

Abstract
Cigarette smoke (CS) exposure is the primary risk factor for the development of chronic obstructive pulmonary disease (COPD). COPD is characterized by chronic peribronchial, perivascular and alveolar inflammation. The inflammatory cells consist primarily of macrophage, neutrophils and lymphocytes. Although myeloid cells are well studied, the role of lymphocyte populations in pathogenesis of COPD remains unclear. Using a mouse model of CS-induced emphysema our laboratory has previously demonstrated that CS exposure causes changes in the T cell receptor repertoire suggestive of an antigen specific response and triggers a pathogenic T cell response sufficient to cause alveolar destruction and inflammation. We extend these findings to demonstrate that T cells from CS-exposed mice of Balb/cJ or C57B6 strain are sufficient to transfer pulmonary pathology to CS-naïve, immunosufficient mice. CS exposure causes a proinflammatory phenotype among pulmonary T cells consistent with from COPD patients. We provide evidence that donor T cells from CS-exposed mice depend on antigen recognition to transfer alveolar destruction using MHC class I deficient recipient mice. Neither CD4+ nor CD8+ T cells from donor mice exposed to CS are alone sufficient to cause inflammation or pathology in recipient mice. We found no evidence of impaired suppression of T cell proliferation among regulatory T cells from CS-exposed mice. These results suggest that CS exposure initiates an antigen specific response that leads to pulmonary destruction and inflammation that involves both CD8+ and CD4+ T cells. These results are direct evidence for an autoimmune response initiated by CS exposure.
Introduction
From 1970 to 2002 the age-adjusted mortality attributable to COPD has doubled and is now the third leading cause of disease in the United States (1, 2). Worldwide, COPD affects an estimated 63 million people and was the cause of 3 million deaths in 2004 (3). Approximately 10% of the global population over the age of 40 exhibits airway obstruction consistent with GOLD stage II or greater (4). The predominant risk factor for COPD is exposure to cigarette smoke (CS), although occupational exposures confer a significant risk for COPD development and indoor burning of biomass fuels is important risk factor in some developing countries (5).
COPD is a progressive disease of airway obstruction characterized by emphysema, airway remodeling, chronic bronchitis and frequent respiratory infections. Inflammatory processes are important drivers of COPD, and previous research has identified a role for virtually all major leukocyte populations in the inflammatory response to CS (6). The majority of studies have focused on macrophage- and neutrophil-derived proteases being causative factors in CS-induced tissue destruction (7, 8). Furthermore, alveolar macrophages from COPD patients exhibit a consistent gene expression pattern that is marked by activation of proteases and proinflammatory cytokines (9).
Recent research has lead to an increased awareness for the role of lymphocyte populations in the development and progression of COPD. Our laboratory has shown that CS exposure induces NKG2D ligands that activate natural killer cells thereby contributing to tissue destruction (10). Persistent activation of natural killer cells through NKG2D during CS exposure leads to hyperreactive viral responses in a mouse model (11). A growing body of research has identified changes among the lymphocytes that make up the adaptive immune system, B cells and T cells, in COPD patients compared to never smokers and smokers without COPD. Circulating T cells in COPD patients produce more IFNγ, and disease severity is correlated with T cell activation (12). CD4+ T cells in the lungs of COPD patients are skewed towards a Th1 phenotype, and T cell-derived cytokines were linked to the expression of proteases by macrophages (13). The number of CD8+ T cells in the lungs of COPD patients correlate with disease severity (14, 15) and they exhibit enhanced expression of cytokines and cytotoxic proteins (16, 17). Furthermore, mice deficient in CD8+ T cells are protected from CS-induced inflammation and emphysema (18). Mice exposed to CS and patients with COPD both exhibit an increase in the number of B cell follicles are increased in the lung (19). These studies point to the involvement of every major lymphocyte population in the inflammatory response to chronic CS exposure.
In 2003, Cosio published an editorial presenting an argument for an autoimmune component to COPD based on the apparent involvement of T and B cells in COPD and other clinical features (20). Since this publication, the investigation of potential autoimmune mechanisms in COPD has rapidly increased. Oligoclonal expansions in CD4+ T cells, indicative of an antigen specific T cell response, have been identified in the lungs of COPD patients (21). CD4+ T cells from the peripheral blood of COPD patients proliferated and produced proinflammatory cytokines when stimulated with elastin fragments (22). A number of recent studies have identified autoantibodies in COPD patients that bind epithelial (23), endothelial (24), smooth muscle (25) or extracellular matrix antigens (22). Autoantibodies against carbonyl modified proteins generated in response to CS exposure have also been identified in COPD patients (26). Our laboratory has avoided many potential confounders associated with the clinical observations of immune function in COPD patients due to concomitant infections and cancer by using a mouse model of CS exposure. We found evidence for an antigen specific response among pulmonary T cells of mice chronically exposed to CS in the form of oligoclonal expansions within the CD4+ and CD8+ T cell receptor repertoire that persisted following smoke cessation (27). We have also demonstrated that T cells from the lungs of CS-exposed mice are sufficient to cause pulmonary pathology in CS-naïve Rag2−/− recipients providing direct evidence for an autoimmune response against lung tissue initiated by CS exposure according to Witebsky’s revised postulates (28, 29).
In this study, we provide a comprehensive characterization of the functional consequences of chronic CS exposure on T cells in a mouse model of COPD. We report the effects of chronic CS exposure on the phenotype and function of CD4+, CD8+, and regulatory T cells and demonstrate that the ability of T cells to drive pulmonary pathology is dependent upon antigen recognition by CD8+ T cells. Specifically, we find that CD4+ and CD8+ T cells from the lungs of CS-exposed mice exhibit enhanced proinflammatory cytokine production. Additionally, we perform the most thorough investigation of the effects of CS on regulatory T cell function. We demonstrate that T cell dysregulation in CS-exposed mice is not accompanied by defects in the regulatory T cell (Treg) population or resistance among CD4+ and CD8+ T cells to Treg control. Finally, we show that the ability of T cells from CS-exposed donors to transfer pathology to recipients requires both CD4+ and CD8+ T cells and is dependent upon antigen presentation on MHC class I in recipient mice. Together, these findings suggest that CS exposure promotes functional changes among T cell populations that are sufficient to cause lung pathology and inflammation that is antigen dependent.
Methods
Mice
Female Balb/cJ and C57B6 wild-type aged 8–12 weeks used in this study were obtained from The Jackson Laboratory (Bar Harbor, ME) and Taconic Farms respectively. C57B6 Ciita−/− and C57B6 B2m−/− were obtained from The Jackson Laboratory and bred in University of Cincinnati facilities. Mice were housed according to institutional guidelines, and the Institutional Animal Care and Use Committee at the University of Cincinnati Medical Center reviewed and approved all experimental protocols. Mice were euthanized by intraperitoneal injection of sodium pentobarbital followed by exsanguination.
Cigarette smoke exposure
Mice were exposed to either filtered air or cigarette smoke generated from burning 3R4F Kentucky Reference Cigarettes (University of Kentucky) using a TE-10z smoking machine as described previously (28) (Teague Enterprises, Woodland, CA). Mice were exposed whole body for 4 hours/day, 5 days/week for 24 weeks. The exposure chambers were maintained at concentration of 150 +/− 15mg/m3 total suspended particulates and CO at this concentration was 400 +/− 30ppm.
Bronchoalveolar lavage
Lungs were lavaged twice with 1ml of Hanks’ balanced salt solution (HBSS). The first bronchoalveolar lavage (BAL) was centrifuged at 300 × g for 10 minutes and the supernatant was recovered and stored at −80C. The cell pellet was then resuspended with the second lavage and centrifuged at 300 × g for 10 minutes. The supernatant was discarded and the cell pellet was resuspended in HBSS/2% fetal bovine serum. Total cells were counted with hemacytometer. For differential leukocyte counts, cells were adhered to slides using a Cytospin3 (Shandon Scientific Ltd, Waltham, MA) and stained with Hemacolor.
Isolation of mouse lung leukocytes
After euthanization, lungs were perfused by injecting 5ml of PBS/0.6mM EDTA through a cannula inserted into the right ventricle. Lungs were dissected away from surrounding tissue and chopped before adding media containing digestion enzymes (RPMI-1640, 20mM HEPES, 10% FCS, 175U/ml Collagenase, 75U/ml Dnase I, 0.2U/ml pancreatic elastase, 35U/ml Hyaluronidase, 100 IU/ml penicillin, and 100ug/ml streptomycin). Lung pieces were incubated in digestion media at 37C for 45 minutes. The resulting suspension was passed through a 19ga needle three times to break up clumps and passed through a 40μm filter to remove debris. Lung leukocytes were then enriched by centrifuging in a discontinuous Percoll gradient and recovering at the interface between the 40% Percoll and the 70% Percoll layers.
Preparation of mouse lungs and histology
Mouse lungs were inflation-fixed with buffered formalin instilled through the trachea at a pressure of 25–30 cm H2O. The trachea was then tied off and the heart lung block was excised and placed in 25ml of buffered formalin. Lungs were embedded in paraffin, sectioned, and slides stained with hematoxylin and eosin at the Cincinnati Childrens’ Hospital Medical Center Pathology Core. Mean linear intercept (MLI) was measured as previously described (30). Focal areas of inflammation were classified as slight (<25 cells), mild (25–100 cells), moderate (7500–20,000μm2) or severe (>20,000μm2) and according to the morphological features the inflammation was associated with. Inflammation scores for individual mice were obtained by summing the instances of inflammation weighted for severity as follows: slight = 1, mild = 2, moderate = 4, and severe = 8.
Measurement of T cell-derived cytokines
Lung leukocytes were isolated from Balb/cJ mice exposed to either FA or CS for six months as described above and either CD4+ or CD8+ T cells were isolated by negative selection using Dynabeads magnetic separation kit (Invitrogen). Isolated CD4+ and CD8+ T cells were stimulated with phorbol myristate acetate (50 ng/ml, Sigma Aldrich) and ionomycin (500 ng/ml, Sigma Aldrich) in the presence of Brefeldin A (3 ug/ml, eBioscience). After 5 hours, cells were recovered and stained for the surface antigens PE-CD4 (clone GK1.5) or PE-CD8a (clone 53-6.7) and then fixed with 3% paraformaldehyde for 15 minutes and stained for intracellular antigens FITC-IL-4 (clone BVD6-24G2), PerCP-Cy5.5 IL-17A (clone eBio17B7), APC-IFNγ (clone XMG1.2), APC-TNFα (clone MP6-XT22) or FITC-IFNγ (clone XMG1.2). All washing and staining steps for detection of intracellular antigens were performed with 1x Permeabilization Buffer (eBioscience). Data was acquired on a BD FACSCalibur and analyzed using FlowJo 7.6.2 (Tree Star).
Isolation and adoptive transfer of mouse pulmonary T cells
Lung leukocytes were isolated from Balb/cJ or C57B6 mice exposed to either FA or CS for six months as described above and then T cells were enriched by negative selection using Dynabeads magnetic separation kit (Invitrogen). Total T cells, CD4+ T cells or CD8+ T cells were then isolated to >99% purity by fluorescence activated cell sorting on the basis of CD3e expression (APC, clone 145-2C11), CD4 expression (FITC, clone GK1.5) or CD8a expression (PE, clone 53-6.7), respectively. Isolated T cells were then expanded ex vivo using Dynabeads T-Activator beads (Invitrogen) coated with antibodies against CD3, CD28, and CD137 for 7–10 days. Expanded T cells were washed twice and resuspended in HBSS and 2 × 106 cells were injected intraperitoneally into WT Balb/cJ, WT C57B6, C57B6 Ciita−/−, or C57B6 B2m−/− mice. Recipient mice were sacrificed 12 weeks after transfer and lungs were inflated and fixed for histology or lavaged. None of the recipient mice died during the 12 weeks before they were sacrificed.
Regulatory T cell suppression assay
Balb/cJ mice exposed to either FA or CS were euthanized and spleens were harvested and pooled from 6 mice per group. Spleens were pressed through 100μm filters and red blood cells were lysed using RBC Lysis Solution (Qiagen). Ninety percent of the splenocytes were used for negative selection of CD4+ T cells using Dynabeads magnetic separation kit. Enriched CD4+ T cells were stained with FITC-CD4 (clone GK1.5) and APC-CD25 (clone PC61.5). Tregs, CD4+CD25+, were isolated from the negatively selected CD4+ T cells by fluorescence activated cell sorting (FACS). Total T cells were enriched by positive selection from the remaining 10% of splenocytes using Dynabeads FlowComp Mouse Pan T kit. Enriched total T cells were stained with FITC-CD4 (clone GK1.5), PE-CD8a (clone 53-6.7), and APC-CD25 (clone PC61.5). T responders, CD8+ or CD4+CD25−, were isolated from the enriched T cells by FACS. Tregs were stained with 5 μM eFluor 670 (eBioscience) and T responders were stained with 7.5 μM CFSE (Invitrogen). Tresponders and Tregs were cultured at ratios ranging from 16:1 to 0.5:1. T responders were stimulated with Dynabeads M-450 Epoxy (Invitrogen) coated with anti-mouse CD3e antibody (clone 145-2C11). Four days later, proliferation among Tresponders was measured by CFSE dilution using a BD FACSCalibur. FlowJo 7.6.2 was used to analyze the results and calculate the division index, which represents the mean number of divisions undergone by the Tresponders. This experiment was independently performed three different times.
Fluorescence activated cell sorting
Fluorescence activated cell sorting was performed at Cincinnati Childrens’ Hospital Medical Center Research Flow Cytometry Core using a BD FACSAria II. Cells were stained with APC-CD3e (clone 145-2C11), PE-CD8a (clone 53-6.7), FITC-CD4 (clone GK1.5), or APC-CD25 (clone PC61.5).
Statistics
Significant differences between groups with respect to MLI, Inflammation Score, and total cells recovered from the BAL were determined by Student’s t test (SigmaPlot 10.0). Inflammation scores were transformed by taking the square root of the raw scores and the total numbers of cells were transformed using Log10 of the raw counts. In every instance the Student’s t test was used, normality was assessed using Kolmogorov-Smirnoff test and homogeneity of variances were tested using SigmaPlot 10 to ensure that the assumptions of the Student’s t test were not violated. A P value of less than 0.05 was considered significant.
For comparisons of the proportion of CD4+ T cells or CD8+ T cells that expressed cytokines by flow cytometry, the odds ratios were calculated to assess the impact of CS exposure on the likelihood of cytokine expression (31). The odds ratio is defined as [PCS/(1−PCS)]/[PFA/(1−PFA)] where PCS is the proportion of cells expressing a cytokine from CS exposed animals and PFA is the proportion of cells expressing a cytokine from FA exposed animals. Odds ratios equivalent to 1 indicate the treatment (CS) has no effect on the proportion. Cytokine expression was measured in T cells pooled from the lungs of 6 mice and repeated in independent experiments. The results from the two independent experiments were combined to create a pooled odd ratio. Pooled odds ratios and 95% confidence intervals were calculated using the meta package in R version 2.15.1.
Results
Lung derived T cells from CS-exposed donor mice transfer pathology and inflammation to CS-naïve, immunocompetent recipients
We previously reported that T cells isolated from the lungs of CS-exposed mice were sufficient to cause airspace enlargement and drive inflammation in immunodeficient recipient mice (28). To further confirm and extend these findings to the use of immunocompetent recipient mice, we transferred total T cells isolated from the lungs of mice exposed to either FA or CS for 6 months into immunosufficient, CS-naïve Balb/cJ mice. Histopathological examination revealed significant airspace enlargement in mice that received T cells from CS-exposed donors compared to recipients of T cells from FA donors (Fig. 1A–C). Gross macroscopic analysis did not indicate inflammation or pathologies in the gastrointestinal tract, skin or eyes. The total number of cells recovered in the bronchoalveolar lavage was elevated in mice that received T cells from CS-exposed donors compared to those that received T cells from FA-exposed donors (Fig 1D). Cell differential analysis of the bronchoalveolar lavage revealed a majority of macrophage and monocytes in both groups (Fig 1E–F). These data confirm and extend our previous findings that CS exposure leads to the generation of pathogenic T cells capable of driving pathology and architectural changes in the lung independent of subsequent smoke exposure in the recipient mouse.
Figure 1. T cells from CS-exposed donor mice cause emphysema and airway inflammation in immunocompetent recipient mice.

Donor Balb/cJ mice were exposed to either FA or CS for 6 months. T cells were isolated by FACS and expanded ex vivo before transferring 2×106 cells per recipient. Recipient mice were sacrificed 12 weeks post transfer. A. Recipients of T cells from CS donors exhibited increased mean linear intercepts indicating alveolar destruction and increased alveolar diameter (N=4 mice/group, P=0.011). B, C. Representative micrographs of H&E stained lung sections from mice that received T cells from either FA-(B) or CS-(C) exposed donor mice showing extent of airspace destruction (scale bars = 100μm). D. T cells from donors exposed to CS caused an increase in the number of inflammatory cells recovered from the bronchoalveolar lavage (N=6 mice/group, P=0.041). E, F. Representative fields showing predominantly macrophages and monocytes recovered from the bronchoalveolar lavage (scale bars = 25μm).
Chronic CS exposure increases the proinflammatory capacity of CD8+ T cells
We examined the cytokine expression profile of CD8+ T cells isolated from the lungs of mice chronically exposed to either FA or CS to better understand the mechanisms through which T cells from CS-exposed mice are able to promote inflammation and alveolar destruction. The proportion of CD8+ T cells that stained double positive for both TNFα and IFNγ was higher among cells isolated from the lungs of CS-exposed mice compared to FA-exposed mice (Fig 2A–B). Furthermore, the geometric mean fluorescent intensity of TNFα staining was higher for CD8+ T cells isolated from the lungs of CS-exposed mice than for those isolated from the lungs of FA exposed mice although the statistical significance of this observation cannot be determined based on the study design (Fig 2C). Similarly, the proportion of CD8+ T cells expressing only TNFα was higher among cells isolated from the lungs of CS-exposed mice compared to those collected from the lungs of FA-exposed mice (Fig 2A–B). Parallel experiments performed without the addition of Brefeldin A also showed a modest increase in IFNγ production by ELISA among CD8+ T cells isolated from the lungs of CS-exposed mice compared cells from FA-exposed mice (Fig 2D). Taken together these results suggest that chronic CS exposure promotes a proinflammatory phenotype among CD8+ T cells in the lung by enhancing expression of IFNγ and TNFα.
Figure 2. Cigarette smoke exposure promotes the expression of proinflammatory cytokines IFNγ and TNFα among CD8a+ T cells in the lung.
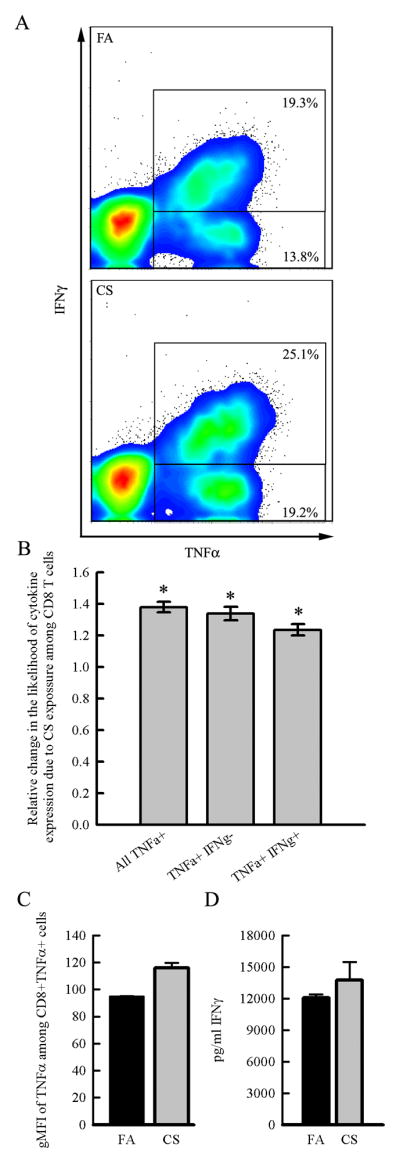
Balb/cJ mice were exposed for 6 months to FA or CS and CD8+ T cells were isolated from digested lungs by negative selection and stimulated with PMA and ionomycin in vitro with or without the presence of Brefeldin A so that cytokine expression could be measured by intracellular flow cytometry or ELISA. CD8+ T cells were pooled from N=6 mice/group and repeated in an independent experiment. A. Representative density plots indicate that chronic CS exposure results in an increase in the proportion of CD8a+ T cells that express TNFα alone or both TNFα and IFNγ compared to the CD8a+ T cells from FA exposed mice. B. Odds ratios comparing the proportion of CD8+ T cells that are TNFα+, TNFα+ IFNγ+, or TNFα+IFNγ− between FA- and CS-exposed mice indicate that CS increases the likelihood that CD8+ T cells express TNFα and IFNγ. Error bars represent the 95% confidence intervals for the pooled odds ratios from two independent experiments. C. The geometric mean fluorescent intensity (gMFI) of TNFα staining is higher among CD8a+TNFα+ cells from CS-exposed mice compared to FA-exposed mice indicating higher production of TNFα. D. CD8+ T cells from CS-exposed mice stimulated in vitro in the absence of Brefeldin A produced more IFNγ as measured by ELISA.
Chronic CS exposure increases the proportion of Th1 and Th17 CD4+ T cells in the lung
We next sought to understand the effects of chronic CS exposure on the accumulation of Th1, Th2 and Th17 polarized CD4+ T cells in lung. CD4+ T cells were isolated from the lungs of mice exposed to either FA or CS and stimulated ex vivo to reveal their cytokine expression profile. Intracellular flow cytometry was used to identify Th1 (CD4+, IFNγ+, IL-4−, IL-17A−), Th2 (CD4+, IL-4+, IFNγ−, IL-17A−), and Th17 (CD4+, IL-17A+, IFNγ−, IL-4−) subsets based on cytokine expression. Chronic CS exposure significantly increased the proportion of pulmonary Th1 and Th17 CD4+ T cells, without any discernible effect on the proportion of Th2 cells (Fig 3A–B). Parallel experiments were performed in the absence of Brefeldin A and ELISA was performed on the cell culture supernatant to measure the production of IFNγ and IL-17A. In accordance with the flow cytometry data, the CD4+ T cells from CS-exposed mice produced more IFNγ and IL-17A in the culture supernatant compared to cells isolated from FA-exposed mice (Fig 3C). These results suggest that chronic CS exposure skews CD4+ T cells towards Th1 and Th17 phenotypes without affecting the Th2 population.
Figure 3. CD4+ T cells in the lung are skewed towards a Th1 and Th17 phenotype in response to chronic cigarette smoke exposure.
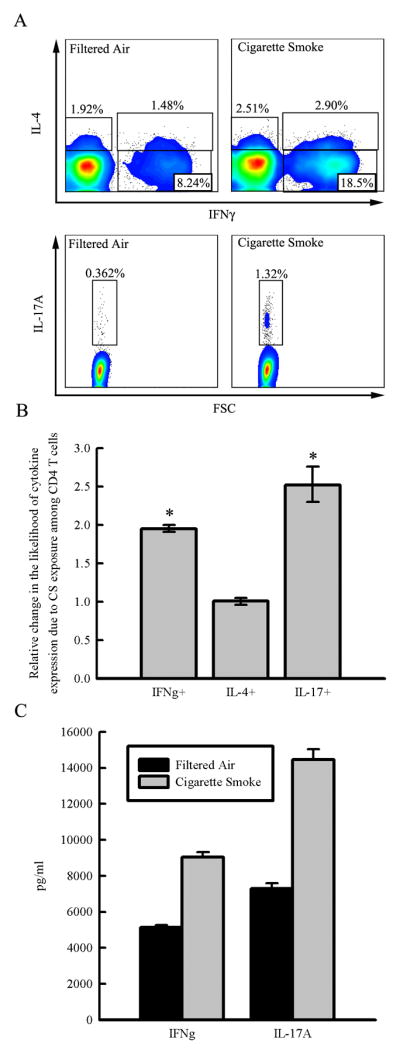
Balb/cJ mice were exposed for 6 months to FA or CS and CD4+ T cells were isolated from lung digestion by negative selection and stimulated with PMA and ionomycin in vitro with or without the presence of Brefeldin A so that cytokine expression could be measured by intracellular flow cytometry or ELISA. CD4+ T cells were pooled from N=6 mice/group and repeated in an independent experiment. A. Representative density plots showing the cytokine expression profile among CD4+ T cells and the gates used to identify the CD4+ subsets. B. Odds ratios comparing the proportion of CD4+ T cells expressing each cytokine demonstrate that CS exposure increases the likelihood that CD4+ T cells express IFNγ and IL-17, but not IL-4. Pooled odds ratios were calculated from two independent experiments and error bars represent the 95% confidence intervals. C. CD4+ T cells isolated from the lungs of FA and CS-exposed mice were stimulated ex vivo with PMA and ionomycin and 48 hours later the production of IFNγ and IL-17A was measured by ELISA in the supernatant.
CS exposure does not reduce the number or suppressive ability of regulatory T cells
We hypothesized that the generation of pathogenic T cells in response to chronic CS exposure may be accompanied by defects in the regulatory T cell (Treg) population given the role of Tregs in peripheral tolerance and immunoregulation. Therefore, we investigated the effects of chronic CS exposure on the number and function of Tregs. We performed flow cytometry to identify the number of Tregs (CD4+, CD25high, FoxP3+) in the lungs and lung-draining lymph nodes of FA- and CS-exposed mice. We found no difference in the proportion or absolute number of Tregs in the lungs and lung draining lymph nodes between FA- and CS-exposed mice (Fig 4A–B).
Figure 4. Cigarette smoke exposure does not alter regulatory T cell number or function.

A.– B. Balb/cJ mice were exposed for 6 months to either CS or FA and single cell suspensions were prepared from lung digestion or mediastinal lymph nodes. Regulatory T cells (CD4+CD25+FoxP3+) were enumerated by flow cytometry. A. Representative dot plots gated on CD4+ T cells showing the percentage of CD4+ T cells that are regulatory T cells (CD4+CD25+FoxP3+) in lungs and mediastinal lymph nodes. B. Regulatory T cells as a proportion of both CD4+ T cells and total cells recovered from either digested lungs or mediastinal lymph nodes of mice exposed to FA or CS for 6 months. Data represented as mean +/− SE; N=3 mice/group. C.– D. Regulatory T cells (CD3e+CD4+CD25high) and conventional T responders (CD3+CD8+ or CD3+CD4+CD25−) were isolated from the spleens of Balb/cJ mice exposed to FA or CS. Tregs and T responders were cocultured and stimulated with beads coated with anti-CD3e antibodies. Cells were pooled from N=6 mice/group and the experiment was repeated twice for a total of three independent experiments. C. Suppression of proliferation was measured by comparing the division index of CD4+ T responders cultured with Tregs to the division index of CD4+ T responders cultured and stimulated in the absence of Tregs. Data shows the mean percent suppression of division index from three independent experiments. D. The percent change in CD4− T cell viability based on 7-AAD staining. Data shows the mean percent of 7-AAD+ CD4− Tresponders from the three independent experiments.
We isolated Tregs (CD4+, CD25high) and T responders (CD8+ or CD4+, CD25−) from the spleens of mice exposed to CS or FA and cocultured them to define the effects of CS exposure on the ability of Tregs to suppress T responders activated in vitro. The four possible combinations of Tregs and T responders from FA and CS-exposed mice (FA Tregs:FA Tresp, FA Tregs:CS Tresp, CS Tregs:FATresp, CS Tregs:CS Tresp) were co-cultured to assess whether differences in suppression of proliferation were due to resistance to suppression by the T responders or defective suppression among Tregs.
Tregs from FA- and CS-exposed mice were not different in their ability to suppress the proliferation of CD4+ T responders from both FA- and CS-exposed mice across a wide range of Treg/T responder ratios (Fig. 4C–D). Furthermore, there did not appear to be any resistance to Treg suppression among the Tresponders based on the combinations of FA Tregs:CS Tresp or CS Tregs:FA Tresp (Fig. 4C).
Interestingly, we found that CD4+CD25− Tresponders and CD8+ Tresponders react differently to coculture with Tregs. Tregs suppress CD4+CD25− Tresponders by reducing the rate cellular division; however, CD8+ Tresponders are suppressed by inducing cell death without affecting the rate of proliferation among the surviving CD8+ Tresponders (data not shown). This suggests that Tregs counter the proliferation of CD4− T cells in vitro by inducing cell death, likely through apoptotic mechanisms. In our experiments the Tregs isolated from mice exposed to CS did not differ in their ability to induce cell death among CD4− Tresponders compared to FA Tregs and Tresponders from CS-exposed mice were as susceptible to induced cell death as Tresponders from FA-exposed mice (Fig 4D). These findings demonstrate that CS exposure does not impair the ability of Tregs to suppress Tresponder activation in vitro nor does it cause resistance among Tresponders to Treg control.
T cells from CS-exposed C57B6 mice transfer alveolar pathology and inflammation to CS-naïve recipients
Pathogenic T cells generated during chronic CS exposure may be responding to a specific antigen or they may be promoting inflammation and damage in a nonspecific manner. We can test if antigen recognition in the recipient is necessary for total T cells to the transfer pathology by using mice that are deficient in either MHC class I or MHC class II. Furthermore we can identify the relative contribution of CD8+ T cells and CD4+ T cells to the inflammation and airspace destruction based on their restriction to MHC class I and MHC class II presentation respectively. Using MHC class I deficient (B2m−/−) and MHC class II deficient recipient mice (Ciita−/−) on the C57B6 background necessitated the validation of our transfer model using donors and recipient mice of the C57B6 strain. We performed the transfer experiment using C57B6 donors exposed to FA or CS and CS-naïve, C57B6 recipient mice. Histological examination of the recipients showed that T cells from CS-exposed donors induce alveolar destruction (Fig 5A–C). We also observed an increase in focal areas of inflammation in the lungs of mice that received T cells from CS-exposed donors (Fig 5D), dominated by perivascular inflammation in recipients of T cells from CS-exposed donors (Fig 5E & 5F). These focal areas of inflammation are a point of divergence between Balb/cJ and C57B6 background. T cells from CS-exposed donors also cause increased recruitment of inflammatory cells, mostly myeloid cells, to the airways as indicated by an increase in the total number of cells recovered from bronchoalveolar lavage (Fig 5G). These results demonstrate that the T cell transfer model can be appropriately applied to the C57B6 mouse strain opening up the possibility of using transgenic mice on the C57B6 background to explore putative mechanisms. Furthermore, these results indicate that the ability of T cells from CS-exposed donors to transfer pathology to CS-naïve recipients is not a phenomenon peculiar to the Balb/cJ strain.
Figure 5. MHC class I expression in recipient mice is required for T cells to transfer emphysema and neither CD4+ nor CD8+ T cells from CS-exposed donor mice are sufficient to cause emphysema when transferred separately.

A.– F. Donor C57B6 mice were exposed to FA or CS for 6 months and CD3e+ T cells were isolated from the lungs. C57B6 recipient mice received 2×106 T cells/mouse and were sacrificed 12 weeks later and lungs were inflated and fixed for histology. A. C57B6 mice that received T cells from CS-exposed donor mice exhibited increased mean linear intercept suggestive of emphysema (P<0.01, n=6–8 mice/group). B.– C. Representative micrographs of H&E-stained lung sections from mice that received T cells from FA donors (B.) and mice that received T cells from CS donors (C.) showing the extent of alveolar damage in the mice that received T cells from CS-exposed donors (scale bars = 100μm). D.– F. Focal areas of inflammation were scored by number of occurrences and severity in H&E-stained lung sections from C57B6 mice that received total T cells from FA- or CS-exposed donor mice. D. Mice that received T cells from CS-exposed donors had a higher inflammation score (p = 0.038, N = 8–9 mice/group). E. Focal areas of inflammation were classified according to nearby pulmonary features and the histology score for each category was summed for all 9 mice that received T cells from CS-exposed donors. F. Representative micrograph of H&E stained lung section showing moderate perivascular inflammation recipient of T cells from CS-exposed donors (scale bar = 50μm). G.– H. CD4+ and CD8+ T cells were isolated separately from C57B6 donor mice exposed to FA or CS for 6 months and injected into recipient mice. Recipient mice were sacrificed 12 weeks post transfer and lungs were inflated and fixed for histology or lavaged and frozen. G. CD8+ T cells from CS-exposed donor mice are sufficient to cause inflammation in the airways as evidenced by an increase in the total cells recovered from bronchoalveolar lavage (p = 0.024, N = 4 mice/group). H. Neither CD4+ T cells nor CD8+ T cells are sufficient to drive airspace enlargement in recipient mice. No significant difference in MLI is evident suggesting that both CD4+ and CD8+ T cells from CS-exposed mice are required to cause airspace enlargement or emphysema (N = 3–7 mice/group). I.– K. Donor mice were exposed to FA or CS and total T cells (CD3e+) were isolated from their lungs and expanded ex vivo. Total T cells from either FA- or CS-exposed donors were transferred into B2m−/− recipient mice at 2×106 cells/mouse. Recipient mice were sacrificed 12 weeks post transfer and lungs were prepared for histology or lavaged and frozen. I. MHC class I expression in recipient mice is required for T cells from CS-exposed mice to cause increased airspace enlargement compared to recipients of T cells from FA exposed donors. There is no difference between the MLI of B2m−/− recipients that received total T cells from FA exposed mice and those that received T cells from CS-exposed mice (N=3–7 mice/group). J. MHC class I expression in recipient mice is required for T cells from CS-exposed mice to transfer airway inflammation. There is no difference in total cells recovered from bronchoalveolar lavage of B2m−/− mice that received T cells from FA exposed donors and those that received T cells from CS-exposed donors (n=3 mice/group). K. MHC class I expression in recipient mice is necessary for T cells from CS-exposed donors to drive focal areas of inflammation. There is no difference in the histological inflammation score of B2m−/− mice that received T cells from FA exposed donors and those that received T cells from CS-exposed donors (n=5–9 mice/group).
Neither CD4+ nor CD8+ T cells from CS-exposed mice are sufficient to transfer alveolar destruction to CS-naïve recipients
We conducted a series of T cell transfer experiments on the C57B6 background to (i) identify whether CD4+ or CD8+ T cells from CS-exposed donors are sufficient to cause pulmonary pathology and inflammation and (ii) identify whether antigen presentation in the recipient is necessary for total T cells from CS-exposed donors to cause lung pathology. CD4+ and CD8+ T cells were isolated from FA- or CS-exposed donors and transferred separately into CS-naïve recipient mice. CD8+ T cells from CS-exposed donors, but not CD4+ T cells, were sufficient to cause pulmonary inflammation assessed by total cells recovered from the bronchoalveolar lavage (Fig 5G). However, upon histological examination, neither CD4+ T cells nor CD8+ T cells from CS-exposed donors were capable of causing alveolar destruction (Fig 5H) or increased leukocytic accumulation in the lungs of recipient mice (data not shown).
Recipient mice deficient in MHC class I are protected from airspace enlargement and inflammation caused by T cells transferred from CS-exposed mice
We used CS-naïve recipient mice deficient in either MHC class I expression (B2m−/−) or mice which lack MHC class II expression (Ciita−/−) to test whether total T cells from CS-exposed mice are responding to a specific antigen (or antigens) in the recipient mice. Unexpectedly, MHC class II deficient mice exhibited severe and profound pulmonary pathology including alveolar destruction, alveolar fibrosis and inflammation regardless of whether the donor T cells were from FA- or CS-exposed mice (data not shown). Ciita−/− is a transcriptional coactivator regulating MHC class II expression, therefore, we hypothesize that the phenomenon we observed when T cells were transferred into Ciita−/− recipients was due to a paucity of CD4+ T cells and Tregs or other unanticipated consequences of Ciita−/− deficiency. As such, we were unable to draw any conclusions based the effect of MHC class II deficiency on the ability of total T cells from CS-exposed donor mice to transfer pathology. When MHC class I deficient mice received T cells from FA-or CS-exposed mice there was no significant increase in alveolar diameter among recipient of T cells from CS-exposed mice compared to recipients of T cells from FA-exposed mice. This suggests that MHC class I expression in recipients is required for T cells from CS-exposed mice to cause alveolar destruction (Fig 5I). Similarly, MHC class I deficiency in recipient mice prevents T cells from CS-exposed donors to elicit an increase in the total cells recovered during bronchoalveolar lavage and in the formation of leukocytic accumulation by histology compared to T cells from FA exposed donors (Fig 5J–K).
Taken together, these results suggest that while CD8+ T cells from CS-exposed donors, alone, are sufficient to recruit immune cells to the airways, they cannot drive alveolar destruction and emphysema. MHC class I deficient hosts are also protected from alveolar airspace enlargement and airway inflammation caused by total T cells from CS-exposed mice. These experiments define the requirement for both CD4+ and CD8+ T cells from CS-exposed donor mice and antigen presentation to CD8+ T cells on MHC class I molecules for transfer of airspace enlargement and inflammation.
Discussion
Recent research using data generated from both patients and mouse models has lead to a greater appreciation of the role of T cells in the progression of COPD. Much of the research based on samples obtained from COPD patients is necessarily associative. Utilizing a mouse model of disease, our laboratory was able to build on our previous finding that T cells from the lungs of mice exposed to CS are sufficient to transfer pulmonary pathology to CS-naïve recipients (28). In the current study, we demonstrate that recipient mice must present antigen to CD8+ T cells and that CD4+ T cells and CD8+ T cells from CS-exposed donor mice are both necessary to transfer pathology to CS-naïve recipients. This is the most direct evidence to date that chronic CS exposure triggers the development of a T cell-mediated autoimmune process that is antigen specific. To our knowledge this is the first report of a murine autoimmune model initiated in response to an environmental insult in two different strains without genetic manipulation or immunization with self antigen. In addition, we confirm previous reports from patients with COPD and mice exposed to CS suggesting that the cytokine expression profiles of both CD4+ and CD8+ T cells are skewed towards a more proinflammatory phenotype after chronic CS exposure (12, 13, 16, 17, 32–34).
Interestingly, we find no evidence for any accompanying defect among Tregs in terms of their numbers or ability to suppress proliferation after chronic CS exposure. These data suggest that autoreactive T cells do not arise during chronic CS exposure as a simple consequence of compromised Treg function. Our results are in contrast to the report by Demoor, et al, which showed an increase in the number of Tregs both in the lungs and in the lung-draining lymph nodes of C57B6 mice after 6 months exposure to CS (35). Differences between Balb/cJ and C57B6 mice or differences in the exposure protocol may account for these discordant results. Additionally, Barcelo et al compared the proportion of CD4 T cells that were Tregs in the BAL of never smokers, COPD patients, and smokers without COPD and found that Tregs were elevated in smokers without COPD, but not in patients with COPD, compared to never-smokers (36). More research needs to be done to better understand the effects of CS on Tregs and how Tregs modulate CS-induced inflammation. However, this manuscript presents the most thorough investigation of the effects of CS on both the number and function of Tregs to date.
CD8+ T cells are increased in the airways of COPD patients, and cytokine and perforin expression among CD8+ T cells in the lungs of COPD patients correlates with disease severity (14–17). Furthermore, CD8-deficient mice on a C57B6 background are resistant to airspace enlargement after six months of CS exposure (18). We report that if total T cells from CS-exposed donor mice are transferred into recipients lacking MHC I expression, effectively blinding CD8+ T cells to antigen, then the ability to transfer airspace enlargement is blocked. This underscores the importance of CD8+ T cells in CS-induced inflammation and suggests that their activity is directed by specific recognition of a lung antigen. However, it is still unclear how CD8+ T cells promote pulmonary pathology after CS exposure, though it is likely through a combination of both direct and indirect mechanisms. CD8+ T cells may directly recognize and eliminate pulmonary epithelial through cytotoxic pathways while also producing IFNγ and TNFα to damage host tissue indirectly through the activation of neutrophils and macrophage. A large body of research describes the role of macrophage and neutrophils in lung destruction following chronic CS exposure (7–9). These results provide an attractive framework for synthesizing the research on adaptive and innate immune function in the progression of COPD by superimposing a role for CD8+ T cells as directors of macrophage and neutrophil activity that persists even after smoking cessation.
Alveolar airspace enlargement and inflammation were not different between MHC class I deficient mice that received T cells from FA- and CS- exposed donors. However, both groups of MHC class I deficient recipients had similar, marginal increases in alveolar airspace and inflammation compared to age-matched B2m−/− mice that did not receive T cells. Graft-versus-host disease (GVHD) between the B2m−/− recipients and the WT donor T cells may be one potential explanation for this artifact of the T cell transfers. We did not observe any gross pathology systemically in MHC class I deficient mice that received either T cells from FA- or CS-exposed mice. Other studies have used B2m−/− mice as recipients in adoptive transfer experiments to determine the requirement for antigen presentation in mouse models of autoimmune disease including non-obese diabetic mice and experimental autoimmune encephalomyelitis without any reported GVHD (37, 38). The amount of inflammation and airsparce enlargement is not severe nor accompanied by systemic effects in the B2m−/− mice that received T cells, therefore, the artifact is unlikely to be GVHD or is at the least very mild GVHD. Ultimately we conclude that T cells from CS-exposed mice were unable to cause an increase in MLI in MHC class I deficient mice compared to recipients of T cells from FA exposed donors suggesting that antigen presentation to CD8 T cells in the recipients is required for alveolar destruction caused by CS T cells.
Although CD8-deficient mice on the C57B6 background were protected from air space enlargement and inflammation after 6 months of CS exposure (18), two other reports demonstrated that the adaptive immune system is not required for the development of emphysema or recruitment of macrophage or neutrophils in response to 6 months of CS exposure using either scid or Rag2−/− mice, which lack both T cells and B cells, on Balb/cJ strain (28, 39). The conflict between these studies may simply be the result of genetic differences between Balb/cJ and C57B6 strains. However, the studies cast doubt on whether the adaptive immune system and, T cells specifically, are necessary for the development of COPD pathologies. Although the present study does not address this specific question, it is clear from these results, and from our previous work, that T cells from CS-exposed mice are sufficient to cause pulmonary pathology in CS-naïve recipient mice.
Awareness of the role of CD4+ T cells, particularly Th17 cells, in COPD and CS-induced inflammation has increased due to recent publications implicating CD4+ T cells and IL-17 in COPD (32–34). As reported previously, we see an increase in the proportion of CD4+ T cells that exhibit a Th17 or Th1 phenotype after CS exposure (32, 34, 40). However, our results indicate that CD4+ T cells may play a supporting role to CD8+ T cells in our model since CD8+ T cells from CS-exposed donors are not sufficient to cause pathology when transferred alone, although total T cells from CS-exposed donors are blocked from driving pathology when recipients lack MHC I expression. Alternatively, the inability of CD8+ T cells from CS-exposed donors to drive pathology when transferred alone may reflect regulatory elements in the host that control the donor CD8+ T cells but are inadequate when CD4+ T cells from the donor are included. CD4+ T cells are necessary for effective and long lasting CD8+ T cell memory (41–43). It is plausible to suggest that transferring CD8+ T cells from CS-exposed donors without the corresponding CD4+ T cells might deprive the CD8+ T cells of the CD4+ T cells necessary to maintain their pathogenic potential. The perpetuation of CD8+ T cell responses against a chronic viral infection, perhaps the infection model most similar to a CD8+ driven autoimmune disease, requires CD4+ T cell help (44). Our results may then suggest that although CD8+ T cells are the primary cell type promoting lung pathology, CD4+ T cells are nevertheless required for perpetuation of the pathogenic response.
Prior research demonstrating that CS exposure leads to the generation of pathogenic T cells suggested that CS exposure causes a breakdown in peripheral tolerance. Tregs are important in maintaining peripheral tolerance; therefore, we were surprised to find that Tregs were unaffected in number or ability to suppress T cell proliferation. Using all possible combinations of responding T cells and Tregs from both FA and CS-exposed mice indicated that the responding T cells from FA and CS-exposed mice were equally susceptible to Treg control. There are two significant implications of these findings: first, there are alternative pathways by which CS exposure causes a breakdown in tolerance and, second, the regulatory pathways activated in Tresponders by Tregs are intact and present a target for therapeutic intervention. However, it must be noted that our in vitro assay to assess Treg suppression only detects the effects of CS exposure Tregs to directly suppress T cell proliferation. A number of studies have shown the Tregs reduce the expression of costimulatory molecules on the surface of dendritic cells thereby preventing activation of naïve T cells (45, 46). Qureshi et al have identified a mechanism whereby CTLA-4 on the surface of Tregs binds to CD80 and CD86 on the surface of dendritic cells and leads to endocytosis of the costimulatory molecules (47). Using flow cytometry we did not find any differences in the expression of CTLA-4 on the surface of Tregs in the lungs or lung draining lymph nodes of mice exposed to CS (data not shown). While we did not find any effects of CS exposure on Treg numbers in the lungs or lung draining lymph nodes or on the ability of Tregs to suppress T cell proliferation, it is possible that CS exposure impairs the ability of Tregs indirectly to suppress activation of naïve T cells by modulating antigen presentation and costimulation by dendritic cells. One potential mechanism whereby CS leads to a breakdown of tolerance is through altering the relationship between dendritic cells and Tregs. Further research is necessary to identify the effects of CS exposure on the complex relationship between Tregs, dendritic cells and naïve T cells.
This report presents evidence that CS exposure drives a pathogenic T cell response that is antigen specific and driven primarily by CD8+ T cells. It details changes in the expression of cytokines by CD4+ and CD8+ T cells in the lungs of mice exposed to chronic CS that are also reflected in T cells recovered from the lungs or sputum of COPD patients. The development of pathogenic T cells in response to CS exposure is not accompanied with any appreciable defect in regulatory T cells indicating other means through which CS exposure causes a breakdown in peripheral tolerance. These findings are presently the most compelling evidence for the ability of chronic CS exposure to cause an antigen-specific T cell response against self tissue. Future studies should focus on translating this research towards therapy or screening tools. The T cell transfer model can also be used to test potential therapies that may slow the progression of COPD in patients whose disease includes an autoimmune component. Identification of the antigen recognized by pathogenic T cells that arise from chronic CS exposure would open the door to study the kinetics of T cell activation and earlier identification of patients with autoreactive T cells, potentially before the onset of clinically relevant symptoms.
Acknowledgments
We are grateful for Dr. Alvaro Puga’s generosity in providing access to the FACS Calibur. We would also like to thank Dr. William Ridgway for thoughtful discussion of the manuscript and Dr. Marepalli Rao for discussion of statistical procedures.
This work was supported by National Institutes of Health research Grants R01 ES015036 and R21 HL109635 (to M.T.B.), T32 ES016646 (to B.L.E.), the University of Cincinnati Center for Environmental Genetics P30 ES006096, and a Flight Attendant Medical Research Institute grant (to M.T.B.),.
Abbreviations
- FA
Filtered air
- CS
Cigarette smoke
- COPD
Chronic obstructive pulmonary disease
- GVHD
Graft versus host disease
- Tregs
Regulatory T cells
Bibliography
- 1.Jemal A, Ward E, Hao Y, Thun M. Trends in the leading causes of death in the United States, 1970–2002. JAMA. 2005;294:1255–1259. doi: 10.1001/jama.294.10.1255. [DOI] [PubMed] [Google Scholar]
- 2.Xu J, Kochanek KD, Murphy SL, Tejada-Vera B. National Vital Statistics Reports. Vol. 58. CDC; 2010. Deaths: Final Data for 2007. [PubMed] [Google Scholar]
- 3.Mathers C, Boerma T, Ma Fat D. The global burden of disease: 2004 update. World Health Organization; 2008. [Google Scholar]
- 4.Buist AS, McBurnie MA, Vollmer WM, Gillespie S, Burney P, Mannino DM, Menezes AMB, Sullivan SD, Lee TA, Weiss KB, et al. International variation in the prevalence of COPD (the BOLD Study): a population-based prevalence study. Lancet. 2007;370:741–750. doi: 10.1016/S0140-6736(07)61377-4. [DOI] [PubMed] [Google Scholar]
- 5.Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379:1341–1351. doi: 10.1016/S0140-6736(11)60968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:2445–2454. doi: 10.1056/NEJMra0804752. [DOI] [PubMed] [Google Scholar]
- 7.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:p2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 8.Russell REK, Thorley A, Culpitt SV, Dodd S, Donnelly LE, Demattos C, Fitzgerald M, Barnes PJ. Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteases. Am J Physiol Lung Cell Mol Physiol. 2002;283:L867–L873. doi: 10.1152/ajplung.00020.2002. [DOI] [PubMed] [Google Scholar]
- 9.Woodruff PG, Koth LL, Yang YH, Rodriguez MW, Favoreto S, Dolganov GM, Paquet AC, Erle DJ. A distinctive alveolar macrophage activation state induced by cigarette smoking. Am J Respir Crit Care Med. 2005;172:1383–1392. doi: 10.1164/rccm.200505-686OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borchers MT, Wesselkamper SC, Curull V, Ramirez-Sarmiento A, Sanchez-Font A, Garcia-Aymerich J, Coronell C, Lloreta J, Agusti AG, Gea J, et al. Sustained CTL activation by murine pulmonary epithelial cells promotes the development of COPD-like disease. J Clin Invest. 2009;119:636–649. doi: 10.1172/JCI34462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wortham BW, Eppert BL, Motz GT, Flury JL, Orozco-Levi M, Hoebe K, Panos RJ, Maxfield M, Glasser SW, Senft AP, et al. NKG2D mediates NK cell hyperresponsiveness and influenza-induced pathologies in a mouse model of chronic obstructive pulmonary disease. J Immunol. 2012;188:4468–4475. doi: 10.4049/jimmunol.1102643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, Gadgil AS, Givelber R, George MP, Stoner MW, Sciurba FC, Duncan SR. Peripheral T cell functions correlate with the severity of chronic obstructive pulmonary disease. J Immunol. 2009;182:3270–3277. doi: 10.4049/jimmunol.0802622. [DOI] [PubMed] [Google Scholar]
- 13.Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis DE, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1:e8. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med. 1997;155:852–857. doi: 10.1164/ajrccm.155.3.9117016. [DOI] [PubMed] [Google Scholar]
- 15.Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, Cavallesco G, Tropeano G, Mapp CE, Maestrelli P, et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:711–717. doi: 10.1164/ajrccm.160.2.9812020. [DOI] [PubMed] [Google Scholar]
- 16.Chrysofakis G, Tzanakis N, Kyriakoy D, Tsoumakidou M, Tsiligianni I, Klimathianaki M, Siafakas NM. Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPD. Chest. 2004;125:71–76. doi: 10.1378/chest.125.1.71. [DOI] [PubMed] [Google Scholar]
- 17.Freeman CM, Han MK, Martinez FJ, Murray S, Liu LX, Chensue SW, Polak TJ, Sonstein J, Todt JC, Ames TM, et al. Cytotoxic potential of lung CD8(+) T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J Immunol. 2010;184:6504–6513. doi: 10.4049/jimmunol.1000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol. 2007;178:8090–8096. doi: 10.4049/jimmunol.178.12.8090. [DOI] [PubMed] [Google Scholar]
- 19.van der Strate BWA, Postma DS, Brandsma CA, Melgert BN, Luinge MA, Geerlings M, Hylkema MN, van den Berg A, Timens W, Kerstjens HAM. Cigarette smoke-induced emphysema: A role for the B cell? Am J Respir Crit Care Med. 2006;173:751–758. doi: 10.1164/rccm.200504-594OC. [DOI] [PubMed] [Google Scholar]
- 20.Agusti A, MacNee W, Donaldson K, Cosio M. Hypothesis: Does COPD have an autoimmune component? Thorax. 2003;58:832–834. doi: 10.1136/thorax.58.10.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sullivan AK, Simonian PL, Falta MT, Mitchell JD, Cosgrove GP, Brown KK, Kotzin BL, Voelkel NF, Fontenot AP. Oligoclonal CD4+ T cells in the lungs of patients with severe emphysema. Am J Respir Crit Care Med. 2005;172:590–596. doi: 10.1164/rccm.200410-1332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, Green L, Hacken-Bitar J, Huh J, Bakaeen F, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13:567–569. doi: 10.1038/nm1583. [DOI] [PubMed] [Google Scholar]
- 23.Feghali-Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, Csizmadia E, Zhang Y, Sciurba FC, Duncan SR. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:156–163. doi: 10.1164/rccm.200701-014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karayama M, Inui N, Suda T, Nakamura Y, Nakamura H, Chida K. Anti-endothelial cell antibodies in patients with COPD. Chest. 2010;138:1303–1308. doi: 10.1378/chest.10-0863. [DOI] [PubMed] [Google Scholar]
- 25.Ñúnez B, Sauleda J, Antó JM, Juliá MR, Orozco M, Monsó E, Noguera A, Gómez FP, Garcia-Aymerich J, Agustí A. Anti-tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183:1025–1031. doi: 10.1164/rccm.201001-0029OC. [DOI] [PubMed] [Google Scholar]
- 26.Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, Triantaphyllopoulos K, Hussain F, Pinart M, Khan Y, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184:796–802. doi: 10.1164/rccm.201010-1605OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Motz GT, Eppert BL, Sun G, Wesselkamper SC, Linke MJ, Deka R, Borchers MT. Persistence of lung CD8 T cell oligoclonal expansions upon smoking cessation in a mouse model of cigarette smoke-induced emphysema. J Immunol. 2008;181:8036–8043. doi: 10.4049/jimmunol.181.11.8036. [DOI] [PubMed] [Google Scholar]
- 28.Motz GT, Eppert BL, Wesselkamper SC, Flury JL, Borchers MT. Chronic cigarette smoke exposure generates pathogenic T cells capable of driving COPD-like disease in Rag2−/− mice. Am J Respir Crit Care Med. 2010;181:1223–1233. doi: 10.1164/rccm.200910-1485OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited) Immunol Today. 1993;14:426–430. doi: 10.1016/0167-5699(93)90244-F. [DOI] [PubMed] [Google Scholar]
- 30.Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, Novak EK, Swank RT. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol. 2003;285:L643–L653. doi: 10.1152/ajplung.00024.2003. [DOI] [PubMed] [Google Scholar]
- 31.Olson WC, Smolkin ME, Farris EM, Fink RJ, Czarkowski AR, Fink JH, Chianese-Bullock KA, Slingluff CL., Jr Shipping blood to a central laboratory in multicenter clinical trials: effect of ambient temperature on specimen temperature, and effects of temperature on mononuclear cell yield, viability and immunologic function. J Transl Med. 2011;9:26–38. doi: 10.1186/1479-5876-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison OJ, Foley J, Bolognese BJ, Long E, 3rd, Podolin PL, Walsh PT. Airway infiltration of CD4+ CCR6+ Th17 type cells associated with chronic cigarette smoke induced airspace enlargement. Immunol Letters. 2008;121:13–21. doi: 10.1016/j.imlet.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Shan M, Cheng HF, Song LZ, Roberts L, Green L, Hacken-Bitar J, Huh J, Bakaeen F, Coxson HO, Storness-Bliss C, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. 2009;1:4ra10. doi: 10.1126/scitranlsmed.3000154. [DOI] [PubMed] [Google Scholar]
- 34.Shan M, Yuan X, Song LZ, Roberts L, Zarinkamar N, Seryshev A, Zhang Y, Hilsenbeck S, Chang SH, Dong C, et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci Transl Med. 2012;4:117ra119. doi: 10.1126/scitranslmed.3003041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demoor T, Bracke KR, Joos GF, Brusselle GG. Increased T-regulatory cells in lungs and draining lymph nodes in a murine model of COPD. Eur Respir J. 2010;35:688–689. doi: 10.1183/09031936.00158509. [DOI] [PubMed] [Google Scholar]
- 36.Barcelo B, Pons J, Ferrer JM, Sauleda J, Fuster A, Agusti AG. Phenotypic characterisation of T-lymphocytes in COPD: abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur Respir J. 2008;31:555–562. doi: 10.1183/09031936.00010407. [DOI] [PubMed] [Google Scholar]
- 37.DiLorenzo TP, Graser RT, Ono T, Christianson GJ, Chapman HD, Roopenian DC, Nathenson SG, Serreze DV. Major histocompatibility complex class I-restricted T cells are required for all but the end stages of diabetes development in nonobese diabetic mice and use a prevalent T cell receptor alpha chain gene rearrangement. Proc Natl Acad Sci USA. 1998;95:12538–12543. doi: 10.1073/pnas.95.21.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol. 2001;166:7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- 39.D’Hulst AI, Maes T, Bracke KR, Demedts IK, Tournoy KG, Joos GF, Brusselle GG. Cigarette smoke-induced pulmonary emphysema in scid-mice. Is the acquired immune system required? Respir Res. 2005;6:147–159. doi: 10.1186/1465-9921-6-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen K, Pociask DA, McAleer JP, Chan YR, Alcorn JF, Kreindler JL, Keyser MR, Shapiro SD, Houghton AM, Kolls JK, et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PloS ONE. 2011;6:e20333. doi: 10.1371/journal.pone.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belz GT, Wodarz D, Diaz G, Nowak MA, Doherty PC. Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J Virol. 2002;76:12388–12393. doi: 10.1128/JVI.76.23.12388-12393.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur J Immunol. 2000;30:1538–1543. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 46.Misra N, Bayry J, Lacroix-Desmazes Sb, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol. 2004;172:4676–4680. doi: 10.4049/jimmunol.172.8.4676. [DOI] [PubMed] [Google Scholar]
- 47.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]