

Abstract
Endophilin is a key protein involved in clathrin-mediated endocytosis. Previous computational and experimental work suggested that the N-terminal helix is embedded into the membrane to induce curvature; however, the role of the SH3 domain remains controversial. To address this issue, we performed computer simulations of the endophilin dimer in solution to understand the interaction between the N-BAR and SH3 domains and its effect on biological function. We predict that the helix binds to the SH3 domain through hydrophobic and salt-bridge interactions. This protects the hydrophobic residues on both domains and keeps the SH3 domain near the end of the N-BAR domain, in agreement with previous experimental results. The complex has a binding strength similar to a few hydrogen bonds (13.0 ± 0.6 kcal/mol), and the SH3 domain stabilizes the structure of the N-terminal helix in solution. Electrostatic calculations show a large region of strongly positive electrostatic potential near the N-terminal that can orient the helix toward the membrane and likely embed the helix into the membrane surface. This predicted mechanism suggests that endophilin can select for both curvature and electrostatic potential when interacting with membranes, highlighting the importance of the SH3 domain in regulating the function of endophilin.
Introduction
Endophilin plays a central role in clathrin-mediated endocytosis (CME) by inducing curvature in the vesicle neck before scission occurs, and by recruiting the key proteins dynamin and synaptojanin (1–4). Endophilin exists biologically as a homodimer. Each monomer consists of two key domains (an N-terminal amphipathic helix Bin-Amphiphysin-Rvs (N-BAR) domain and an SRC Homology 3 (SH3) domain) joined by a flexible linker. The N-BAR domain is able to induce curvature by wedging the N-terminal amphipathic helix (the H0 helix) and insert helices into the bilayer (5–9). The SH3 domain includes a peptide-binding region that is able to bind to dynamin and synaptojanin (10,11). Although the functions of the N-BAR and SH3 domains are understood individually, the mechanism by which full endophilin is involved in endocytosis is controversial. For instance, Bai et al. (4) suggested that the key function of endophilin is to deform the membrane in an SH3-independent manner before membrane scission of the vesicle. However, Milosevic et al. (12) concluded that endophilin is not required for vesicle scission but is necessary for uncoating of the clathrin-coated vesicle (CCV), and that the SH3 domain is required to recruit synaptojanin for this process (12). These two groups drew different major conclusions from their studies, leaving the question as to what role the SH3 domain plays in the endocytotic mechanism of endophilin unanswered.
Solution-phase small-angle x-ray scattering (SAXS) measurements in another study (13) suggested that each SH3 domain is located near either distal end of the N-BAR dimer. Atomic-level structures of the N-BAR dimer and the SH3 domains were manually docked into the electron density calculated from the SAXS measurements. The best fit was found by placing the SH3 domains at the ends of the N-BAR dimer.
The structure derived from the electron density data suggests that an interdomain interaction may exist between the N-BAR and SH3 domains. Interestingly, an electrostatic interdomain interaction between the SH3 and Fer-CIP4 homology-BAR (F-BAR) domains in syndapin was found to regulate protein function by protecting the hydrophobic residues in the peptide-binding groove while the protein is in solution (14). Similar work was done to investigate whether a similar interaction exists between the SH3 domain and positively charged residues on the N-BAR distal tip, but it was concluded that the interaction between the two domains was not the same as in syndapin (15).
Although the SAXS data suggest that there is an interaction between the N-BAR and SH3 domains, the nature of these interactions remains unknown. Looking beyond a direct interaction between the tips of the N-BAR core, a visual inspection of both the N-terminal helix and the SH3 domain peptide-binding groove suggests that an interaction may exist between the two moieties. Specifically, the location of the SH3 domain at the distal end of the BAR domain suggests that it may be near the H0 helix, and both the helix and the SH3 domain can be aligned such that the hydrophobic residues on the helix are placed in the so-called peptide-binding groove of the SH3 domain (16), protecting nonpolar residues on both domains. If this interaction were to occur, it would also lead to the formation of salt bridges between the helix and the loops surrounding the groove that could further stabilize the complex. Although this interaction is not directly suggested by experiments, a computational study of this possible structure could be used to determine whether this H0-SH3 interaction reasonably explains the previous observed results, and whether this new structure warrants further experimental investigation.
However, previous experimental and computational work suggested that in the absence of the SH3 domain, a single free N-terminal helix of the endophilin N-BAR domain is unstructured in solution. Electron paramagnetic resonance (EPR) (6) and circular dichroism (5) spectroscopy experiments showed that a single H0 helix only folds when in the presence of an appropriately curved membrane. Additionally, computer simulation studies of the folding energetics of a single H0 helix showed that the unfolded state was favored in solution, whereas the folded state was favored when the helix was in the top bilayer of a curved membrane (8). However, the formation of an H0/SH3 complex in endophilin could serve to stabilize the helix structure when the protein is in solution, and control when the helix interacts with curved membranes as opposed to other proteins.
In this work, we used fully atomistic molecular-dynamics (MD) simulations combined with metadynamics for enhanced sampling (17–19) to investigate whether endophilin can autoregulate its function in solution by forming a complex between the H0 helix and the SH3 domain. The formation of this H0/SH3 complex could then protect hydrophobic residues on both domains, and may also explain how the SH3 domain can be held at the distal end of the N-BAR domain. These simulations of the full endophilin protein in solution show that the H0/SH3 complex forms hydrophobic interactions and salt bridges between the helix and the peptide-binding groove, and that the complex is stable over a 100 ns simulation. We used umbrella-sampling simulations to probe the binding energy of the helix and SH3 domain and found that the predicted binding strength is on the order of a few hydrogen bonds, mostly dominated by a series of hydrophobic interactions between the amphipathic helix and the peptide-binding groove. We also performed metadynamics simulations (17–19) to probe the folding landscape of the H0 helix in the peptide-binding groove and found that the SH3 domain is able to stabilize the H0 helix and keep it from unfolding in solution. Cui et al. (8) performed similar simulations on the H0 helix in solution and in the presence of a curved membrane, and found that while it is unstable in solution, the membrane can stabilize the helical structure. Lastly, we calculated the electrostatic potential of the full equilibrated endophilin protein and found that the H0/SH3 complex has a region of large positive electrostatic potential concentrated on the helix and negative electrostatic potential concentrated on the SH3 domain, with a region of neutral electrostatic potential separating the two regions.
Our simulation data lead us to propose a new model for the role of the SH3 domain while endophilin is in solution. While in solution, the SH3 binds with the amphipathic helix, forming an interdomain complex that protects the hydrophobic residues on both domains and serves to autoregulate the function of the protein. When the endophilin reaches the membrane surface, if the membrane has an appropriate negative electrostatic potential, the SH3 domain will point away from the membrane surface and orient the helix toward the surface. Eventually, the H0 helix unbinds from the SH3 domain and inserts into the membrane without the need to unfold in solution and refold in the lipid bilayer, and remodels the membrane surface (presumably into membrane-curvature-induced defects) (8). After this process is completed, the SH3 domain is free to move away from the end of the N-BAR dimer and recruit other proteins to the membrane surface. This mechanism may explain how the SH3 domains are held near the distal ends of the BAR domain, as suggested by the SAXS data. It also suggests that the SH3 domain plays a role in modulating the endocytotic function of the full endophilin protein by creating a structure that can electrostatically screen which types of membranes the H0 helix interacts with, as well as by avoiding interactions with hydrophobic patches of other proteins.
Materials and Methods
Simulation of the full endophilin in solution
The CHARMM 22 force field (20) with CMAP corrections (21) was used for all MD simulations. Simulations of the full endophilin A1 in solution were performed using NAMD 2.7 (22) (http://www.ks.uiuc.edu/Research/namd/) and were run on the Texas Advanced Computing Center Ranger supercomputer cluster. The structure of the N-BAR domain was taken from Masuda et al. (7) (PDB ID: 1X03) and the SH3 domain structure was taken from Trempe et al. (10) (PDB ID: 31QL). The H0 helix was manually placed into the peptide-binding groove and aligned such that the hydrophobic residues were protected from the solvent. Each SH3 domain was placed so that it would be in contact with its monomer’s corresponding helix. The protein was solvated on all sides with at least 15.0 Å of TIP3P water and 0.15 M NaCl using VMD (24) (http://www.ks.uiuc.edu/Research/vmd/). The system was simulated using isothermal and isobaric constant NPT conditions with an integration time step of 2 fs. Bonds between heavy atoms and hydrogen atoms were constrained using SHAKE/SETTLE (25). The temperature was gradually heated to 310 K and held constant using a Langevin thermostat (26) with a dampening coefficient of 0.5 ps−1. The pressure was held constant at 1 bar using a Langevin barostat (27) with a piston period of 0.2 ps and a damping frequency of 0.2 ps−1. Periodicity was imposed in all directions and long-range electrostatics were accounted for using the particle mesh Ewald method (28). The system was initially minimized with 100 kcal/(mol Å) harmonic restraints placed on the Cα backbone atoms. The system was minimized using 100,000 conjugate-gradient steps and the restraints were gradually released over 800 ps of simulation time. The system was then equilibrated for 101 ns.
The salt-bridge distance distributions for each monomer chain were calculated with VMD using the last 20 ns of the simulation, with frames recorded every 2 ps. The hydrophobic residue distance distributions were calculated by measuring the distance between the side-chain centers of mass, using the same simulation data set employed for the salt bridges. The electrostatic potential of the full endophilin A1 protein was calculated by placing 0.1 kcal/(mol Å) restraints on the Cα backbone atoms and continuing to run the simulation for 20 ns, with frames recorded every 2 ps. The calculation was performed using the PMEPot plugin in VMD, which uses a particle mesh Ewald routine.
H0/SH3 binding free energy
Umbrella-sampling simulations were performed using the colvar module in NAMD 2.9 to explore the binding strength between the H0 helix and the SH3 domain in the presence of the full endophilin protein. The equilibrated full protein structure was prepared in the same way as the full protein equilibrium studies. The system was then simulated for an extra 5 ns to ensure that the density of the solvent had relaxed. The distance between the helix atoms (residues 4–21) and the residues in the peptide-binding groove (residues 299, 304, 324, 327, 338, 340, and 343) on monomer A was used as a collective variable. Sampling windows, spaced 0.25 Å apart and spanning from 4.0 Å to 30.0 Å, were used to calculate the binding potential of mean force between the helix and SH3 domain. The individual windows were prepared by moving the initial starting configuration to the corresponding distance by using a moving harmonic restraint of 25.0 kcal/(mol Å) over 500 ps. The window was then relaxed for 500 ps. The starting configuration for each window was the window immediately preceding it or, in the case of the windows below the original configuration, the window immediately after it. The individual windows were run for 4 ns under NPT conditions as previously described with a 25.0 kcal/(mol Å) harmonic restraint on the collective variable distance. The potential of mean force was calculated using the weighted histogram analysis method (WHAM) (29).
Simulations of the H0/SH3 complex in solution
Metadynamics folding simulations were done using a single H0 helix (residues 1–21) and SH3 domain (residues 292–352) taken from the equilibrated full endophilin simulation, where the terminal residue on the helix (residue 21) was patched with a methylamide cap to reduce electrostatic interactions between the helix C-terminus and the peptide-binding pocket. The preequilibrated H0/SH3 complex was prepared in the same way as the full protein, and the system was minimized with 0.1 kcal/(mol Å) restraints on the Cα backbone atoms using 50,000 conjugate gradient steps, after which the restraints on the protein Cα backbone atoms were reduced over 800 ps.
H0 helix folding in the presence of the SH3 domain
The folding free-energy landscape of the H0 helix in the peptide-binding groove was calculated using metadynamics to probe whether the SH3 domain stabilized the helical structure of the N-terminal helix in solution. The metadynamics simulations were performed using NAMD 2.9 with PLUMED (30) in explicit solvent to explore the folding space using the well-tempered metadynamics method with multiple walkers (17–19). Two collective variables were used to describe to folding of the H0 helix, the α-β similarity of the Ramachandran dihedral angles to ideal angles, and the number of backbone 1-4 hydrogen bonds formed. These are the same collective variables that were used by Cui et al. (8) to describe the folding of the H0 helix in a similar study. All of the folding simulations were performed under previously described conditions, except for the use of constant volume instead of constant pressure. The simulation was prepared by relaxing the system at constant volume for 2 ns. The initial hill height was set to 0.5 and the bias factor was set to 12. Hills were added every 200 fs. Five metadynamics walkers were used for a total simulation time of 478 ns.
Results and Discussion
Interdomain interactions keep SH3 at the distal ends of N-BAR
A fully atomistic MD simulation of full endophilin in solution, which was prepared by placing the H0 helix into the peptide-binding groove, formed a stable interdomain complex between the N-terminal helix and the SH3 domain. The helix and SH3 domain were manually aligned so that the hydrophobic domains on the helix and peptide-binding groove were hidden from the solvent, and even after the protein was allowed to relax for 101 ns, the H0/SH3 complex was found to stay at the distal ends of the endophilin dimer, which is where the SAXS data suggest the SH3 domains are located (Fig. 1). Some asymmetry is seen between both monomers, but they both maintain the interaction between the H0 helix and the SH3 domain and keep the helix stabilized while in solution. The helix and SH3 domains on both chains form a hydrophobic interaction between the Phe10 residue on the helix and the Phe338 residue on the SH3 domain (Fig. 2), resulting in a pincer-like gripping of the helix Phe10 residue by the SH3 residues. In addition to the hydrophobic interactions, the helix and SH3 domain form salt bridges between oppositely charged residues on either domain, specifically between Lys7-Asp324 and Lys16-Glu304 (Fig. 3), that further stabilize the structure. Although Fig. 3 shows what appears to be an interaction corresponding to Lys16-Glu308, the distance between the two residues is too large for an actual electrostatic interaction to occur without being shielded by the solvent. Instead, what appears to be occurring is that a kink in the helix corresponding to a metastable folding state for the helix (see subsection titled “The SH3 domain stabilizes the H0 helix structure in solution” below) causes the distance between the residues to remain fixed due to the Lys16-Glu304 salt bridge.
Figure 1.
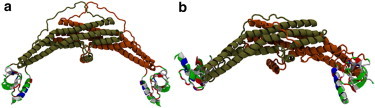
(a) A full endophilin protein was initially prepared by placing the N-terminal helix into the peptide-binding region of the SH3 domain with the hydrophobic residues of the helix pointed toward the groove to hide them from the solvent. (b) The protein was relaxed in solution for 101 ns and the H0/SH3 complex was found to be stable. The interaction between the helix and the SH3 domain was found to keep the SH3 domains at the distal end of the BAR domain and keep hydrophobic residues on both domains hidden from the solvent. The proteins are shown here in the New Cartoon representation. The BAR domain and linker are colored according to the protein chain, and the H0/SH3 complex is colored according to the residue type (hydrophobic residues are white, polar residues are green, acidic residues are red, and basic residues are blue).
Figure 2.
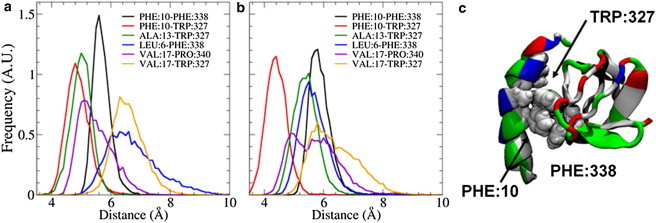
(a and b) The distributions of distances between hydrophobic residues on the helix and SH3 domain for the first (a) and second (b) endophilin monomer chains show that the strongest interactions exist between the Phe10 residue on the helix and residues Trp327 and Phe338 on the SH3 domain. (c) Inspection of the equilibrated H0/SH3 complex shows that the Trp327 and Phe338 residues form a pincer-like structure around the Phe10 residue, leading to a strong interaction. The H0/SH3 complex is shown using the New Cartoon and van der Waals representations and is colored according to residue type (hydrophobic residues are white, polar residues are green, acidic residues are red, and basic residues are blue).
Figure 3.
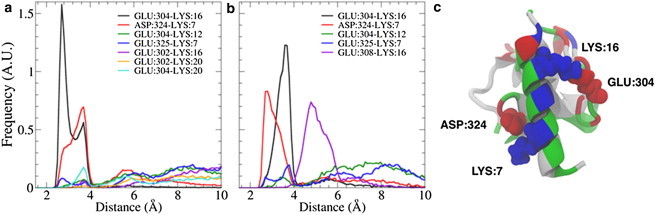
The distance distributions of the salt bridges that formed between the H0 helix and the SH3 domain for the first (a) and second (b) endophilin monomer chains show that the long-lived salt bridges are formed between the Lys7 and Asp324 residues and the Lys16 and Glu304 residues. (c) The equilibrated H0/SH3 structure shows that when the helix hides its hydrophobic residues from the solvent, the oppositely charged residues are aligned to form salt bridges that further stabilize the H0/SH3 complex. The H0/SH3 complex is shown using the New Cartoon and Van der Waals representations and is colored according to residue type (hydrophobic residues are white, polar residues are green, acidic residues are red, and basic residues are blue).
To further justify our initial choice of the H0/SH3 structure, we also simulated four configurations in which the H0 helix was rotated azimuthally around the vector of the helix to change the initial interactions between the two domains (Fig. S1 of the Supporting Material). These additional simulations (detailed in the Supporting Material) show that when the helix is rotated, the helix and SH3 domain become disordered; however, the original configuration maintains its structure over 200 ns of simulation (Fig. S2).
Umbrella-sampling simulations were used to calculate the binding free energy of the H0/SH3 complex in the full protein, and showed that most of the interaction strength in between the two domains is due to hydrophobic interactions. As the helix is moved farther away from the peptide-binding groove, the hydrophobic interactions are broken until only transient salt-bridge interactions are able to occur (Fig. 4), resulting in a binding energy of 13.0 ± 0.6 kcal/mol, a binding strength on the order of a few hydrogen bonds. The free-energy minimum was also found to correspond to the structure seen in monomer A, which further suggests that monomer B is in a metastable state that is similar to monomer A in function but different in energy. This is most likely because fully atomistic MD simulations are limited to shorter timescales and length scales than are seen in real biological systems; however, the binding free energy suggests that the H0/SH3 structure is still thermodynamically stable and worthy of further experimental investigation.
Figure 4.
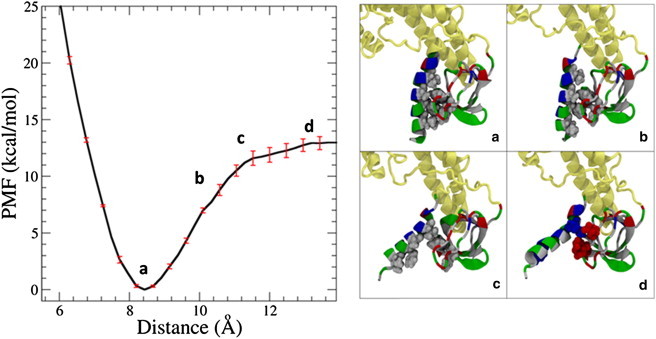
The binding energy of the H0/SH3 complex in the full endophilin protein is 13.0 ± 0.6 kcal/mol, on the order of several hydrogen bonds. (a) The stability of the complex is driven by the hiding of the hydrophobic residues on both domains from the solvent. (b) As the distance is increased between the helix and the SH3 domain, more hydrophobic residues are exposed to the solvent, and the structure becomes less energetically stable. (c) Eventually, the hydrophobic residues on both structures are fully exposed to the solvent, leading to an unfavorable structure. (d) A further increase in distance leads to some transient salt-bridge formation, until there is no interaction between either of the domains. The proteins are represented here using the New Cartoon and van der Waals representation. The BAR domain and linker are colored tan and the H0/SH3 complex is colored according to residue type (hydrophobic residues are white, polar residues are green, acidic residues are red, and basic residues are blue).
The SH3 domain stabilizes the H0 helix structure in solution
Because previous work showed that a single H0 helix is unstructured in solution (5,8), we used metadynamics simulations to explore the helix folding landscape. Our calculations show that the SH3 domain’s peptide-binding region is able to stabilize the H0 helical structure in solution. When the amphipathic helix is in solution alone, it becomes unstructured due to exposure of the hydrophobic residues to the solvent. The SH3 domain, however, allows the helix to hide its hydrophobic residues, namely Phe10, in the peptide-binding groove. The folding free-energy landscape calculated from metadynamics shows that a mostly helical H0 structure is energetically favorable when the H0/SH3 complex is in solution, although some of the residues are unstructured (Fig. 5). The barrier to (mostly) unfold the helix is 7.0 kcal/mol, which is greater than the thermal energy by an order of magnitude (kBT = 0.68 kcal/mol, where kB is the Boltzmann constant and T = 310 K). The convergence of the metadynamics simulations is discussed in the Supporting Material and illustrated in Fig. S3.
Figure 5.
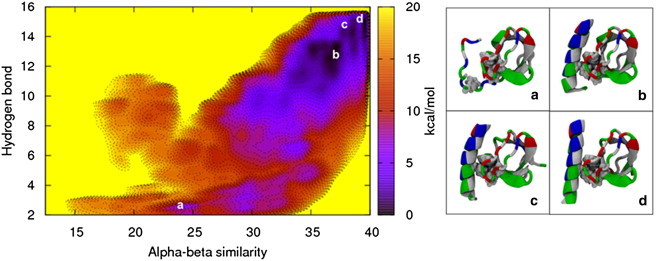
The folding free-energy landscape of the H0/SH3 complex in solution shows that the presence of the SH3 domain stabilizes the N-terminal helix. (a–d) The free-energy minimum (d), along with the other energetically stable folded states (b and c), keeps the Phe10 residue on the helix hidden from the solvent, whereas the unfolded state (a) exposes the residue to the solvent, resulting in a higher free-energy state. The H0/SH3 complex is shown using the New Cartoon and Van der Waals representations and is colored according to residue type (hydrophobic residues are white, polar residues are green, acidic residues are red, and basic residues are blue).
Electrostatic potential of the H0/SH3 complex
An electrostatic analysis of the equilibrated endophilin protein shows a sharp separation between the positively and negatively charged electrostatic potentials on the H0/SH3 complex. The volume slice of the electrostatic potential through the H0/SH3 complex shows that the negative electrostatic potential is concentrated on the SH3 domain and the positive electrostatic potential is concentrated on the exposed region of the N-terminal helix, with a region of neutral electrostatic potential between the two domains (Fig. 6). This is further illustrated by the surface representations of the full endophilin electrostatic potential (Fig. 7). The positive electrostatic potential is concentrated on the exposed regions of the amphipathic helices and the negative electrostatic potential resides in the interior of the protein. The electrostatic surface suggests that when the protein is in contact with the membrane surface, which will have a negative electrostatic potential, the H0/SH3 complex will be oriented such that the helix will be pointed toward the membrane and the SH3 domain will be oriented away from the membrane.
Figure 6.
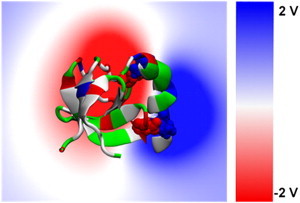
A volume slice of the electrostatic potential through the H0/SH3 complex shows the separation in electrostatic potential between the helix and the SH3 domain. The negative electrostatic potential resides solely on the SH3 domain, whereas the positive potential is on the helix. Separating both is a region of neutral electrostatic potential. The H0/SH3 complex is shown using the New Cartoon and van der Waals representations and is colored according to residue type (hydrophobic residues are white, polar residues are green, acidic residues are red, and basic residues are blue).
Figure 7.
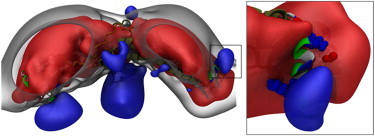
Electrostatic potential surfaces of the full endophilin protein. The red surface is the electrostatic potential at −2 V, the clear surface is the electrostatic potential at 0 V, and the blue surface is the electrostatic potential at 2 V. The positive electrostatic potential is concentrated on the amphipathic helices of the protein and is separated from the rest of the protein by the neutral potential surface. The separation of electrostatic potential between the SH3 domain and the N-terminal helix suggests that when the protein is on the membrane surface, the H0/SH3 complex will be oriented such that the helix will be pointed toward the membrane and the SH3 domain will be repulsed away from the bilayer surface. The protein is shown using the New Cartoon and van der Waals representations. The BAR domain and linker are colored according to the protein chain, and the H0/SH3 complex is colored according to residue type (hydrophobic residues are white, polar residues are green, acidic residues are red, and basic residues are blue).
Conclusions
The simulations presented here predict that at least one role of the SH3 domain is to regulate endophilin function in solution by binding with the N-terminal helix on the N-BAR domain. Our computational studies suggest that the stability of the H0/SH3 complex and the folded character of the helix in the peptide-binding groove are governed by hydrophobic interactions between both domains, hiding the hydrophobic residues from the solvent while allowing the charged residues on the helix and the SH3 domain to form salt bridges. The electrostatic potential resulting from the H0/SH3 complex shows that the positive electrostatic potential is concentrated on the outer helix while the negative potential is concentrated on the SH3 domain. This separation of the positive and negative electrostatic potential suggests that when the full protein is on or near the membrane surface, the SH3 domain will point away from the membrane and the outer helix will point toward the membrane.
The importance of the SH3 domain has been unclear, and different studies have yielded contradicting conclusions regarding its necessity for the function of endophilin in CME (4,12). According to the model we have presented here, the SH3 domain can autoregulate endophilin protein function when the protein is in solution through the formation of an interdomain complex between the N-terminal helix and the SH3 domain peptide-binding region. This complex ensures that the protein will protect the hydrophobic residues that are required for inducing curvature on the H0 helix and keep the SH3 domain from recruiting proteins while in solution. The new model also helps to explain how the SH3 domain is held in place at the distal ends of the N-BAR domain, as suggested by previous SAXS studies (13).
The electrostatic potential of the H0/SH3 complex also suggests that the mechanism for insertion of the N-terminal helix is changed by the interaction between the SH3 domain and the H0 helix. Because the helix is unstructured in solution, it is thought that the H0 helix inserts into the membrane by refolding into areas of the membrane where defects are found. Although this is the case for the N-BAR domain, the stabilization of the helix and the change in electrostatic potential caused by the formation of the H0/SH3 complex would allow the folded helix to instead orient itself toward the membrane surface and possibly insert without having to first adopt an unfolded state. Additionally, because of the electrostatic potential separation between the H0 helix and the SH3 domain, insertion of the H0 helix would most likely favor membranes with a higher concentration of negatively charged lipids. The autoinhibited form of the full endophilin protein would allow the protein to screen for the correct combination of electrostatic surface potential and packing defects before insertion of the helix occurs.
Our simulations do not predict the explicit role of the membrane environment in disassembling the H0/SH3 complex; however, they do suggest a model for how the full protein interacts with the membrane. The binding energy of the complex is on par with a hydrogen bond when the protein is in solution, but it seems likely that when endophilin is on the membrane surface and the helix is oriented toward the bilayer, the unbinding free energy would be lowered to facilitate the insertion of the helix. The lowering of the unbinding energy would also likely be correlated with the concentration of negatively charged lipids, due to the electrostatic potential of the complexed protein. Additionally, membrane curvature, and specifically membrane defects, may play a role in aiding the insertion of the helix into the membrane (5,8). Future work to answer these questions will involve calculating the free energy required to move the H0 helix from the SH3 domain to the lipid bilayer for the full endophilin protein—clearly, a computationally demanding task. It will also be important to compare lipid bilayers with different levels of curvature to understand the role membrane defects play in the insertion of the N-terminal helix. Such future work, combined with the simulations we have already performed and new experimental work probing the interaction between the H0 and SH3 domains with methods such as EPR measurements of the H0 helicity, fluorescence resonance energy transfer studies of the H0/SH3 binding kinetics, and cross-linking studies investigating how biological function is affected, will help further validate or refine the proposed autoregulation model for endophilin in solution.
Acknowledgments
This work was funded by National Institutes of Health grants GM063796 (F.X.V. and G.A.V.) and GM094479 (V.M.U.), and Public Health Service grant DA24101 (V.M.U.). The research was supported by an allocation of advanced computing resources provided by the National Science Foundation. The calculations were performed on Ranger at the Texas Advanced Computing Center, Kraken at the National Institute for Computational Sciences, and with resources provided by the Research Computing Center, University of Chicago.
Supporting Material
References
- 1.Schmidt A., Wolde M., Söling H.D. Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature. 1999;401:133–141. doi: 10.1038/43613. [DOI] [PubMed] [Google Scholar]
- 2.Farsad K., Ringstad N., De Camilli P. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J. Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMahon H.T., Gallop J.L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 4.Bai J., Hu Z., Kaplan J.M. Endophilin functions as a membrane-bending molecule and is delivered to endocytic zones by exocytosis. Cell. 2010;143:430–441. doi: 10.1016/j.cell.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatia V.K., Madsen K.L., Stamou D. Amphipathic motifs in BAR domains are essential for membrane curvature sensing. EMBO J. 2009;28:3303–3314. doi: 10.1038/emboj.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallop J.L., Jao C.C., McMahon H.T. Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 2006;25:2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masuda M., Takeda S., Mochizuki N. Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J. 2006;25:2889–2897. doi: 10.1038/sj.emboj.7601176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui H., Lyman E., Voth G.A. Mechanism of membrane curvature sensing by amphipathic helix containing proteins. Biophys. J. 2011;100:1271–1279. doi: 10.1016/j.bpj.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mim C., Cui H., Unger V.M. Structural basis of membrane bending by the N-BAR protein endophilin. Cell. 2012;149:137–145. doi: 10.1016/j.cell.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trempe J.-F., Chen C.X.-Q., Fon E.A. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol. Cell. 2009;36:1034–1047. doi: 10.1016/j.molcel.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 11.Ringstad N., Gad H., De Camilli P. Endophilin/SH3p4 is required for the transition from early to late stages in clathrin-mediated synaptic vesicle endocytosis. Neuron. 1999;24:143–154. doi: 10.1016/s0896-6273(00)80828-4. [DOI] [PubMed] [Google Scholar]
- 12.Milosevic I., Giovedi S., De Camilli P. Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron. 2011;72:587–601. doi: 10.1016/j.neuron.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q., Kaan H.Y.K., Hooda R.N., Goh S.L., Sondermann H. Structure and plasticity of endophilin and sorting nexin 9. Structure. 2008;16:1574–1587. doi: 10.1016/j.str.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 14.Rao Y., Ma Q., Haucke V. Molecular basis for SH3 domain regulation of F-BAR-mediated membrane deformation. Proc. Natl. Acad. Sci. USA. 2010;107:8213–8218. doi: 10.1073/pnas.1003478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gortat A., San-Roman M.J., Schmidt A.A. Single point mutation in Bin/Amphiphysin/Rvs (BAR) sequence of endophilin impairs dimerization, membrane shaping, and Src homology 3 domain-mediated partnership. J. Biol. Chem. 2012;287:4232–4247. doi: 10.1074/jbc.M111.325837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayer B.J. SH3 domains: complexity in moderation. J. Cell Sci. 2001;114:1253–1263. doi: 10.1242/jcs.114.7.1253. [DOI] [PubMed] [Google Scholar]
- 17.Barducci A., Bussi G., Parrinello M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008;100:020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 18.Laio A., Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raiteri P., Laio A., Parrinello M. Efficient reconstruction of complex free energy landscapes by multiple walkers metadynamics. J. Phys. Chem. B. 2006;110:3533–3539. doi: 10.1021/jp054359r. [DOI] [PubMed] [Google Scholar]
- 20.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 21.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 22.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reference deleted in proof.
- 24.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 25.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 26.Grest G.S., Kremer K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A. 1986;33:3628–3631. doi: 10.1103/physreva.33.3628. [DOI] [PubMed] [Google Scholar]
- 27.Feller S.E., Zhang Y., Brooks B.R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 28.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 29.Grossfield, A. WHAM: the weighted histogram analysis method, version 2.0.6, http://membrane.urmc.rochester.edu/content/wham.
- 30.Bonomi M., Branduardi D., Parrinello M. PLUMED: a portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009;180:1961–1972. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.