

Abstract
Recent studies suggest that certain mitochondrial haplogroup markers and some specific variants in mitochondrial haplogroup may also influence the phenotypic expression of particular mitochondrial disorders. In this report, the clinical, genetic, and molecular characterization were identified in a Chinese pedigree with the aminoglycoside antibiotic (AmAn)-induced deafness and nonsyndromic hearing loss (NSHL). The pathogenic gene responsible for this hereditary NSHL pedigree was determined by Microarray chip, which possessed the nine NSHL hot-spot mutations, including GJB2 (35delG, 176dell6bp, 235de1C, and 299delAT), GJB3 (538C>T), SLC26A4 (IVS7-2A>G and 2168A>G), and mitochondrial DNA (mtDNA) 12S rRNA (C1494T and A1555G). Only the homoplasmic A1555G mutation was detected, which was confirmed by direct sequencing. Also, real-time amplification refractory mutation system quantitative polymerase chain reaction methodology was performed to calculate the A1555G mutation load. The proband's complete mtDNA genome were amplified and direct sequencing was performed to determine the mitochondrial haplogroup and private mutations. The proband's mitochondrial haplogroup belonges to M7b1 and a private mutation MTCOX2 G7598A (p.Ala 5 Thr) is found. Phylogenetic analysis of COX2 polypeptide sequences demonstrates that the alanine residue is relatively conserved, but owing to the missense mutation (p.Ala 5 Thr), its side chain hydrophobicity will be changed, and what is more, as it is adjacent to a glutamine residue, which is highly conserved and hydrophilic, in an evolutionary stable domain; G7598A (p.Ala 5 Thr) may alter the protein secondary structure and physiological function of COX2 and, thus, aggravate the mitochondrial dysfunction conferred by the A1555G mutation. Furthermore, the G7598A mutation is absent in 100 unrelated healthy controls; therefore, G7598A (p.Ala 5 Thr) in the mitochondrial haplogoup M7b1 may have a modifying role, enhancing its penetrance and severity, in the AmAn-induced deafness and NSHL associated with 12S rRNA A1555G mutation in the Han Chinese pedigree.
Introduction
Mutations in mitochondrial DNA (mtDNA), especially in the 12S rRNA gene, are important causes of maternal hereditary hearing loss (Prezant et al., 1993; Fischel-Ghodsian, 2005). Of these, the A1555G mutation has been associated with both aminoglycoside antibiotic (AmAn)-induced deafness and nonsyndromic hearing loss (NSHL) in many families worldwide (Guan, 2005; Jacobs et al., 2005). Matrilineal relatives within and among families carrying the A1555G mutation exhibit a wide range of penetrance, severity, and age-of-onset in hearing loss (Malik et al., 2003; Young et al., 2005; Dai et al., 2006). Functional characterization of cell lines derived from matrilineal relatives of a large Arab-Israeli family or one large Chinese family demonstrated that the A1555G mutation caused mild mitochondrial dysfunction and more sensitivity to AmAn (Guan et al., 1996, 2000). These findings strongly indicated that the A1555G mutation by itself is insufficient to produce the various deafness phenotypes. Therefore, other modifier factors, including AmAn, nuclear modifier genes, and mitochondrial variants/haplotypes, may also modulate the expressivity and penetrance of hearing loss associated with the A1555G mutation (Hutchin et al., 1993; Bykhovskaya et al., 2000; Guan et al., 2001, 2006; Tang et al., 2007).
Recent studies suggest that certain mutations with phylogeographic importance as haplogroup markers and some mutations in certain haplogroups may also influence the phenotypic expression of particular mitochondrial disorders (Walt van der et al., 2003; Zhadanov et al., 2005). In particular, the haplogroup J-specific variants 4216T>C and 13708G>A may increase the penetrance of vision loss associated ND4 (Torroni et al., 1997) and hearing loss associated with tRNASer(UCN) 7445A>G mutation (Guan et al., 1998), while the haplogroup L1b-specific variants ND1 3308T>C and tRNAAla 5655T>C likely caused higher penetrance of deafness in an African pedigree than in Japanese and French families carrying the 7511T>C mutation (Li et al., 2004).
In the previous studies of mitochondrial genes, tRNACys T5802C, tRNACys G5821A, tRNASer(UCN) G7444A, tRNAArg T10454C, ND5 T12338C, tRNAGlu A14693G, tRNAThr T15908C, and tRNAThr G15927A, defined as the secondary mutations, have been reported to possibly influence or worsen the clinical phenotypic manifestation of hearing loss associated with the A1555G mutation in Chinese NSHL pedigrees (Zhao et al., 2005; Chen et al., 2008; Wang et al., 2008).
In the present study, we performed clinical, molecular, and genetic characterization of a maternal hereditary pedigree in subjects from the Fujian Province of China carrying the A1555G mutation. A wide range of penetrance and severity of hearing loss were observed in the matrilineal relatives compared to the previous report in this region (Ou et al., 2009, 2011). Secondary mutations in the mitochondrial genome, as mentioned above, may modulate the phenotypic manifestation of hearing loss absent in our pedigree. As a result, to further investigate the molecular mechanism of this hearing loss pedigree, we performed whole mitochondrial genome amplification and subsequently DNA direct sequencing. Phylogenetic analysis was performed to determine the mitochondrial haplogroup and private mutations.
Furthermore, it has been suggested that GJB2 mutations may modulate phenotypic manifestation of hearing loss associated with the A1555G mutation (Abe et al., 2001). To further examine the role of the GJB2 gene in the phenotypic expression of the A1555G mutation, we sequenced the coding region of the GJB2 gene in this pedigree.
Materials and Methods
Subjects and audiological examinations
All investigations, as shown in Figure 1, were conducted with the approval of the ethics committee of The 1st affiliated Hospital of Fujian Medical University and according to the tenets of the Declaration of Helsinki. Informed consent was obtained from all individuals before donation of blood samples and genetic analysis of their mtDNAs. A comprehensive history and physical examination were performed to identify any syndromic findings, the history of the use of AmAn and genetic factors related to the hearing impairment in this pedigree. An age-appropriate audiological examination was performed, which included pure-tone audiometry (PTA) and/or auditory brainstem response, immittance testing, and distortion product otoacoustic emissions. The PTA was calculated from the sum of the audiometric thresholds at 500, 1000, 2000, and 4000 Hz. The severity of hearing impairment was classified into five grades: normal (<26 dB), mild (26–40 dB), moderate (41–70 dB), severe (71–90 dB), and profound (>90 dB).
FIG. 1.

The pedigree with aminoglycoside antibiotic (AmAn)-induced deafness and nonsyndromic hearing loss. Hearing-impaired individuals are indicated by filled symbols. Plus symbol denotes individuals who had a history of exposure to AmAn. Proband is indicated by the square box and arrow.
Molecular genetic studies by deafness gene microarray
Genomic DNA was extracted from peripheral blood of the family by Tiangen Genomic DNA (Beijing, China). The deafness gene microarray diagnostic kit of the CapitalBio Corporation was applied to detect the nine NSHL hot-spot mutations, including GJB2 (35delG, 176dell6bp, 235de1C, and 299delAT), GJB3 (538C>T), SLC26A4 (IVS7-2A>G and 2168A>G), and mtDNA 12S rRNA (C1494T and A1555G). The procedure of deafness gene microarray analysis was performed as per the CapitalBio Corporation and a previous report (Li et al., 2008).
Heteroplasmy of A1555G by real-time amplification refractory mutation system quantitative polymerase chain reaction
Primers for ARMS
To measure low proportions of mutant heteroplasmy, we designed mismatched primers for real-time amplification refractory mutation system quantitative polymerase chain reaction (RT-ARMS-qPCR). The introduction of one or two mismatched nucleotides immediately 5' to the mutation site greatly increased the binding specificity of the allele-specific modified primers toward either the wild-type or the mutant sequence targets. In RT-ARMS-qPCR, the forward primer was at nucleotide positions 1420–1432 for the amplification of both wild-type mtDNA and the A1555G mutation. The reverse primers for RT-ARMS-qPCR analysis of the A1555G mutation are listed in Table 1.
Table 1.
Primers for Real-Time Amplification Refractory Mutation System Quantitative Polymerase Chain Reaction
| Forward primers | 5′-TGG ATT TAG CAG T-3′(1420–1432) |
| ARMS reverse primer for wild-type A1555 | 5′-CTT ACC ATG TTA CGA CTT GT-3′(1574–1555) |
| ARMS reverse primer for mutant A1555G | 5′-CTT ACC ATG TTA CGA CTT CC-3′(1574–1555) |
The underlined uppercase bases indicate the mismatches.
RT-qPCR
The RT-ARMS-qPCR assay was performed in triplicate for each wild-type and mutant target sequence. The 20 μL PCR reaction mixture contained 10 μL SYBR ® Premix Ex Taq™ (Takara), 10 nM each primer, and 4 ng of total genomic DNA. Real-time PCR conditions were 30 s at 95°C, followed by 40 cycles of denaturation for 5 s at 95°C, and annealing/extension for 5 s at 58°C. During PCR, the fluorescent signal intensities were recorded and analyzed on an Applied Biosystems StepOnePlus™ Instrument. Dissociation curves for the amplicons were generated after PCR to confirm that the increased fluorescence intensities were not attributed to nonspecific signals (primer-dimers). If a sample contained >1,000,000 or <100 copies based on the CT numbers, the assay was repeated at a higher or lower dilution of the DNA extract so that the measurement would fall within a linear DNA copy number range (Bai and Wong, 2004; Ou et al., 2011).
Preparation of DNA for calibration curves
DNA for the wild-type and mutant target sequences was generated from cloned plasmid DNA containing the pMD18-T vector (Invitrogen) and PCR products of primers mtF1420 and mtR1774. The copy numbers of the wild-type and mutant DNA sequences were calculated based on the size and molecular weight of the plasmid DNA. Serial dilutions were made, and RT-ARMS-qPCR reactions were performed to construct the calibration curves for the wild-type and mutant A1555G DNA sequences.
Measurement of mutant heteroplasmy
Calibration curves for both wild-type and mutant mtDNA were always included in each run. The copy number of the target sequence in the sample was calculated from the CT number and the calibration curve. The proportion of the mutant A1555G sequence was calculated from the copy number of the wild-type and mutant sequences. Alternatively, the proportion of the mutant mtDNA could be calculated from ΔCt=(Ctwild−Ctmutation), proportion of mutant=1/(1+1/2ΔCt).
Mutational analysis of mitochondrial genome
The entire mitochondrial genomes of the proband were PCR amplified in 24 overlapping fragments by use of sets of the light-strand and the heavy-strand oligonucleotide primers, as described elsewhere (Rieder et al., 1998). Each fragment was purified and subsequently analyzed by direct sequencing. The resultant sequence data were blasted with the updated consensus Cambridge sequence (GeneBank accession number: NC_00129209) on the NCBI Website. Mitochondrial hapologroup and mtDNA variants were determined according to the PhyloTree Build 14 (www.phylotree.org) and MitoTool (www.mitotool.org). mtDNA variants in the complete mtDNA were scored as novel or reported and polymorphism or deleterious mutation according to MITOMAP database (www.mitomap.org/MITOMAP). Phylogenetic analysis for the private mutation was performed to explore whether it was deleterious or neutral to the mitochondrial genome and the diseases.
Mutational analysis of GJB2 gene
The DNA fragments spanning the entire coding region of the GJB2 gene were amplified applying the following primers: forward: 5′CCTGTTCTGTCCTAGCTAGTGA3′ and reverse: 5′CTCCATTGTGGCATCTGGAGTT3′. PCR amplification and subsequent sequencing analysis were performed. The sequences were blasted with the GJB2 gene (GeneBank accession number: NC_000013.10) to identify the mutations on the NCBI Website.
Results
Clinical features and audiology evaluation of the Chinese pedigree
The pedigree was diagnosed as AmAn-induced deafness or NSHL without any clinical abnormalities, including diabetes, muscular diseases, visual dysfunction, and neurological disorders by the ear-nose-throat (ENT) department. Of these, the proband (III-3) of the pedigree received streptomycin (0.75 g/dose for 7 days) for hyperpyrexia at 12 months. He exhibited bilateral hearing impairment 2 weeks later after the drug administration. As illustrated in Table 2, audiological evaluation showed that he had profound hearing impairment (98 dB at both ears, with a slopes pattern). Further, comprehensive family history and physical examination as well as audiological examination in members of the three-generation family showed that five (two male and three female) of seven matrilineal relatives exhibited hearing defect, ranging from profound (II3, III3), to severe (II4, III1), to moderate (I2), to mild (II2), and normal (III2). The total penetrance of this pedigree was 71% (affected matrilineal relatives/total matrilineal relatives).
Table 2.
The Clinical Features and Mitochondrial DNA Loads of the Chinese Pedigree
| |
|
|
|
The A1555G copy numbers |
|
|
|---|---|---|---|---|---|---|
| Subject | Gender | Age of onset | AmAn exposure | Wild-type | Mutant | Hearing-loss level |
| I1 | M | – | No | 1.31E6 | – | Normal |
| I2 | F | 50 | No | – | 1.36E6 | Moderate |
| II1 | M | – | No | 3.44E5 | – | Normal |
| II2 | F | – | No | – | 3.14E5 | Mild |
| II3 | F | <1 | Yes | – | 1.16E6 | Profound |
| II4 | M | 3 | No | – | 8.82E5 | Severe |
| II5 | F | – | No | 1.18E5 | – | Normal |
| III1 | F | 1–2 | Yes | – | 4.45E5 | Severe |
| III2 | M | – | No | – | 5.47E6 | Normal |
| III3 | M | <1 | Yes | – | 1.44E6 | Profound |
| III4 | F | – | No | 1.19E6 | – | Normal |
| III5 | M | – | No | 8.64E5 | – | Normal |
AmAn, aminoglycoside antibiotic.
Molecular genetic studies by deafness gene microarray
The deafness gene microarray contained the nine NSHL hot-spot mutations, including GJB2 (35delG, 176dell6bp, 235de1C, and 299delAT), GJB3 (538C>T), SLC26A4 (IVS7-2A>G and 2168A>G), and mtDNA 12S rRNA (C1494T and A1555G). On the Micoarray, the two parallel five-dots represented the wild type and mutation, respectively. When the wild and/or mutation sequences existed, the corresponding dots would have the green fluorescent light. As shown in Fig. 2, the square areas represent the mtDNA A1555G, the upper dots were the wild-dots, which had no fluorescence, meaning the wild-type sequence and the parallel five green dots below the represented mutated sequence. Therefore, Microarray results indicated that the pathogenic gene was a homoplasmic A1555G mutation.
FIG. 2.

Mitochondrial DNA (mtDNA) 12S rRNA A1555G homoplasmic mutation. The square box indicates the mitochondrial DNA (mtDNA) 12S rRNA A1555G square areas. The upper dark dots indicate the wild-type is absent and the white dots below indicate the A1555G homoplasmic mutation.
Heteroplasmy of A1555G by RT-ARMS-qPCR
For RT-ARMS-qPCR analysis, calibration curves for the wild-type A1555 and mutant A1555G sequences were constructed (Fig. 3). There was an excellent correlation between the cycle number and mtDNA copy number with correlation coefficients of 0.998 and 0.994 for the wild-type and mutant target sequences, respectively. Melting curves were obtained after the RT-ARMS-qPCR, the peaks were sharp and significant for the wild type and mutant. All the maternal members showed positive A1555G mutant signals and the proportion of mutant was 100%.
FIG. 3.

(A) Calibration curves for the wild-type A1555; (B) calibration curves for the mutant A1555G; (C) standard curve of the wild-type: Y=− 3.331X+34.137 (R2=0.996); (D) standard curve of the mutant: Y=− 3.052X+31.642 (R2=0.988); (E) melting peaks for A1555; (F) melting peaks for A1555G.
Mutational analysis of mitochondrial genome
The proband's complete mtDNA genome was amplified and sequenced, all the mtDNA variances, as shown in Table 3, were obtained after being blasted to the updated consensus Cambridge sequence (GeneBank accession number: NC_00129209) on the NCBI website. There were 47 mutations in total, including 19 nonsense mutations and 11 missense mutations. According to the PhyloTree Build 14 and MitoTool, the mitochondrial haplogroup of this pedigree belonged to M7b1 (Fig. 4) and 4 private mutations T204C, G5252A, G7598A, A11800G as well as A1555G were obtained. Of the four private mutations, according to the Mitomap, only MTCOX2 G7598A (p.Ala 5 Thr) was found to lead to a nonsynomous mutation (Fig. 5), the others were identified as polymorphisms.
Table 3.
Mitochondrial DNA Variances of the Proband in P206 Pedigree
| Locus name | Position | Mutation type | CI | AA position | AA change | haplogroupSNPa | Previously reportedb |
|---|---|---|---|---|---|---|---|
| D-loop | 73 | A to G | 0.4 | Yes | Yes | ||
| D-loop | 150 | C to T | 0.59 | Yes | Yes | ||
| D-loop | 199 | T to C | 0.82 | Yes | Yes | ||
| D-loop | 204 | T to C | 0.78 | NO | Yes | ||
| D-loop | 248 | 248d | 0 | Yes | Yes | ||
| D-loop | 263 | A to G | 0.95 | Yes | Yes | ||
| D-loop | 302 | 302d | 0 | NO | Yes | ||
| D-loop | 310 | 310d | 0 | NO | Yes | ||
| D-loop | 489 | T to C | 0.22 | Yes | Yes | ||
| D-loop | 16129 | G to A | 0.53 | Yes | Yes | ||
| D-loop | 16192 | C to T | 0.8 | Yes | Yes | ||
| D-loop | 16223 | C to T | 0 | Yes | Yes | ||
| D-loop | 16297 | T to C | 0.97 | Yes | Yes | ||
| 12S rRNA | 750 | A to G | 0.95 | Yes | Yes | ||
| 12S rRNA | 1438 | A to G | 0.87 | Yes | Yes | ||
| 12S rRNA | 1555 | A to G | 0.99 | NO | Yes | ||
| 16S rRNA | 2706 | A to G | 0.37 | Yes | Yes | ||
| ND1 | 4048 | G to A | 0.97 | 248 | D to N | Yes | Yes |
| ND1 | 4071 | C to T | 0.95 | 255 | Syn(Y) | Yes | Yes |
| ND1 | 4164 | A to G | 0.98 | 286 | Syn(M) | Yes | Yes |
| ND2 | 4769 | A to G | 0.93 | 100 | Syn(M) | Yes | Yes |
| ND2 | 5252 | G to A | 0.96 | 261 | Syn(L) | NO | Yes |
| ND2 | 5351 | A to G | 0.98 | 294 | Syn(L) | Yes | Yes |
| ND2 | 5460 | G to A | 0.03 | 331 | A to T | Yes | Yes |
| CO1 | 6455 | C to T | 0.92 | 184 | Syn(F) | Yes | Yes |
| CO1 | 6680 | T to C | 0.93 | 259 | Syn(T) | Yes | Yes |
| CO1 | 7028 | C to T | 0.39 | 375 | Syn(A) | Yes | Yes |
| CO2 | 7598 | G to A | 0.91 | 5 | A to T | NO | Novel |
| CO2 | 7684 | T to C | 0.98 | 33 | Syn(L) | Yes | Yes |
| CO2 | 7853 | G to A | 0.95 | 90 | V to I | Yes | Yes |
| ATP6 | 8701 | A to G | 0.07 | 59 | T to A | Yes | Yes |
| ATP6 | 8860 | A to G | 0.98 | 112 | T to A | Yes | Yes |
| CO3 | 9540 | T to C | 0.08 | 112 | Syn(L) | Yes | Yes |
| CO3 | 9824 | T to C | 0.86 | 206 | Syn(L) | Yes | Yes |
| ND3 | 10398 | A to G | 0.01 | 114 | T to A | Yes | Yes |
| ND3 | 10400 | C to T | 0.16 | 114 | T to A | Yes | Yes |
| ND4 | 10873 | T to C | 0.08 | 38 | Syn(P) | Yes | Yes |
| ND4 | 11719 | G to A | 0.28 | 320 | Syn(G) | Yes | Yes |
| ND4 | 11800 | A to G | 0.99 | 347 | Syn(G) | NO | Yes |
| ND5 | 12405 | C to T | 0.98 | 23 | Syn(L) | Yes | Yes |
| ND5 | 12705 | C to T | 0 | 123 | Syn(I) | Yes | Yes |
| ND5 | 12811 | T to C | 0.98 | 159 | Y to H | Yes | Yes |
| Cytb | 14766 | C to T | 0.27 | 7 | T to I | Yes | Yes |
| Cytb | 14783 | T to C | 0.16 | 13 | Syn(L) | Yes | Yes |
| Cytb | 15043 | G to A | 0.13 | 99 | Syn(G) | Yes | Yes |
| Cytb | 15301 | G to A | 0.08 | 185 | Syn(L) | Yes | Yes |
| Cytb | 15326 | A to G | 0.95 | 194 | T to A | Yes | Yes |
The highest CI is 1, which means this site is completely conservative in 43 primate species. A CI value of 0.91 indicates 39 of 43 primate species have the same allele with the queried variant.
Haplogroup M7b1's SNPs.
See the online MITOMAP database.
CI, conservation index; AA, amino acid.
FIG. 4.
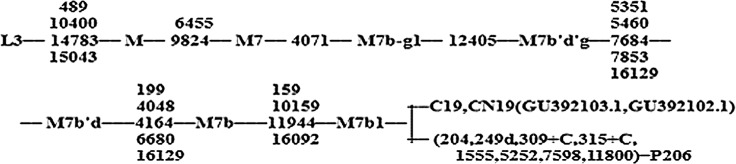
Classification tree of the proband's complete mtDNA sequence. Nucleotide position numbers are relative to the RSRS and rCRS. The mutations 309.1C(C), 315.1C, AC indels at 515–522, 16182C, 16183C, 16193.1C(C), and 16519 were not considered for phylogenetic reconstruction and are, therefore, excluded from the tree. The GeneBank Accession number for C19 and CN19 is GU392103.1 and GU392102.1, respectively.
FIG. 5.
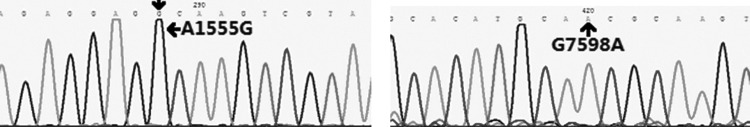
Chromatogram of the sequencing of A1555G and G7598A, as indicated by the arrows.
According to the Phylotree, G7598A as a mitochondrial haplogroup marker was found in six haplogroups: M31b, C1a, C7ala2, E, M49a′b, and Hv1b1, but was absent in M7b1 (van Oven and Kayser 2009). Meanwhile, for the haplogroup M7b1, a total of 27 mitochondrial queries of M7b1 were retrieved from the Mitotool database, but only two queries harbored the G7598A, which were both correlated with colorectal cancer, as shown in Table 4.
Table 4.
Mitochondrial DNA Queries of Haplogroup M7b1 Harbor G7598A
| Haplogroup M7b1 | Accession number | Disease |
|---|---|---|
| G7598A | GU392103.1 | Colorectal cancer |
| G7598A | GU392102.1 | Colorectal cancer |
| G7598A | P206 (Present study) | Deafness |
The search is based on 9,328 human mitochondrial DNA sequences from NCBI (dated on September 6, 2011) by MitoTool database.
MTCOX2 G7598A (p.Ala 5 Thr) was further evaluated by phylogenetic analysis from another 43 primate organisms and a conservative index (CI) was calculated. The phylogenetic analysis was performed on on-line Clustaw2 software from EMBL-EBI (www.ebi.ac.uk/Tools/msa/clustalw2/). The result (Fig. 6) showed that the CI of 7598 nucleotide position of human mitochondrial was 91% (39/43), which meant that the position was highly conserved, but as shown by Fig. 7, the protein translated by the G7598A (p.Ala 5 Thr) was relatively conserved.
FIG. 6.

Alignment result of 43 primate organisms' mitochondrial genome (partly shown) by ClustlW2 Software; arrow indicates the position of the 7598 nucleotide of homosapiens' mitochondrial.
FIG. 7.

Alignment result of 43 primate organisms' MTCOX2 protein, partly shown, by ClustlW2 Software; arrow shows position of the fifth protein of COX2. Outlined circle shows the Ala, the fifth MTCOX2 protein in homosapiens' mitochondrial. Outlined square shows the Thr, the fifth MTCOX2 protein in Hylobates lar's mitochondrial.
Mutational analysis of GJB2 gene
To examine the role of the GJB2 gene in phenotypic expression of the A1555G mutation, we performed mutational screening of the GJB2 gene in the pedigree. No variants of the GJB2 gene were found in this Chinese pedigree.
Discussion
In the present study, we have performed clinical, genetic, and molecular characterization of the Chinese pedigree with both AmAn-induced deafness and NSHL. Hearing impairment as a sole clinical phenotype is only present in the maternal lineage of this pedigree. This pedigree exhibits a much higher penetrance and severity of hearing loss than the WZD 27 pedigree, which has the same haplogroup M7b1 carrying the A1555G mutation (Lu et al., 2010). In particular, the penetrance of hearing loss in the WZD27 pedigree is 42.9%, much lower than the 71% in our study. To further explore the explanation for the variable clinical phenotypic manifestation, we performed a more detailed genetic and molecular research on this pedigree.
Owing to the special matrimonial pattern among the deafness community and the many recessive carriers, which exist among healthy people, the pathological factors for deafness become more complicated in the hereditary or sporadic deafness pedigree; therefore, we performed the deafness gene Microarray assay to identify the pathological gene for this NSHL pedigree. Only the homoplasmic A1555G mutation was detected, which indicates that the deafness is monogenic in this pedigree excluding a synergetic role for the other common NSHL genes.
None of the variants in the GJB2 gene are found in this Chinese pedigree which indicates that the GJB2 gene may not be serving as a nuclear modified gene to affect the phenotype of the A1555G mutation in those subjects.
As the threshold effect in mitochondrial disease, the number of mutant mtDNAs is relatively correlated with the penetrance and severity of the clinical expression (Dimauro and Davidzon, 2005). There have been reports that the heteroplasmy A1555G mutation may account for the variable clinical phenotype in the Spanish deafness pedigree (Ballana et al., 2008) and it is suggested that heteroplasmic point mutations with a 10%–20% or a lower percentage for the minor allele were likely to be missed during direct sequencing (Bendall et al., 1996). In this regard, due to the detective limitation of direct sequencing as well as the deafness gene Microarray technology, RT-ARMS-qPCR, a more sensitive and specific methodology, was applied to detect the A1555G mutation load. However, we failed to identify any heteroplasmic A1555G mutations in this pedigree. Therefore, the threshold effect was not attributed to elucidate the variable severity and penetrance of the pedigree.
Mutational analysis of the complete mitochondrial genomes demonstrates that this pedigree belongs to the haplogroup M7b1, in addition to the identical homoplasmic A1555G mutation. There are 47 mutations in total in the proband's mitochondrial genome, including 19 nonsense mutations and 11 missense mutations. However, according to the PhyloTree, only four private mutations, including T204C, G5252A, G7598A, and A11800G are found and the others are identified as haplogroup markers of M7b1, which are supposed to be benign in the mitochondrial genome. Of the four private mutations, according to the MITOMAP, only MTCOX2 G7598A (p.Ala 5 Thr) is found to lead to a nonsynonmous mutation and the others are identified as polymorphisms. What is more, the G7598A mutation is detected in all the maternal members of the pedigree indicating that the G7598A mutation is not a somatic mutation and it can potentially be transmitted to the next generation by maternal inheritance.
According to the Phylotree, G7598A works as a certain mitochondrial haplogroup marker found in 6 haplogroups: M31b, C1a, C7ala2, E, M49a′b, and Hv1b1, but is absent in M7b1. Meanwhile, for the haplogroup M7b1 to harbor the G7598A as well, has been reported in colorectal cancer, which means that G7598A may have a special role in colorectal cancer (Wang et al., 2011). What is more, G7598A works as a deleterious variant in a LHON maternal lineage of Caucasians, as has been reported (Zhadanov et al., 2006); besides, the secondary deafness-associated mutations, such as tRNACys T5802C, tRNACys G5821A, tRNASer(UCN) G7444A, tRNAArg T10454C, ND5 T12338C, tRNAGlu A14693G, tRNAThr T15908C, and tRNAThr G15927A are absent in the proband, therefore, G7598A may also modulate the variable clinical manifestation seen in the present deafness pedigree.
Phylogenetic analysis of MTCOX2 G7598A (p.Ala 5 Thr) is preformed and the CI of the 7598 nucleotide position is 91% (39/43), which means that the position is highly conserved and G7598A may be a candidate pathological mutation.
Interestingly, phylogenetic analysis of COX2 polypeptide sequences demonstrates that the alanine residue (G7598A) is relatively conserved and the mtDNA of the white-handed gibbon (Hylobates lar) shows the same amino acid sequence at the beginning of the COX2 gene as well as the proband (Fig. 7). However, given the significant evolutionary distance between the gibbon and modern humans (Hall et al., 1998; Ingman and Gyllensten, 2003), it is highly unlikely that the 7598A mutation would have the same significance for this hominoid ape species.
The G7598A leads to a nonsynonymous mutation and substitutes a small hydrophobic neutral alanine for a hydrophilic polar threonine at the fifth amino acid position of the COX2 subunit, thereby changing its side chain hydrophobicity. Though the alanine residue is relatively conserved, but owing to the nonsynonymous mutation (p.Ala 5 Thr), its side chain hydrophobicity will be changed, and what is more, as it is adjacent to the glutamine residue, highly conserved and hydrophilic, in an evolutionarily stable domain, thereby enhancing the hydrophilicity of the adjacent polar glutamine. This amino acid change has been predicted to interfere with the anchoring of the N-terminal transmembrane helix-hairpin of COX2 within the mitochondrial membrane (Tsukihara et al., 1996), which, in turn, would compromise the assembly or stability of the COX holoenzyme. In this regard, G7598A (p.Ala 5 Thr) may alter the protein secondary structure and physiological function of COX2 and, thus, aggravate the mitochondrial dysfunction conferred by the A1555G mutation.
Further, the G7598A mutation is absent in 100 unrelated healthy controls in this region; therefore, the G7598A (p.Ala 5 Thr) in the mitochondrial haplogroup M7b1 may have a modifying role, enhancing its penetrance and severity, in the AmAn-induced deafness associated with the 12S rRNA A1555G mutation in the Han Chinese pedigree. If that is so, exposure to the AmAn is much more dangerous for the pedigree and it should keep in mind that they would never be treated with the AmAn. Furthermore, to prevent AmAn-induced deafness, it is necessary to diagnose the G7598A, as a risk to be profound hearing loss, for the A1555G carriers especially whose haplogroup is M7b1.
Neverthless, more pedigrees of the haplogroup M7b1 with G7598A need to be collected and more investigating and study should be further done to validate the role of G7598A in the haplogroup M7b1 in the AmAn-induced deafness and NSHL associated with the A1555G mutation.
Acknowledgments
We thank all the deaf patients and their family members for their kindly cooperation in this study. This work was supported by a grant from the National Science Fund (81041108) and Natural fund of Fujian Province (2012J01342).
Author Disclosure Statement
No competing financial interest exists.
References
- Abe S. Kelley PM. Usami SI, et al. Connexin 26 gene (GJB2) mutation modulates the severity of hearing loss associated with the 1555A>G mitochondrial mutation. Am J Med Genet. 2001;103:334–338. [PubMed] [Google Scholar]
- Bai RK. Wong LJ. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin Chem. 2004;50:996–1001. doi: 10.1373/clinchem.2004.031153. [DOI] [PubMed] [Google Scholar]
- Ballana E. Govea N. Estivill X, et al. Detection of unrecognized low-level mtDNA heteroplasmy may explain the variable phenotypic expressivity of apparently homoplasmic mtDNA mutations. Hum Mutat. 2008;29:248–257. doi: 10.1002/humu.20639. [DOI] [PubMed] [Google Scholar]
- Bendall KE. Macaulay VA. Baker JR. Sykes BC. Heteroplasmic point mutations in the human mtDNA control region. Am J Hum Genet. 1996;59:1276–1287. [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaya Y. Estivill X. Fischel-Ghodsian N, et al. Candidate locus for a nuclear modifier gene for maternally inherited deafness. Am J Hum Genet. 2000;66:1905–1910. doi: 10.1086/302914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B. Sun D. Yang L, et al. Mitochondrial ND5 12338T>C, tRNACys 5802T>C, and tRNAThr15927G>A variants may have a modifying role in the phenotypic manifestation of deafness-associated 12S rRNA 1555A>G mutation in three Han Chinese pedigrees. Am J Med Genet. 2008;146A:1248–1258. doi: 10.1002/ajmg.a.32285. [DOI] [PubMed] [Google Scholar]
- Dai P. Liu X. Guan MX, et al. Extremely low penetrance of deafness associated with the mitochondrial 12S rRNA mutation in 16 Chinese families: implication for early detection and prevention of deafness. Biochem Biophys Res Commun. 2006;340:194–199. doi: 10.1016/j.bbrc.2005.11.156. [DOI] [PubMed] [Google Scholar]
- Dimauro S. Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–232. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- Fischel-Ghodsian N. Genetic factors in aminoglycoside toxicity. Pharmacogenomics. 2005;6:27–36. doi: 10.1517/14622416.6.1.27. [DOI] [PubMed] [Google Scholar]
- Guan MX. Prevalence of mitochondrial 12S rRNA mutations associated with aminoglycoside ototoxicity. Voltra Rev. 2005;105:211–237. doi: 10.1016/j.mito.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Guan MX. Enriquez JA, et al. The deafness associated mtDNA 7445 mutation, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase ND6 subunit gene expression. Mol Cell Biol. 1998;18:5868–5879. doi: 10.1128/mcb.18.10.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan MX. Fischel-Ghodsian N. Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet. 1996;5:963–971. doi: 10.1093/hmg/5.7.963. [DOI] [PubMed] [Google Scholar]
- Guan MX. Fischel-Ghodsian N. Attardi G. A biochemical basis for the inherited susceptibility to aminoglycoside ototoxicity. Hum Mol Genet. 2000;9:1787–1793. doi: 10.1093/hmg/9.12.1787. [DOI] [PubMed] [Google Scholar]
- Guan MX. Fischel-Ghodsian N. Attardi G. Nuclear background determines biochemical phenotype in the deafness associated mitochondrial 12S rRNA mutation. Hum Mol Genet. 2001;10:573–580. doi: 10.1093/hmg/10.6.573. [DOI] [PubMed] [Google Scholar]
- Guan MX. Yan Q. del Castillo I, et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am J Hum Genet. 2006;79:291–302. doi: 10.1086/506389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ML. Jones DS. Wood BA. Evolution of the gibbon subgenera inferred from cytochrome b DNA sequence data. Mol Phylogenet Evol. 1998;10:281–286. doi: 10.1006/mpev.1998.0539. [DOI] [PubMed] [Google Scholar]
- Hutchin TP. Stoneking M. Cortopassi G, et al. Association of a particular point mutation of the mitochondrial DNA with aminoglycoside-induced deafness. Am J Hum Genet. 1993;53(Suppl.):A20. (abstract). [Google Scholar]
- Ingman M. Gyllensten U. Mitochondrial genome variation and evolutionary history of Australian and New Guinean aborigines. Genome Res. 2003;13:1600–1606. doi: 10.1101/gr.686603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs HT. Mueller RF, et al. Mitochondrial DNA mutations in patients with postlingual, nonsyndromic hearing mpairment. Eur J Hum Genet. 2005;13:26–33. doi: 10.1038/sj.ejhg.5201250. [DOI] [PubMed] [Google Scholar]
- Li CX. Cheng J, et al. Construction of a multiplex allele-specific PCR-based universal array (ASPUA) and its application to hearing loss screening. Hum Mutat. 2008;2:306–414. doi: 10.1002/humu.20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Fischel-Ghodsian N. Guan MX, et al. Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res. 2004;32:867–877. doi: 10.1093/nar/gkh226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J. Guan MX, et al. Mitochondrial haplotypes may modulate the phenotypic manifestation of the deafness-associated 12S rRNA 1555A>G mutation. Mitochondrion. 2010;10:69–81. doi: 10.1016/j.mito.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S. Sudoyo H. Sasmono T, et al. Nonsyndromic sensorineural deafness associated with the A1555G mutation in the mitochondrial small subunit ribosomal RNA in a Balinese family. J Hum Genet. 2003;48:119–124. doi: 10.1007/s100380300018. [DOI] [PubMed] [Google Scholar]
- Ou QS. Cheng ZJ. Yang B, et al. Mitochondrial DNA A1555G mutation of seven families with nonsyndromic hearing loss. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2009;26:550–554. doi: 10.3760/cma.j.issn.1003-9406.2009.05.017. . (In Chinese.) [DOI] [PubMed] [Google Scholar]
- Ou QS. Cheng ZJ. Yang B, et al. Analysis of the ratio of mitchondrial DNA with A1555G mutant to wild type in deaf patients of Fujian province in China by a new method and its relationship with the severity of hearing loss. Chin Med J (Engl) 2011;124:3347–3352. [PubMed] [Google Scholar]
- Prezant TR, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- Rieder MJ. Taylor SL. Tobe VO. Nickerson DA. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 1998;26:967–973. doi: 10.1093/nar/26.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X. Yang L. Guan MX, et al. Very low penetrance of hearing loss in seven Han Chinese pedigrees carrying the deafness-associated 12S rRNA A1555G mutation. Gene. 2007;393:11–19. doi: 10.1016/j.gene.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Torroni A. Petrozzi M. D'Urbano L, et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet. 1997;60:1107–1121. [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T. Aoyama H. Yoshikawa S, et al. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- van Oven M. Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- Walt van der JM. Nicodemus KK. Vance JM, et al. Mitochondrial polymorphis significantly reduce the risk of Parkinson disease. Am J Hum Genet. 2003;72:804–811. doi: 10.1086/373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C-Y. Li H. Hao X-D, et al. Uncovering the profile of somatic mtDNA mutations in Chinese colorectal cancer patients. PLoS One. 2011;6:e21613. doi: 10.1371/journal.pone.0021613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Lu J. Zhu Y, et al. Mitochondrial tRNAThr 15927G>A mutation may modulate the phenotypic manifestation of ototoxic 12S rRNA 1555A>G mutation in four Chinese families. Pharmacogenet Genome. 2008;18:1059–1070. doi: 10.1097/FPC.0b013e3283131661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young W-Y. Zhao L. Guan MX, et al. Extremely low penetrance of hearing loss in four Chinese families with the mitochondrial 12S rRNA A1555G mutation. Biochem Biophys Res Commun. 2005;328:1244–1251. doi: 10.1016/j.bbrc.2005.01.085. [DOI] [PubMed] [Google Scholar]
- Zhadanov SI. Atamanov VV. Schurr TG, et al. A novel mtDNA ND6 gene mutation associated with LHON in a Caucasian family. Biochem Biophys Res Commun. 2005;332:1115–1121. doi: 10.1016/j.bbrc.2005.05.059. [DOI] [PubMed] [Google Scholar]
- Zhadanov SI. Schurr TG, et al. De novo COX2 mutation in a LHON family of Caucasian origin: implication for the role of mtDNA polymorphism in human pathology. J Hum Genet. 2006;51:161–170. doi: 10.1007/s10038-005-0340-y. [DOI] [PubMed] [Google Scholar]
- Zhao L. Wang Q. Guan MX, et al. Clinical evaluation and mitochondrial genome sequence analysis of two Chinese families with aminoglycoside-induced and nonsyndromic hearing loss. Biochem Biophys Res Commun. 2005;336:967–973. doi: 10.1016/j.bbrc.2005.08.199. [DOI] [PubMed] [Google Scholar]