

Abstract
Oligomerization has important functional implications for many membrane proteins. However, obtaining structural insight into oligomeric assemblies is challenging, as they are large and resist crystallization. We focus on proteorhodopsin (PR), a protein with seven transmembrane α-helices that was found to assemble to hexamers in densely packed lipid membrane, or detergent-solubilized environments. Yet, the structural organization and the subunit interface of these PR oligomers were unknown. We used site-directed spin-labeling together with electron spin-resonance lineshape and Overhauser dynamic nuclear polarization analysis to construct a model for the specific orientation of PR subunits within the hexameric complex. We found intersubunit distances to average 16 Å between neighboring 55 residues and that residues 177 are >20 Å apart from each other. These distance constraints show that PR has a defined and radial orientation within a hexamer, with the 55-site of the A-B loop facing the hexamer core and the 177-site of the E-F loop facing the hexamer exterior. Dynamic nuclear polarization measurements of the local solvent dynamics complement the electron spin-resonance-based distance analysis, by resolving whether protein surfaces at positions 55, 58, and 177 are exposed to solvent, or covered by protein-protein or protein-detergent contacts.
Introduction
Oligomerization is an important functional state of many membrane proteins (1). G-protein coupled receptors (GPCRs), which make up the largest family of receptors, are frequently found as oligomers in native as well as artificial lipid environments (2,3). There is new evidence that oligomerization enhances protein function by allosteric modulation of their signaling function, or by changes in ligand recognition or the efficiency of G-protein binding (1). This study specifically deals with the oligomerization of proteorhodopsin (PR), a newly discovered seven transmembrane protein (7TM) (4). Microbial rhodopsins, such as PR, sensory rhodopsin II, and bacteriorhodopsin (BR) are viable experimental models for GPCRs because they share a 7TM structure and bind a retinal chromophore, although they share no sequence homology with GPCRs, such as visual rhodopsins. Recently, BR/rhodopsin and sensory rhodopsin II/rhodopsin chimeras with the third cytoplasmic loop of bovine rhodopsin were constructed and found to activate the G-protein transducer (5,6). The activation of the G-protein by these chimeras, albeit at lower activation levels, suggests that there is a conservation in the function-relevant structural rearrangements and conformational motions between the microbial 7TMs and the GPCRs. PR is an excellent model for studying oligomerization of 7TM, considering the growing evidence of common mechanisms for protein activation in both mammalian and microbial rhodopsins and given the ease of expression and stability of PR in a variety of synthetic detergent and lipid membrane environments.
Despite the expected functional significance of membrane protein oligomerization, there are significant challenges to existing analytical techniques for providing structural insight into large protein assemblies. Crystallization to obtain x-ray diffraction of membrane proteins and their assembly is exceedingly difficult because detergent micelles can mask hydrophilic regions necessary for crystal contacts (7). Solution NMR structure studies do not require crystals, but are limited by broad lines of slowly tumbling, large protein complexes and insufficient chemical shift resolution and dispersion. Solid-state NMR spectroscopy offers unique opportunities to study membrane proteins with significant disorder and constituted in detergents or lipid membrane environments.
Still, to obtain structural insights into the complexes of large membrane proteins is extremely challenging, and in particular of proteins with high α-helical content, as is the case with 7TM proteins, including microbial rhodopsins and GPCRs. This is, in part, because large interhelical distances are difficult to measure, precluding the assignment of important tertiary contacts, let alone intersubunit distances of oligomers of these membrane proteins (8). A new mass spectrometry technique called laser-induced liquid-bead ion desorption can be used to measure intact oligomeric protein complexes, although it is not yet a widely available method (9). These experimental limitations underscore a need for different strategies to obtain structural details of membrane protein oligomer assemblies, such as the relative orientation of the protein in the assembly or defining the solvent-, protein-, or detergent-exposed protein interface.
In this work, we build on recent literature that probed the assembly of PR by atomic force microscopy (AFM) and mass spectrometry (10,11). By varying the composition of the synthetic membrane environment, PR was found in a distribution of oligomeric states from monomer to hexamer by mass spectrometry (10). AFM of PR in lipid bilayers showed that the majority of assemblies are radially arranged hexamers (11), but crucial structural details remain unknown:
-
1.
Which environmental condition produces a given assembly?
-
2.
Where is the subunit interface?
-
3.
What is the relative orientation of PR within the hexameric structure?
We show how site-directed spin-labeling combined with continuous-wave (CW) electron spin resonance (ESR) and Overhauser dynamic nuclear polarization (DNP) methods, further enhanced with standard biochemical separation and characterization tools, can provide unique structural insights into the specific subunit packing of PR in its oligomeric assembly.
We used fast protein liquid chromatography (FPLC) to separate detergent-solubilized, spin-labeled PR by size and therefore, the oligomeric state. Next, we determined the size of the different oligomers by size-exclusion chromatography coupled with static light scattering, ultraviolet, and refractive index detection (SEC-LS/UV/RI), which is an emerging tool for determining the oligomeric state of membrane proteins (12–14). The usual methods for determining the molecular mass from the hydrodynamic radii of proteins, such as size-exclusion chromatography, native gel electrophoresis, and dynamic light scattering may provide an estimate of the total molecular weight of a membrane protein complex. However, unless one has explicit knowledge of the amount of bound detergent and thus the molecular weight of the protein complex including the surrounding detergent molecules, the subunit stoichiometry cannot be determined (15).
In contrast, SEC-LS/UV/RI permits the measurement of the molecular weight of the protein in the protein/detergent micelle complex, by determining the contributions of the detergent. We found that PR exists predominantly as a hexamer when reconstituted in 0.05% n-dodecyl-β-maltoside (DDM) detergent, and that increasing DDM detergent concentration significantly shifts the population toward monomeric PR in solution. Thus, by controlling the detergent concentration, we were able to harvest sufficient concentrations of monomeric versus hexameric PR by FPLC separation for their characterization.
The different oligomeric states of PR were first characterized by CW ESR spectral lineshape analysis, which is a powerful biophysical tool because it allows for distance measurement of high-molecular-weight proteins under physiologically relevant sample conditions and at much lower sample concentrations (μM), compared to x-ray crystallography or NMR (16). CW ESR lineshape analysis permits the measurement of close distances up to 25 Å, which are crucial length scales for characterizing the subunit interfaces of large protein complexes (17). Although ESR does not provide a complete structure, a discrete number of strategic distance measurements between spin-labeled residues can confer structural information about proteins or the organization of protein assemblies that is otherwise difficult to obtain.
CW ESR has been widely used to characterize light-induced conformational changes in BR where both transient ESR detection and distance measurements have allowed for site-specific monitoring of conformational changes (18–22) or determine structural features of important 7TMs (18–20,22–26). This study, however, provides for the first time (to our knowledge) structural insight into an oligomeric state of 7TM, specifically of PR. We used an approach that has been previously employed to characterize protein and peptide multimers, e.g., membrane-modifying peptides, proteins, and amyloid protein fibers (27–29), where the ratio of spin-labeled and unlabeled protein constituting the oligomer is varied. By mixing unlabeled (cysteine-free) and labeled PR before oligomerization, we could systematically modulate the average distances across spin-labeled subunits. This allowed us to differentiate spectral signatures originating from intersubunit interactions versus intraprotein contacts, as well as determine the orientation of the subunits within the hexameric assembly.
For a closer look at the solvent accessibility and detergent environment of interfacial and surface sites of PR, we turned to the recently developed Overhauser DNP technique (30–34), to directly probe the site-specific hydration water dynamics of PR at the loop or interfacial sites of 55, 58, and 177. The goal was to find the PR-solvent or PR-detergent interface, whose accurate determination is critical to confirm the state of PR oligomers. Overhauser DNP methods have already been used to characterize the hydration dynamics of various biomolecular surfaces, including apo-myoglobin (35), aggregated tau protein (36), and lipid vesicles (37,38), but not of protein oligomers or complexes.
Taken together, we were able to conclude that the structure and orientation of PR is defined and radial, not random, within the hexameric oligomeric state. These findings complement and extend previous structural studies of PR oligomers, by providing information about the PR-detergent-solvent interface and distance constraints critical for identifying the specific orientation of PR subunits within their complexes—all of which was previously unknown.
Materials and Methods
PR sample preparation
The expression and purification of PR were performed as described previously (39,40). A gene (provided by Gregg Whited from Genencore, Palo Alto, CA) encoding for cysteine-free, green proteorhodopsin with 6× histidine tags at the C-terminus was subcloned into a pTricHis2 plasmid. The naturally occurring cysteines at positions 107, 156, and 175 were replaced with serines. We confirm that these mutations have not affected the overall function of the channel as they still bind retinal and give similar UV absorption spectra, as also discussed by others for similar mutations (41). This plasmid was further mutagenized to change the glutamic acid at position 108 to a glutamine in order to remove the proton acceptor site and prolong the intermediate M state in the PR photocycle (42,43), which will facilitate future characterization in different functional states.
Single cysteine mutants were prepared from this PR plasmid using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA), modified for a two-stage polymerase chain reaction. BL21(DE3) cells transformed with mutant expression plasmids were incubated in LB media with 1 mg/mL ampicillin at 37°C with rotary shaking. Protein expression was induced at an optical density of A600 = 0.8–1 of cell growth by adding IPTG and retinal to final concentrations of 1 mM and 10 μM, respectively. The cells were harvested after 5 h of protein expression, by centrifugation at 2000 × g for 20 min at 4°C. Each preparation produces ∼20–30 mg of purified PR from a 1-L cell suspension.
Purification and spin-labeling of PR
Cells were suspended in a lysis buffer containing 50 mM KPO4, 150 mM KCl, 0.02 mg/mL lysozyme, 0.1 mg/mL DNase I, and 2 mM MgCl2, as well as antiproteolytics, and were lysed by probe-tip sonication. The membranes containing PR were separated from the cytosolic fraction by ultracentrifugation, and solubilized overnight in 2% DDM. Solubilized proteins were incubated overnight with a His-tag affinity resin. Unbound proteins were removed by washing, and bound PR was reacted with methanethiosulfonate spin-label at a 10:1 label/protein ratio. The labeled protein was eluted with a buffer containing 0.05% DDM, 50 mM KPO4, 150 mM KCl, and 250 mM imidazole, and subsequently concentrated with 50-kDa MWCO centrifugal filter tubes. The protein was further purified and oligomers separated by size-exclusion chromatography using a Sephadex 200 column on an FPLC instrument (Akta; GE Healthcare, Wauwatosa, WI). Peak fractions were collected and concentrated with 50-kDa MWCO centrifugal filter tubes.
For some experiments, we prepared spin-diluted oligomers wherein unlabeled protein with no cysteines is mixed with spin-labeled PR to reduce the number of spin-labeled PR per oligomeric complex. As our unlabeled protein contains no free cysteines, any potential for cross-linking between protein subunits is avoided. While the protein was bound to the affinity column, the concentration of the DDM detergent was increased to 1.0% to disrupt the oligomers, and unlabeled PR added at a mole fraction of 1/6 to a 5/6 mol fraction of spin-labeled PR-55R1. A 1:5 ratio leads to an average of no more than one spin-labeled 55R1 (or ∼17%) per complex. Next, the resin was washed repeatedly with 0.05% DDM to decrease the detergent concentration and promote higher-order oligomerization. The sample was applied to the FPLC SEC column for separation by size, as with the previous samples.
Determining molecular weight of the protein/detergent assembly by SEC-LS/UV/RI
The mass of PR oligomers was measured in solution using a miniDAWN TREOS three-angle static light-scattering detector, an Optilab rEX refractive index detector (both by Wyatt Technology, Santa Barbara, CA), and a model No. 600-HPLC equipped with a model No. 996 photodiode array detector (both by Waters, Milford, MA). We injected purified protein samples (100 μL) onto a silica SEC column (300 Å pore size and 5000–1,250,000 protein MW range; Wyatt) with a running buffer, including 0.03% DDM, at pH 8.2 and a rate of 0.5 mL/min. The light scattering and refractive index detectors were calibrated according to the manufacturer’s guidelines.
The mathematical basis for determining the subunit stoichiometries of transmembrane proteins in a detergent solution is described in detail by Slotboom et al. (15). Briefly, the molecular weight (MW) of the protein can be measured using the relationship
| (1) |
where LS is the signal from the light-scattering detector; ε280, protein is the molar extinction coefficient of the protein; UV280 is the light absorbed by the protein at 280 nm; K is a constant; and RI is the signal from the refractive index detector. The MW of proteorhodopsin was extracted using the protein conjugate template in the ASTRA V software (Wyatt Technology, Santa Barbara, CA). The dn/dc values used in the ASTRA analysis are 0.185 mL/g for proteins (44), which is a value is independent of amino-acid composition, and 0.143 mL/g for DDM detergents (15). The extinction coefficient at 280 nm, ε280, protein, for proteorhodopsin is 2507 mL g−1 cm−1, as calculated from the amino-acid sequence using the ProtParam tool on the ExPaSy server (expasy.org). Three separate preparations of the proteorhodopsin/DDM complex were run and the average molecular weight computed.
CW ESR spectroscopy and intersubunit distance determination
ESR spectra were collected at room temperature (RT) and 140 K on an EMX X-band spectrometer (Bruker, Billerica, MA) using a cylindrical cavity tuned to a microwave frequency of 9.8 GHz, on resonance at a center field of 3330 G. The field modulation frequency was set at 100 kHz, with a field-modulation amplitude of 1 G for experiments at RT and 2 G at 140 K. Samples with volumes of 3–4 μL were loaded into quartz capillaries with 0.6 mm ID and 0.8 mm OD and sealed with beeswax.
Intersubunit distances around site 55R1 were determined from the low-temperature spectra collected at 140 K. The dipolar-broadened spectrum was fit to simulated powder patterns using a program based on a convolution method, similar to that introduced by Steinhoff et al. (45). This method is essentially the inverse of the deconvolution method (46,47). Both are well-established and comparable techniques with subtle differences. In our case, the g and A tensors of the broadened and unbroadened spectra are slightly different, which would manifest itself in a slightly incorrect Pake pattern obtained by deconvolution, possibly leading to artifacts in the determined distance distribution. For the studied system here, we determined the convolution method by assuming a single distance and width to be a more robust approach. We used a single distance distribution despite the presence of several spin-pair interactions with three distinct distances, because the smallest distance will have by far the largest effect on the broadened spectrum of nitroxide spin-labels on PR surfaces. Our experimental results will further validate the assumption of a single mean distance, given that the two possible longer distances present within the oligomer arrangements, that will turn out to be radially arranged PR hexamers, will be beyond the dipolar-broadening limit of CW ESR lineshape (>25 Å).
A spin-diluted hexamer with approximately one spin-labeled cysteine per oligomer was used as a reference spectrum to determine the line-width of the spectrum with no dipolar broadening. The monomer PR sample provides a less suitable reference spectrum, given small deviations in the g and A tensors to that of the hexamer PR. The unbroadened absorption spectrum was then convoluted with a dipolar Pake pattern corresponding to a Gaussian distance distribution with one mean distance r and a distance distribution width σ, and the convoluted spectrum then fit to the broadened 55C hexamer spectrum by least-square optimization, by either varying the mean distance r or the width of the distribution σ.
DNP experiments
DNP-enhanced NMR experiments were performed using the same ESR setup as described above. A quartz-capillary (outer diameter = 0.8 mm) with the sample was placed in a tight-fit, home-built, U-shaped NMR coil that was inserted into a cylindrical ESR cavity (the same cavity used for ESR experiments) and connected via a coaxial cable to a simple LC tuning box tuned to 14.8 MHz and connected to a broadband channel of an Avance NMR spectrometer (Bruker). Throughout the DNP measurements, the ESR cavity was tuned at a quality factor (Q) of ∼15,000. The central ESR transition was irradiated with a home-built 8–10 GHz microwave source, amplified with solid-state sources to provide several Watts of power as described previously in Armstrong et al. (48). Sample heating during irradiation was minimized by maintaining the highest possible microwave B1 conversion at a high Q factor, and by continuously flowing gaseous nitrogen over the sample.
The DNP-enhanced NMR signal, E, is related to the interaction between the electron spin on the nitroxide spin-label and the nuclear spin of water protons by
| (2) |
where ξ is the coupling factor which describes the electron-nuclear spin dipolar cross-relaxation efficiency; ƒ is the leakage factor which scales for differences in the concentration of electron spins and accounts for relaxation of the protons by other mechanisms than the time-dependent dipolar field of the electron spin; s is the saturation factor that describes the saturation of the electronic Zeeman transition; and γs and γI are the gyromagnetic ratios of the electron and proton respectively, where γs/γI = 658. The values for E and ƒ were extracted from the raw data using a home-built software program (J. Franck, University of California Santa Barbara, personal communication, 2011). The parameter γs/γI is a constant, s can be assumed to be close to 1 under the present experimental condition (33), and E and ƒ are measured experimentally, so the parameter ξ can be determined. The hydration water diffusion-dynamics contained in the parameter, ξ, can be extracted as the translational correlation time, τ, for the diffusion of local hydration water in the vicinity of the spin-label, using a force-free hard-sphere model (see 56), as described previously in Armstrong et al. (33).
Given that τ = d2/D, where d is the distance of closest approach between the electron spin and the proton of water, and D is the local translational diffusion coefficient of water of the spin-label, the translational diffusion coefficient of the tethered spin-label can be safely neglected. Thus, without explicitly computing d, one can approximate long τ as slow diffusivity and short τ as fast diffusivity for hydration water, if contributions of slow, bound water are not significant.
Results and Discussion
Spin-labeled sites of PR
For this study, three single cysteine mutants of PR were constructed. Fig. 1 shows the residues chosen for site-directed mutagenesis and spin-labeling. Given that Klyszejko et al. (11) established that the PR subunits are arranged radially, and supported by the presence of a common oligomer interface motif of GXXXG along the B helix (49), we hypothesized that helices A and B or E and H may be pointing toward the center of the complex. Some of these residues, closest to the center of the complex, would likely be close enough within 20–25 Å, giving rise to dipolar broadening visible in the CW ESR spectrum. Therefore, we screened many mutants on these helices and loop regions for dipolar broadening in the CW ESR spectra (e.g., sites S61, W83, V92 Y164, S173, C175, and M189) that would be indicative of multimeric interface interactions, of which three contrasting residues of interest (S55, W58, and T177) were chosen for detailed analysis as presented here.
Figure 1.
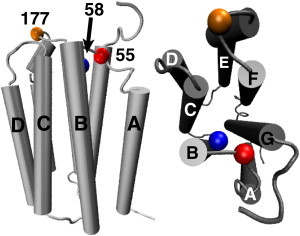
Location of single cysteine mutants for site-directed spin-labeling. The NMR structure of PR (PDB:2L6X) (53) was used to determine the locations of the three residues.
There are several partial structures of PR by NMR (50–52), as well as a recent, complete, detergent-solubilized structure of PR (53). The locations of the residues S55, W58, and T177 (Fig. 1) were determined using the solution NMR structure of PR (PDB:2L6X (53)). Residue 55 is on the A-B loop, 58 is near the terminus of the B-helix, and 177 is on the E-F loop. Each of these residues is on the cytoplasmic face of the PR pump. The cysteine mutants were expressed and purified in either 0.05% or 1.0% DDM. Purifying the protein in 1.0% DDM allowed us to increase the concentration of monomeric PR in solution, as well as modulate the coverage of solvent-exposed residues by detergent molecules compared to using 0.05% DDM.
Stoichiometry of PR oligomers determined by SEC-LS/UV/RI detection
To characterize the effects of oligomerization on the structure of PR, the spin-labeled complexes were separated by size using an FPLC with a size-exclusion column. Fig. 2 shows the elution profile of PR purified in 0.05% DDM. The majority of the protein elutes within one broad component, but there is also a small peak at ∼20-min elution time. The broad peak and small monomer peak are characteristic and representative for PR in 0.05% DDM, for different mutants, as well as in mixtures of unlabeled and labeled protein. The average molecular weight of the protein/detergent complexes, and therefore the degree of PR oligomerization, was determined using the so-called three-detector method, wherein the sample is applied to a size-exclusion column connected in-line with the LS, RI, and UV detectors. This technique allows for the average molecular weight to be calculated at each point of the chromatogram, as shown (black dots) in Fig. 2. The Protein Conjugate analysis package within the ASTRA software was used to determine the average molecular weight of the entire protein/detergent complex, as well as the contributions to the molecular weight by only the protein for the regions shaded gray in Fig. 2.
Figure 2.
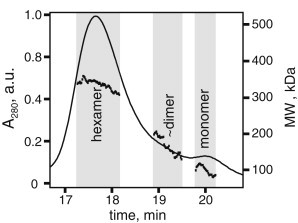
Oligomers of PR solubilized in 0.05% DDM detergent solutions were separated by FPLC and the approximate sizes of the assemblies measured by static light scattering. The ASTRA analysis of the SEC-LS/UV data is shown. Molecular weight of the PR/detergent assemblies at each point (dotted line); fractions collected for ESR and DNP (gray).
Although there is no complete separation of the individual oligomers, the average molecular weight of the protein or protein complex can be used to compute the oligomerization state of PR at each peak along the elution profile. Table 1 presents the computed average molecular weight for the PR-detergent assembly corresponding to each peak (gray regions in Fig. 2). Given the molecular weight for the protein and all bound detergent molecules, the contributions to the molecular weight only from the protein can be determined. The protein molecular weight was divided by the mass of one PR (29,000 kDa) to give the approximate oligomeric assembly for each peak. Although it is possible that some fraction of other multimers exist in the sample, we consistently find that hexameric PR dominates the population in 0.05% DDM solution, followed by dimers and monomers.
Table 1.
Summary of SEC-LS/UV/RI data and determination of the PR oligomerization using the Protein Conjugate template in the program ASTRA
| Peak 1 | Peak 2 | Peak 3 | |
|---|---|---|---|
| Total MW protein+detergent (g/mol) | 339,800 | 207,870 | 69,640 |
| Protein MW (g/mol) | 187,030 (3%) | 66,390 (16%) | 38,390 (35%) |
| No. of spin-labeled PR per complex | 6.4 | 2.3 | 1.3 |
The relative SD is given for the MW of the protein.
Hexamers and dimers, beside monomers, seem to also dominate the PR population in other detergent micelle environments. If anything, the protein number in the larger multimer population tends to be slightly larger than 6 (i.e., between 6 and 6.5) (see Table 1), which makes it less likely for a mixture of dominant pentamers and hexamers to constitute the main population. Also, ongoing nanometer distance measurements by pulsed DEER experiments suggest that hexamers are the dominant multimers, given that at least two distinct distance populations are observed with mean distances of ratio 1:1.7. Still, some small fraction of other oligomers cannot be excluded. The fractions from the FPLC separation that correspond to hexameric, dimeric, and monomeric complexes (Fig. 2) were combined into three batches of samples to conduct electron-paramagnetic resonance (EPR) and DNP experiments on each fraction separately.
Changes in CW ESR lineshape at RT as a function of PR oligomer size
CW ESR spectra were taken of samples 55R1, 58R1, and 177R1, each in the monomeric, dimeric, and hexameric oligomer states (Fig. 3). At room temperature (RT), the ESR lineshape is strongly affected by the motion of the nitroxide linker, which is related to the interaction of the nitroxide with the tertiary structure protein environment, as well as the flexibility of the protein backbone. The monomer fraction of each spin-labeled mutant shows two spectral components with different mobilities for the nitroxide, labeled i and m for immobile and mobile, respectively. It is possible that some of the motional broadening in the monomeric fraction of 55R1 may be due to incomplete separation from dimers by FPLC.
Figure 3.
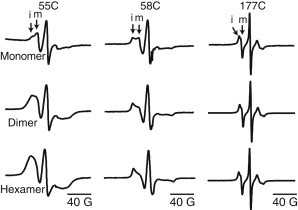
Room temperature CW ESR spectra of monomeric, dimeric, and hexameric fractions of 55C, 58C, and 177C in 0.05% DDM. The 55C monomeric spectrum is significantly narrower than the corresponding hexameric spectrum. In the 58C spectra, the ratio of the immobile (i) and mobile (m) components of the spectra varies as the size of the assembly increases. The lineshape of the 177C assemblies does not change depending on the size of the assembly.
This is supported by a slightly higher protein mass (38,390 g/mol in Table 1) in the monomeric fraction. As the size of the complex increases, the spectra for 55R1 and 58R1 change significantly. The spectra for the 55R1 dimer and hexamer are dramatically broadened compared to that of the monomer, suggesting dipolar broadening between neighboring 55 residues. The spin-labeled residues must be <25 Å apart for the distance-dependent dipolar broadening to be visible by CW ESR, providing an important clue about the location of residue 55 at the oligomeric interfaces. For 58R1, the ratio of the immobile to mobile component increases with the degree of oligomerization, but there is no apparent increase in dipolar broadening effects on the lineshape.
The increase in the immobile ESR spectral component indicates that the spin-label is experiencing a more restricted environment as the size of the complex increases, which is due to an increase in tertiary protein contacts. We can conclude that residue 58 is likely at or near the oligomer interface and experiences an increase in inter- or intra-PR contact when the protein is in a hexamer, while neighboring 58 residues across two different PR within the complex must be >25 Å apart. In contrast, there is no change in the CW ESR spectra of 177R1 between any of the three PR populations. This together with the overall narrow ESR line-widths confirms that residue 177 is located on a surface- and solvent-accessible site and is not participating in protein contacts. Also, because there is no observable dipolar broadening in any of the spectra, the spin-labels must be >25 Å apart. Thus, residue 177 is not affected by the oligomeric state and is not at or near the oligomeric interface.
CW ESR lineshape at RT of PR complexes mixed with unlabeled protein
The RT CW ESR analysis of spin-diluted complexes further substantiates the nature of the dipolar interaction between 55R1 within the PR hexamers. We prepared hexameric 55R1 PR with only one PR subunit spin-labeled on average by mixing 1/6 mol fraction of spin-labeled with 5/6 mol fraction of unlabeled (cysteine-free) PR (Fig. 4 a). Fig. 4 b shows the RT CW ESR spectra of the 55R1/unlabeled PR (17% 55C-labeled PR) mixture in monomeric, dimeric, and hexameric complexes overlaid on that of 100% 55R1 PR hexamer samples.
Figure 4.
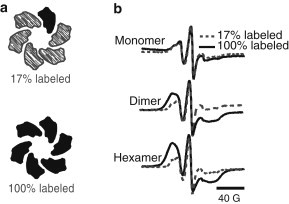
Cartoon of labeled and spin-diluted PR hexamers shown together with room temperature CW ESR spectra. (a) 55C PR was mixed with unlabeled WT PR to yield oligomers with ∼17% spin-labeled 55C residues. (b) Room-temperature CW ESR spectra comparing fully labeled 55C with oligomeric assemblies containing ∼17% spin-labeled residues. The broadening in the hexameric and dimeric fractions is significantly decreased.
Compared to the fully labeled sample, the spectrum of the spin-diluted hexamer sample is significantly narrower. Equally revealing is that the spectra of the spin-diluted PR samples now do not change with the complex size, unlike the fully labeled sample, and are nearly identical to that of the 100% 55R1 monomer, shown in Fig. 4. Together, this shows that spin-diluting the PR oligomers can eliminate the dipolar broadening of the spectrum to reconstructs a spectrum that is very close to that of the monomer 55R1 spectrum, except for small changes in the g and A tensor values. The line broadening of 55R1 can thus be attributed to mostly dipolar inter-PR spin-label interactions.
Determination of intersubunit distances by CW ESR lineshape analysis at 140 K
At temperatures <200 K, the motion of the nitroxide linker is arrested and the dipolar interaction becomes the dominant source of line broadening. The ESR spectra of frozen samples for 55R1 and 58R1 are shown in Fig. 5. The spectrum of the 55R1 hexamer is still significantly broader than that of the 55R1 monomer, and the ratio of the amplitudes of the center to the low field peak is decreased, which are characteristic features of dipolar-broadened spectra (54). This confirms that neighboring 55 residues within the hexameric complex are <25 Å apart. The spectral lineshapes of the 58R1 hexamer and the monomer are, however, now indistinguishable at 140 K.
Figure 5.
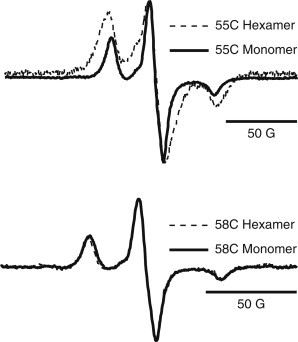
Rigid-limit CW ESR spectra at 140 K of 55C and 58C in both the hexameric and monomeric assemblies. The 55C hexamer is dipolar-broadened by neighboring 55C residues within the complex. The 58C hexameric spectrum is not broader than the respective monomer spectrum, suggesting that 58 residues are >∼2 nm apart.
The distance between two spin-labels can be quantified by lineshape analysis of the dipolar broadened CW ESR spectrum of the frozen sample (45,47). We determined the distance between neighboring 55R1 residues using a program based on the convolution method of Steinhoff et al. (45), as discussed in detail in Materials and Methods. The spin-diluted 55R1 hexamer spectrum was used as the unbroadened reference spectrum, convoluted with a known Gaussian distance distribution of a single mean distance and width to match the dipolar broadened 55R1 spectra. Evaluation according to residual fits present excellent agreement (see Fig. S2 A in the Supporting Material). A symmetric hexameric assembly would lead to three distances as shown in the inset of Fig. 6 a: r, 1.7r, and 2r. Therefore, we can assume that the neighboring residues with the distance r will by far dominate the line broadening in the CW ESR spectrum, as long as the shortest distance r is larger than ∼14 Å. Due to the general complexity of resolving three distances and distributions in a hexameric sample and the limitation of CW-based EPR methods in resolving distances beyond 25 Å, we sought to measure only the distance between neighboring 55 residues.
Figure 6.
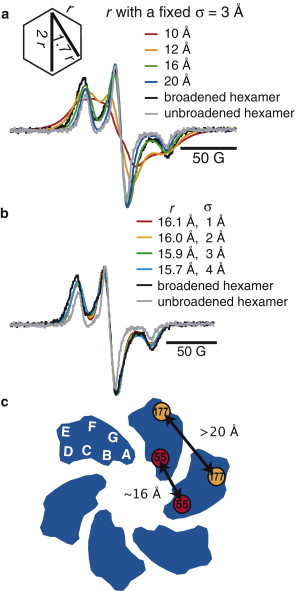
Rigid-limit CW ESR spectra at 140 K for the hexameric, dipolar-broadened 55C (black) and spin-diluted, unbroadened spin-diluted hexamer (gray). (a) Simulated spectra for distances, r, between 10 and 20 Å with a fixed width of the distance distribution, σ = 3 Å. (Inset) Relationship of the three possible distances in a symmetric hexamer. (b) Optimized distances, r, are shown for fixed widths of the Gaussian distribution, σ = 1–4 Å. (c) A distance constraint of ∼16 Å between neighboring 55C residues suggests an orientation where the A-B loop points toward the center of the complex.
Fig. 6 a presents simulated powder spectra for interspin distances between 10 and 20 Å and a fixed distribution width, σ, of 3 Å. Based on qualitative comparison between the convoluted spectra for several distances and the experimentally observed one, we could rule out distances >16 Å and <12 Å to be feasible as closest inter-55R1 distances. In a first approximation and given σ = 3 Å, the simulated spectrum of 16 Å gives the best approximate fit to the broadened spectrum of the hexameric 55R1. However, because σ is still unknown, we now repeated the simulation first with a series of fixed σ-values and optimized the simulated distance, r, for each respective σ-value. Fig. 6 b shows the simulated spectra for σ between 1 and 4 Å.
The resulting simulated distances are given for each σ, which lie between 15.7 and 16.1 Å. The best fits are those with a σ of 3–4 Å. Distributions >4 Å are not physically reasonable given that the spin-label is bound and restricted on a defined biological system, and larger distributions do not lead to a significantly better fit. The determined distance of 15–16 Å confirms our assumption that the distance r is the dominant source of broadening, as this corresponds to a next-neighbor distance that would cause strong dipolar broadening in the CW ESR spectrum, while the other two distances, 1.7r and 2r, would be outside the measurable distance ranges by CW ESR lineshape analysis, given 1.7r > 25.5 Å and 2r > 30 Å. The experimental finding of a relatively narrow width (i.e., 3–4 Å) for the distance distribution further validates the approximation of one mean distance to be reasonable.
Given that Klyszejko et al. (11) established that PR hexamers assemble radially, the only orientation that would place neighboring 55R1 residues within ∼16 Å distances is one where the A-B loop faces the center of the complex (Fig. 6 c). Furthermore, the only orientation that would place residue 58R1 >25 Å apart is if the B-Helix is tilted away from the center of the complex. Both of these distance constraints are plausible, given the scale bar on the radially assembled hexamers in the AFM study by Klyszejko et al. (as seen in Fig. 4 B of Klyszejko et al. (11)).
CW ESR lineshape at RT of PR in excess detergent concentration
To further characterize the environments of the three residues 55, 58, and 177 and, specifically, to determine the influence of the detergent molecules on their ESR spectra, we purified the same three mutants in 1.0% DDM concentration, and separated the sample as before using the FPLC method (see Fig. S3). The spectrum for the 55R1 monomer changes slightly from 0.05% to 1%, likely because increasing the detergent concentration increases the monomeric PR population and distribution. This is likely because there is more detergent available to cover the hydrophobic regions of the protein to provide more stability for the monomeric PR configuration. The dimer and hexamer, as measured at site 55, are virtually the same in 1.0% vs. 0.05% DDM concentration.
For 58 in 0.05% DDM, an increase in the immobile component was observed from the monomer to hexamer PR (Fig. 3). This would suggest that 58 is positioned at a slightly more-detergent–exposed oligomer interface, while clearly experiencing interprotein interactions. This is further supported by the observation that for a monomer sample that should not experience interprotein contacts, the immobile component is increased in 1% DDM (see Fig. S3) compared to 0.05% DDM. In fact, at 1% DDM, the ESR spectrum of 58R1 does not change very much from monomer to hexamer, suggesting that 58R1 is more sensitive to oligomer size in 0.05% DDM than in 1% DDM, where 58R1 may be more covered by detergent contacts. At position 177R1, there are minimal differences in the spectra for different multimers and DDM concentration. The hexamer is slightly broader at 1%, particularly for the hexamer, where there is a low field hump.
The spectra at 0.05% and 1.0% for mutant 58R1 give important clues about the environment near this site. We conclude that 58R1 is simultaneously at the PR multimer and PR-detergent interface. At low detergent concentrations, the detergent preferentially covers the hydrophobic helical regions of the protein, while leaving site 58 (which is at the top of the helix) more exposed to water, so that changes in oligomerization make a bigger impact on the mobility of the nitroxide linker. When the sample is in 1% DDM, we suggest site 58 is covered in excess detergent, so that the PR-detergent interaction dominates the lineshape.
Site-specific hydration dynamics by Overhauser DNP
Finally, we sought to directly quantify the surface hydration dynamics around the three spin-labeled sites in the different detergent environments using Overhauser DNP measurements to evaluate the site-specific solvent accessibility. There are several examples of using water-soluble (e.g., Ni-EDTA) versus lipid-soluble (e.g., oxygen) spin relaxants to determine whether particular residues of membrane proteins are bulk solvent-exposed or buried within the lipid bilayer (55), but it will be difficult to use this method to characterize residues on the membrane interface formed by loop sites of PR, as the difference in their solvent accessibility is too subtle to be studied by existing techniques. Table 2 shows the DNP parameters, f, ξ, and the computed τ for the three hexameric mutants in either 0.05% or 1.0% DDM, with the experimental approach for Overhauser DNP described in more detail in Materials and Methods.
Table 2.
The DNP parameters
| Sample | Hexamer in 0.05% |
DDM |
Hexamer in 1.0% |
DDM |
||
|---|---|---|---|---|---|---|
| f | ξ | τ(ps) | f | ξ | τ(ps) | |
| 55C | 0.337 | 0.0322 | 412 | 0.495 | 0.0378 | 404 |
| 58C | 0.488 | 0.0284 | 464 | 0.368 | 0.0315 | 437 |
| 177C | 0.184 | 0.105 | 171 | 0.597 | 0.0393 | 376 |
Leakage factor ƒ, coupling factor ξ, and the translational correlation time τ, for 55C, 58C, and 177C. The error in τ is within 10%.
Notably, Table 2 shows that in 0.05% DDM, 55R1 and 58R1 have very long translational correlation times (412 and 462 ps) when compared to 177 R1 (171 ps), indicative of more restricted solvent dynamics, which would be the expected outcome given 55R1 and 58R1 are found to be at or near subunit interfaces. However when the mutants are purified in 1.0% DDM, all sites assume a similarly long τ (376–437 ps), as found around the more buried 55R1 and 58R1 sites of PR in 0.05% DDM. Thus, the τ-value indicates slowed solvent dynamics for 55R1 and 58R1 hexamers, in either 0.05% or 1.0% DDM. Residue 177R1 in 0.05% DDM has a τ-value consistent with a highly solvent-accessible spin-labeled site, but when the detergent concentration is increased to 1.0% DDM, τ increases significantly from 171 to 376 ps. The results for 177R1 show an interesting contrast to those of 55R1 and 58R1.
The change in hydration dynamics shows that when this functionally relevant loop region becomes buried in excess detergent, this may have functional implications for PR. The DNP results also confirm that 55R1 and 58R1 in the hexamers are clearly buried in tertiary interprotein contacts, where the spin-label is not exposed to solvent or detergent environments. Given that loop regions of membrane proteins often play important functional roles, Overhauser DNP may offer insights to why there are significant differences in the structure and function of proteins solubilized in different types of detergents, detergent at different concentrations, or in reconstituted liposomes Instead.
Conclusion
In this work, we show how intersubunit distance constraints from CW ESR lineshape analysis combined with site-specific surface hydration dynamics measurements by Overhauser DNP can determine the orientation of proteorhodopsin in its oligomeric assembly. We found that the dominant oligomeric state of proteorhodopsin solubilized in either 0.05% or 1.0% DDM detergent is a hexamer (Fig. 2), and that PR is oriented with the A-B loop, including site 55, pointing toward the center of the complex within 16 Å (Fig. 6 c). These results are consistent with the two-dimensional AFM results of Klyszejko et al. (11), where a radial arrangement of PR hexamers in dense lipid patches was found. ESR lineshape analysis at RT further suggests that 58C is positioned near an oligomeric interface, as there is an increase in tertiary contacts when the size of the complex increases from monomer to hexamer.
The complementary Overhauser DNP analysis provides us with a unique opportunity to probe the local hydration dynamics selectively around the spin-labeled residue, and to determine which residues were solvent-accessible versus those buried in tertiary protein contacts. Somewhat expectedly, residue 177 is on a surface-exposed loop, while both sites 55 and 58 are buried within protein contacts. Taken together, the spectroscopic results allow us to conclude that the PR units within a hexameric assembly have distinctly defined radial orientation. The site-specific hydration landscape around PR oligomers provided further clues as to why there may be functional differences between PR constituted in different detergent or lipid membrane environments. In fact, ongoing studies in our group show that the oligomerization of PR clearly affects the functional properties of PR.
Given the importance of membrane protein oligomer states for the study of their function and structure, this approach may prove useful for the study of other membrane protein systems, especially because both the ESR- and DNP-based measurements are not limited by the size and complexity of the protein or proteoliposomes, and can be carried out in solution state under physiological conditions.
Acknowledgments
We thank many co-workers and students who greatly assisted in handling of proteorhodopsin samples. Initially Gregg Whited from Genencore, Galen Stucky, and Hongjun Liang helped us obtain the PR genes, while Sunyia Hussain, Chris Carnabatu, and Christopher Knoll heavily contributed to optimizing the biosynthesis protocol for PR subsequently. We are also grateful to John Perona, Armand Vartanian, and Andrew Hadd for helping us access and use the static-light scattering instrument with SEC-LS/UV/RI detection capabilities, as well as Jerry Hu and the Materials Research Laboratory at the University of California at Santa Barbara for support accessing EPR instrumentation.
This work was supported by the National Science Foundation through the Materials Research Science and Engineering Centers Program No. DMR-1121053 (Materials Research Laboratory at the University of California at Santa Barbara), the Institute for Collaborative Biotechnologies through grant No. W911NF-09-0001 from the U.S. Army Research Office (the content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred) for K.M.S. and S.H., the Packard Fellowship for Science and Engineering for S.H. and A.P., and the Feodor Lynen Research Fellowship of the Alexander von Humboldt Foundation for M.J.N.J. M.K. was also supported by the CAMP program of the University of California at Santa Barbara’s Materials Research Laboratory and the Beckman Scholars program awarded to the University of California at Santa Barbara’s California NanoSystems Institute. This work utilized the Materials Research Laboratory Central Facilities (a member of the National Science Foundation-funded Materials Research Facilities Network, www.mrfn.org) supported by the Materials Research Science and Engineering Centers Program of the National Science Foundation under grant No. DMR-1121053.
Supporting Material
References
- 1.Ferré S., Franco R. Oligomerization of G-protein-coupled receptors: a reality. Curr. Opin. Pharmacol. 2010;10:1–5. doi: 10.1016/j.coph.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurevich V.V., Gurevich E.V. GPCR monomers and oligomers: it takes all kinds. Trends Neurosci. 2008;31:74–81. doi: 10.1016/j.tins.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henderson R., Unwin P.N.T. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 4.Béjà O., Spudich E.N., DeLong E.F. Proteorhodopsin phototrophy in the ocean. Nature. 2001;411:786–789. doi: 10.1038/35081051. [DOI] [PubMed] [Google Scholar]
- 5.Geiser A.H., Sievert M.K., Ruoho A.E. Bacteriorhodopsin chimeras containing the third cytoplasmic loop of bovine rhodopsin activate transducin for GTP/GDP exchange. Protein Sci. 2006;15:1679–1690. doi: 10.1110/ps.062192306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakatsuma A., Yamashita T., Kandori H. Chimeric microbial rhodopsins containing the third cytoplasmic loop of bovine rhodopsin. Biophys. J. 2011;100:1874–1882. doi: 10.1016/j.bpj.2011.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobe B., Guncar G., Forwood J.K. Crystallography and protein-protein interactions: biological interfaces and crystal contacts. Biochem. Soc. Trans. 2008;36:1438–1441. doi: 10.1042/BST0361438. [DOI] [PubMed] [Google Scholar]
- 8.Lacapère J.-J., Pebay-Peyroula E., Etchebest C. Determining membrane protein structures: still a challenge! Trends Biochem. Sci. 2007;32:259–270. doi: 10.1016/j.tibs.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Morgner N., Kleinschroth T., Brutschy B. A novel approach to analyze membrane proteins by laser mass spectrometry: from protein subunits to the integral complex. J. Am. Soc. Mass Spectrom. 2007;18:1429–1438. doi: 10.1016/j.jasms.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann J., Aslimovska L., Brutschy B. Studying the stoichiometries of membrane proteins by mass spectrometry: microbial rhodopsins and a potassium ion channel. Phys. Chem. Chem. Phys. 2010;12:3480–3485. doi: 10.1039/b924630d. [DOI] [PubMed] [Google Scholar]
- 11.Klyszejko A.L., Shastri S., Glaubitz C. Folding and assembly of proteorhodopsin. J. Mol. Biol. 2008;376:35–41. doi: 10.1016/j.jmb.2007.11.030. [DOI] [PubMed] [Google Scholar]
- 12.Dorwart M.R., Wray R., Blount P. S. aureus MscL is a pentamer in vivo but of variable stoichiometries in vitro: implications for detergent-solubilized membrane proteins. PLoS Biol. 2010;8:e1000555. doi: 10.1371/journal.pbio.1000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waight A.B., Love J., Wang D.-N. Structure and mechanism of a pentameric formate channel. Nat. Struct. Mol. Biol. 2010;17:31–37. doi: 10.1038/nsmb.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yernool D., Boudker O., Gouaux E. Trimeric subunit stoichiometry of the glutamate transporters from Bacillus caldotenax and Bacillus stearothermophilus. Biochemistry. 2003;42:12981–12988. doi: 10.1021/bi030161q. [DOI] [PubMed] [Google Scholar]
- 15.Slotboom D.J., Duurkens R.H., Erkens G.B. Static light scattering to characterize membrane proteins in detergent solution. Methods. 2008;46:73–82. doi: 10.1016/j.ymeth.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 16.Hemminga M., Berliner L. Springer; New York: 2007. ESR Spectroscopy in Membrane Biophysics. [Google Scholar]
- 17.Berliner L.J., Eaton S.S., Eaton G.R., editors. Biological Magnetic Resonance. Vol. 19: Distance Measurements in Biological Systems by EPR. Kluwer; New York: 2000. [Google Scholar]
- 18.Steinhoff H.J., Mollaaghababa R., Hubbell W.L. Time-resolved detection of structural changes during the photocycle of spin-labeled bacteriorhodopsin. Science. 1994;266:105–107. doi: 10.1126/science.7939627. [DOI] [PubMed] [Google Scholar]
- 19.Thorgeirsson T.E., Xiao W., Shin Y.K. Transient channel-opening in bacteriorhodopsin: an EPR study. J. Mol. Biol. 1997;273:951–957. doi: 10.1006/jmbi.1997.1362. [DOI] [PubMed] [Google Scholar]
- 20.Mollaaghababa R., Steinhoff H.J., Khorana H.G. Time-resolved site-directed spin-labeling studies of bacteriorhodopsin: loop-specific conformational changes in M. Biochemistry. 2000;39:1120–1127. doi: 10.1021/bi991963h. [DOI] [PubMed] [Google Scholar]
- 21.Rink T., Pfeiffer M., Steinhoff H.-J. Unraveling photoexcited conformational changes of bacteriorhodopsin by time-resolved electron paramagnetic resonance spectroscopy. Biophys. J. 2000;78:1519–1530. doi: 10.1016/S0006-3495(00)76704-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinhoff H.-J., Mollaaghababa R., Hubbell W.L. Site directed spin labeling studies of structure and dynamics in bacteriorhodopsin. Biophys. Chem. 1995;56:89–94. doi: 10.1016/0301-4622(95)00019-t. [DOI] [PubMed] [Google Scholar]
- 23.Pfeiffer M., Rink T., Steinhoff H.J. Site-directed spin-labeling reveals the orientation of the amino acid side-chains in the E-F loop of bacteriorhodopsin. J. Mol. Biol. 1999;287:163–171. doi: 10.1006/jmbi.1998.2593. [DOI] [PubMed] [Google Scholar]
- 24.Steinhoff H.J., Savitsky A., Möbius K. High-field EPR studies of the structure and conformational changes of site-directed spin labeled bacteriorhodopsin. Biochim. Biophys. Acta. 2000;1457:253–262. doi: 10.1016/s0005-2728(00)00106-7. [DOI] [PubMed] [Google Scholar]
- 25.Wegener A.A., Chizhov I., Steinhoff H.J. Time-resolved detection of transient movement of helix F in spin-labeled Pharaonis sensory rhodopsin II. J. Mol. Biol. 2000;301:881–891. doi: 10.1006/jmbi.2000.4008. [DOI] [PubMed] [Google Scholar]
- 26.Radzwill N., Gerwert K., Steinhoff H.J. Time-resolved detection of transient movement of helices F and G in doubly spin-labeled bacteriorhodopsin. Biophys. J. 2001;80:2856–2866. doi: 10.1016/S0006-3495(01)76252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milov A.D., Tsvetkov Y.D., Raap J. The secondary structure of a membrane-modifying peptide in a supramolecular assembly studied by PELDOR and CW-ESR spectroscopies. J. Am. Chem. Soc. 2001;123:3784–3789. doi: 10.1021/ja0033990. [DOI] [PubMed] [Google Scholar]
- 28.Margittai M., Langen R. Template-assisted filament growth by parallel stacking of tau. Proc. Natl. Acad. Sci. USA. 2004;101:10278–10283. doi: 10.1073/pnas.0401911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haemers S., Koper G.J.M., Frens G. Effect of oxidation rate on cross-linking of mussel adhesive proteins. Biomacromolecules. 2003;4:632–640. doi: 10.1021/bm025707n. [DOI] [PubMed] [Google Scholar]
- 30.Hausser K.H., Stehlik D. Dynamic nuclear polarization in liquids. Adv. Magn. Reson. 1968;3:79–139. [Google Scholar]
- 31.Armstrong B.D., Han S. A new model for Overhauser enhanced nuclear magnetic resonance using nitroxide radicals. J. Chem. Phys. 2007;127:104508. doi: 10.1063/1.2770465. [DOI] [PubMed] [Google Scholar]
- 32.Höfer P., Parigi G., Bennati M. Field dependent dynamic nuclear polarization with radicals in aqueous solution. J. Am. Chem. Soc. 2008;130:3254–3255. doi: 10.1021/ja0783207. [DOI] [PubMed] [Google Scholar]
- 33.Armstrong B.D., Han S. Overhauser dynamic nuclear polarization to study local water dynamics. J. Am. Chem. Soc. 2009;131:4641–4647. doi: 10.1021/ja809259q. [DOI] [PubMed] [Google Scholar]
- 34.Sezer D., Prandolini M.J., Prisner T.F. Dynamic nuclear polarization coupling factors calculated from molecular dynamics simulations of a nitroxide radical in water. Phys. Chem. Chem. Phys. 2009;11:6626–6637. doi: 10.1039/b905709a. [DOI] [PubMed] [Google Scholar]
- 35.Armstrong B.D., Choi J., Han S. Site-specific hydration dynamics in the nonpolar core of a molten globule by dynamic nuclear polarization of water. J. Am. Chem. Soc. 2011;133:5987–5995. doi: 10.1021/ja111515s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavlova A., McCarney E.R., Han S. Site-specific dynamic nuclear polarization of hydration water as a generally applicable approach to monitor protein aggregation. Phys. Chem. Chem. Phys. 2009;11:6833–6839. doi: 10.1039/b906101k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kausik R., Han S. Dynamics and state of lipid bilayer-internal water unraveled with solution state 1H dynamic nuclear polarization. Phys. Chem. Chem. Phys. 2011;13:7732–7746. doi: 10.1039/c0cp02512g. [DOI] [PubMed] [Google Scholar]
- 38.Kausik R., Han S. Ultrasensitive detection of interfacial water diffusion on lipid vesicle surfaces at molecular length scales. J. Am. Chem. Soc. 2009;131:18254–18256. doi: 10.1021/ja9060849. [DOI] [PubMed] [Google Scholar]
- 39.Krebs R.A., DeVita A.M., Braiman M.S. Purification and spectroscopic studies of proteorhodopsin. Biophys. J. 2002;82:224a. [Google Scholar]
- 40.Parthasarathy R.D., Caterino T., Braiman M.S. Site-directed mutagenesis of proteorhodopsin: Study of light activated proton transfer. Biophys. J. 2002;82:226A–227A. [Google Scholar]
- 41.Hillebrecht J.R., Galan J., Birge R.R. Structure, function, and wavelength selection in blue-absorbing proteorhodopsin. Biochemistry. 2006;45:1579–1590. doi: 10.1021/bi051851s. [DOI] [PubMed] [Google Scholar]
- 42.Bangwei X., Tetley W., Stuart J. Evaluation of blue and green absorbing proteorhodopsins as holographic materials. J. Phys. Chem. B. 2008;112:2524–2532. doi: 10.1021/jp0740752. [DOI] [PubMed] [Google Scholar]
- 43.Dioumaev A.K., Wang J.M., Lanyi J.K. Proton transport by proteorhodopsin requires that the retinal Schiff base counterion Asp-97 be anionic. Biochemistry. 2003;42:6582–6587. doi: 10.1021/bi034253r. [DOI] [PubMed] [Google Scholar]
- 44.Folta-Stogniew E., Williams K.R. Determination of molecular masses of proteins in solution: Implementation of an HPLC size exclusion chromatography and laser light scattering service in a core laboratory. J. Biomol. Tech. 1999;10:51–63. [PMC free article] [PubMed] [Google Scholar]
- 45.Steinhoff H.J., Radzwill N., Wollmer A. Determination of interspin distances between spin labels attached to insulin: comparison of electron paramagnetic resonance data with the x-ray structure. Biophys. J. 1997;73:3287–3298. doi: 10.1016/S0006-3495(97)78353-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altenbach C., Oh K.-J., Hubbell W.L. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- 47.Rabenstein M.D., Shin Y.-K. Determination of the distance between two spin labels attached to a macromolecule. Proc. Natl. Acad. Sci. USA. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Armstrong B.D., Lingwood M.D., Han S. Portable X-band system for solution state dynamic nuclear polarization. J. Magn. Reson. 2008;191:273–281. doi: 10.1016/j.jmr.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 49.Russ W.P., Engelman D.M. The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 50.Pfleger N., Wörner A.C., Glaubitz C. Solid-state NMR and functional studies on proteorhodopsin. Biochim. Biophys. Acta. 2009;1787:697–705. doi: 10.1016/j.bbabio.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 51.Hempelmann F., Hölper S., Glaubitz C. His75-Asp97 cluster in green proteorhodopsin. J. Am. Chem. Soc. 2011;133:4645–4654. doi: 10.1021/ja111116a. [DOI] [PubMed] [Google Scholar]
- 52.Pfleger N., Lorch M., Glaubitz C. Characterization of Schiff base and chromophore in green proteorhodopsin by solid-state NMR. J. Biomol. NMR. 2008;40:15–21. doi: 10.1007/s10858-007-9203-5. [DOI] [PubMed] [Google Scholar]
- 53.Reckel S., Gottstein D., Dötsch V. Solution NMR structure of proteorhodopsin. Angew. Chem. Int. Ed. Engl. 2011;50:11942–11946. doi: 10.1002/anie.201105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jeschke G. Determination of the nanostructure of polymer materials by electron paramagnetic resonance spectroscopy. Macromol. Rapid Commun. 2002;23:227–246. [Google Scholar]
- 55.Hubbell W.L., Cafiso D.S., Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 56.Hwang L.-P., Freed J.H. Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. J. Chem. Phys. 1975;63:4017–4026. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.