, Angelis YS, Panderi I, Koupparis M, Tsantili-Kakoulidou A, Peters RJB, Nielen MWF, Georgakopoulos C. Preventive doping control screening analysis of prohibited substances in human urine using rapid-resolution liquid chromatography/high-resolution time-of-flight mass spectrometry. Rapid Communications in Mass Spectrometry. 2010;24:1595–1609. doi: 10.1002/Rcm.4554. Available at <Go to ISI>://000278661200012. [DOI] [PubMed] [Google Scholar]
- Wang J, Leung D. Analyses of macrolide antibiotic residues in eggs, raw milk, and honey using both ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry and high-performance liquid chromatography/tandem mass spectrometry. Rapid Communications in Mass Spectrometry. 2007;21:3213–3222. doi: 10.1002/Rcm.3207. Available at <Go to ISI>://000249613000008. [DOI] [PubMed] [Google Scholar]
- Wewer V, Dombrink I, vom Dorp K, Dormann P. Quantification of sterol lipids in plants by quadrupole time-of-flight mass spectrometry. Journal of Lipid Research. 2011;52:1039–1054. doi: 10.1194/jlr.D013987. Available at http://www.ncbi.nlm.nih.gov/pubmed/21382968. DOI jlr.D013987 [pii] 10.1194/jlr.D013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelan P, Zirrolli JA, Murphy RC. Low-energy fast atom bombardment tandem mass spectrometry of monohydroxy substituted unsaturated fatty acids. Biological Mass Spectrometry. 1993;22:465–473. doi: 10.1002/bms.1200220808. Available at http://www.ncbi.nlm.nih.gov/pubmed/8357860. DOI 10.1002/bms.1200220808. [DOI] [PubMed] [Google Scholar]
- Wu Z, Gao W, Phelps MA, Wu D, Miller DD, Dalton JT. Favorable effects of weak acids on negative-ion electrospray ionization mass spectrometry. Analytical Chemistry. 2004;76:839–847. doi: 10.1021/ac0351670. Available at http://www.ncbi.nlm.nih.gov/pubmed/14750883. DOI 10.1021/ac0351670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Itoh N, Hayakawa M, Habuchi Y, Inoue R, Chen ZH, Cao J, Cynshi O, Niki E. Lipid peroxidation in mice fed a choline-deficient diet as evaluated by total hydroxyoctadecadienoic acid. Nutrition. 2006;22:303–311. doi: 10.1016/j.nut.2005.07.020. Available at http://www.ncbi.nlm.nih.gov/pubmed/16500556. DOI S0899-9007(05)00318-7 [pii] 10.1016/j.nut.2005.07.020. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Itoh N, Hayakawa M, Piga R, Cynshi O, Jishage K, Niki E. Lipid peroxidation induced by carbon tetrachloride and its inhibition by antioxidant as evaluated by an oxidative stress marker, hode. Toxicology and Applied Pharmacology. 2005;208:87–97. doi: 10.1016/j.taap.2005.01.015. Available at http://www.ncbi.nlm.nih.gov/pubmed/16164964. DOI S0041-008X(05)00040-2 [pii] 10.1016/j.taap.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Yoshikawa A, Kinumi T, Ogawa Y, Saito Y, Ohara K, Yamamoto H, Imai Y, Niki E. Hydroxyoctadecadienoic acid and oxidatively modified peroxiredoxins in the blood of alzheimer’s disease patients and their potential as biomarkers. Neurobiology of Aging. 2009;30:174–185. doi: 10.1016/j.neurobiolaging.2007.06.012. Available at http://www.ncbi.nlm.nih.gov/pubmed/17688973. DOI S0197-4580(07)00252-7 [pii] 10.1016/j.neurobiolaging.2007.06.012. [DOI] [PubMed] [Google Scholar]