

Abstract
Halorhodopsin from Natronomonas pharaonis (pHR), a retinylidene protein that functions as a light-driven chloride ion pump, is converted into a proton pump in the presence of azide ion. To clarify this conversion, we investigated light-induced structural changes in pHR using a C2 crystal that was prepared in the presence of Cl− and subsequently soaked in a solution containing azide ion. When the pHR-azide complex was illuminated at pH 9, a profound outward movement (∼4 Å) of the cytoplasmic half of helix F was observed in a subunit with the EF loop facing an open space. This movement created a long water channel between the retinal Schiff base and the cytoplasmic surface, along which a proton could be transported. Meanwhile, the middle moiety of helix C moved inward, leading to shrinkage of the primary anion-binding site (site I), and the azide molecule in site I was expelled out to the extracellular medium. The results suggest that the cytoplasmic half of helix F and the middle moiety of helix C act as different types of valves for active proton transport.
Introduction
Halorhodopsin (HR), a seven-transmembrane-helix retinylidene protein, utilizes light energy to transport chloride ions from the extracellular side to the cytoplasmic side (1–4). Currently, two homologs of HR, i.e., HR from Halobacterium salinarum (sHR) and HR from Natronomonas pharaonis (pHR), have been crystallized, and their crystal structures have been determined (5,6). Although the anion-binding site (site I) in the vicinity of the retinal Schiff base is conserved between sHR and pHR, their structures are different in many aspects. For example, sHR possesses a second anion-binding site between site I and the extracellular surface, whereas the second anion-binding site in pHR is located at a different position (7). sHR contains a fatty acid at a threonine near the Schiff base (5), but there is no fatty acid in the pHR structure (6,7). When the low sequence identity between sHR and pHR is taken into account, it may be more accurate to consider pHR and sHR as belonging to different subfamilies of HR. Indeed, there is a significant difference in the photochemical reaction between sHR and pHR (8,9). More curiously, their photoreactions are perturbed differently by azide (10–15): whereas pHR is converted into a proton pump that is driven by a single photon (12), the sHR-azide complex utilizes two photons to pump a proton from the extracellular to the cytoplasmic side (11).
The photoreaction cycle of the pHR-azide complex resembles the trans photocycle of bacteriorhodopsin (BR), a light-driven proton pump found in Halobacterium salinarum (16). This similarity is interesting because there is a significant difference in the distribution of ionizable residues between BR and pHR, i.e., Asp-85 and Asp-96 in BR are replaced by nonionizable residues in pHR. In the proton-pumping cycle of BR, Asp-85 receives a proton from the Schiff base, and subsequently Asp-96 mediates a proton transfer from the extracellular medium to the deprotonated Schiff base (17,18). To explain the proton-pumping activity of the pHR-azide complex, it has been postulated that an azide ion bound to site I has a role similar to that of Asp-85 in BR, and another azide molecule is inserted into the cytoplasmic half to shuttle a proton from the cytoplasmic medium to the Schiff base (13). It was previously observed that BR is converted into a light-driven chloride pump when Asp-85 is replaced with threonine (19). Based on this observation, Facciotti et al. (20) proposed the intriguing hypothesis that BR transports hydroxyl ions from the extracellular side to the cytoplasmic side. More recently, Kouyama and Murakami (21) proposed a revised version of this hypothesis: BR functions as a water/proton antiporter, and pHR functions as an HCl/proton antiporter. In this scenario, one would expect pHR and BR to share a common structural motif for regulation of intraprotein proton movements.
In this study, we investigated light-induced structural changes in the pHR-azide complex using a C2 crystal that was prepared in the presence of chloride ion and subsequently soaked in a solution containing azide ions. Although no reaction state was efficiently trapped at neutral pH, large light-induced structural changes were observed when a long-lived M state was generated at pH 9. Owing to different protein-protein contacts, the three subunits contained in the asymmetric unit underwent different light-induced structural changes. In a subunit with the EF loop facing an open space, a profound outward movement of the cytoplasmic half of helix F was induced, creating a long water channel between the retinal Schiff base and the extracellular surface. A similar structural change has been suggested to occur during the proton-pumping cycle of BR, although its detailed features remain to be determined (22–26). The results presented here provide insight into the ion-pumping mechanism of not only pHR but also BR and other rhodopsins.
Materials and Methods
Preparation and crystallization of pHR
Natronomonas pharaonis strain KM-1, which was generated by UV mutagenesis from the type strain DSM 2160T, was grown as previously described (27). The harvested cells were suspended in 3 M NaCl, and after two cycles of freeze-thaw, the cell membranes were washed three times with 100 mM NaCl and twice with distilled water. The crystallization of pHR was performed as previously described (6).
Measurement of absorption spectra and kinetics
We acquired transient transmission data from the pHR-azide complex using a computer-controlled experimental setup with a digital oscilloscope and a frequency-doubled Nd-YAG laser (28). We then analyzed the absorption kinetics measured at various wavelengths using the singular value decomposition method (29).
Data collection and scaling
For structural determination of the azide-bound form of pHR, a single crystal of pHR was picked and transferred into a postcrystallization solution containing 0.2–0.5 M NaN3, 3 M (NH4)2SO4, 0.1 M pH buffer (HEPES at pH 7 or glycine at pH 9), and 30% trehalose. After incubation for 20 min, the crystal was flash-cooled with liquid propane at its melting temperature in dim light. To trap a reaction state with a long lifetime at room temperature, we transiently warmed a frozen crystal of the pHR-azide complex to room temperature by blocking the flow of cooled nitrogen gas and then rapidly cooled it under illumination with orange light (>540 nm, ∼100 mW/cm2).
X-ray diffraction measurements were performed at SPring-8 beamlines BL26B2 and BL38B1, where a frozen crystal kept at 100 K was exposed to a monochromatic x-ray beam at wavelength of 1.0 Å with an x-ray flux rate of ∼2 × 1012 photons/mm2/s. Diffraction data were collected using a CCD detector, with an oscillation range of 1° and an x-ray flux of 2 × 1013 photons/mm2 per image. Indexing and integration of diffraction spots were carried out with Mosflm 6.1 (30). The data were scaled using SCALA in the CCP4 program suites (31).
Structural refinement
We built a model of the azide-bound purple form of pHR according to the molecular-replacement method using the previously reported model of pHR (PDB ID: 3A7K) as an initial search model. Structure refinement was done using CNS-1.2 (32) and XtalView-4.0 (33).
To build a model of the reaction state of the pHR-azide complex, we compared the diffraction data from the crystal that was frozen under illumination (Flight) with those from a crystal that was flash-cooled in dim light (Fdark). In the first round of refinement, the structure of the reaction state was modified on the basis of the |Flight| − |Fdark| difference map. We further refined the structure by comparing the observed amplitude |Flight| with a weighted average of the calculated amplitudes of the unphotolyzed state and a reaction state in each subunit. The occupancy of the reaction state in each subunit was adjusted independently, i.e., the observed amplitude |Flight| was compared with the following calculated amplitude: |Fcalc| = Σi {αi × |Fi_React| + (1 − αi) × |Fi_Ground|}, where αi (i = 1, 2, 3) is the occupancy of the reaction state in subunits A, B, and C, and Fi_React and Fi_Ground are the structure factors of the reaction state and the unphotolyzed state, respectively, in the i-th subunit. The structure of the reaction state in each subunit was refined by the simulated annealing method, and the structure of the other conformer was assumed to be identical to that of the unphotolyzed state in a crystal that was flash-cooled in dim light. In this refinement, we estimated the optimal values of αi by investigating the crystallographic R values evaluated at various αi values (Fig. S1 in the Supporting Material). The crystallographic R-value decreased to 0.209 when the optimal values of αi (0.52, 0.64, and 0.56 for subunits A, B, and C, respectively) were used (Table S1).
Results
Photochemical reactions of the pHR-azide complex
Spectroscopic titrations of pHR with various ions showed that the binding affinity of azide ion to pHR was 10 times lower than that observed for chloride ion (Fig. S2). Similar differences in the affinities were previously reported (13,34). Owing to the strong absorption band of the second chromophore bacterioruberin, the absorption band of retinal in the pHR-azide complex appeared as a shoulder in the long wavelength region. However, it is apparent that the absorption band of retinal in the pHR-azide complex is slightly blue-shifted as compared with that in the pHR-chloride complex. A similar spectroscopic property of retinal was previously observed for a solubilized sample of the pHR-azide complex (34).
Fig. 1 a shows the absorption changes observed when the pHR-azide complex in a membrane suspension at pH 6.0 and in the presence of 1 M sodium azide was excited with light pulses at 532 nm. In the investigated time region, the absorption kinetics data were fitted with four exponential components. In Fig. 1 b, the amplitudes of the individual components are plotted against the wavelength of the measuring light. The P1 component, which is characterized by a negative value at 410 nm, is attributable to the L-to-M transition. The P2 component, which has a negative value at 620 nm, is attributable to the rising phase of the O state with a red-shifted absorption spectrum. Because the P2 component has a twin peak at 410 nm and 500 nm, it is suggested that an equilibrium state between L and M is established at a much faster rate than the decay rate of M. Meanwhile, the difference spectrum associated with the P3 component has a positive peak at 640 nm and a negative peak at 550 nm. This component is attributable to the decay of the O intermediate into the initial state. Thus, it appears that the absorption kinetics at pH 6 are described by the previously reported reaction scheme: K → L → Mcis → O → pHR’ → pHR (13).
Figure 1.
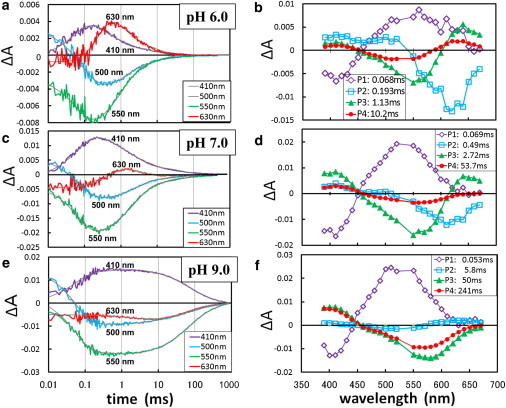
Light-induced absorption changes in the pHR-azide complex at various pH levels. (a, c, and e) An aqueous suspension of claret membrane in 1 M NaN3 at pH 6 (a), pH 7 (c), or 0.5 M NaN3 at pH 9. (e) was excited with light pulses at 532 nm and absorption changes were recorded at various wavelengths. (b, d, and f) The absorption kinetics in the time range of 0.01 ms to 1 s were fitted with four components, and the amplitude of each component was plotted against the wavelength of the measuring light.
To explain the absorption kinetics data collected at high pH (Fig. 1, c–e), however, we need to modify the above reaction scheme. At pH 7, a positive peak at 640 nm is still seen in the P3 component, but the corresponding peak is not seen in the P4 component with a longer time constant. The difference spectrum associated with the P4 component is characterized by a positive peak at 410 nm and a negative peak at 560 nm, suggesting that some fraction of M has a much longer lifetime than that of the O state. One possible explanation is that a long-lived M-like state (we refer to it as Malk) is generated by a branching reaction that occurs before the formation of the O state (possibly from Mcis; Fig. 2). Because the formation of O was not detected at pH 9, the branching reaction leading to Malk was suggested to become the major reaction pathway in the alkaline pH region. It should be mentioned that the decay kinetics of Malk at pH 9 is described by two time constants. Although the P3 and P4 components are characterized by a positive peak at 410 nm and a negative peak at ∼570 nm, the negative peak at ∼570 nm is more significant in the P3 component than in the P4 component. The profile of the P4 component can be explained by supposing that Malk is in dynamic equilibrium with an N state (Nalk), as was previously proposed to explain the photoreaction kinetics of the D96N mutant of BR (17).
Figure 2.
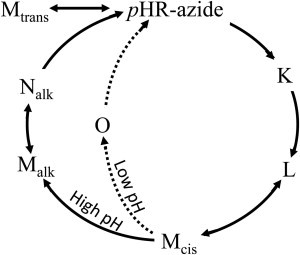
Photochemical reactions of the pHR-azide complex. A bypass reaction, indicated by the dashed lines, is observed at pH 6 in the presence of 1 M NaN3. At alkaline pH, a long-lived M-like state (Malk) is generated by a branching reaction from the M state (Mcis). In the absence of small anions and at high pH, a yellow form (Mtrans) containing all-trans retinal is generated in the dark.
Structure of the unphotolyzed state of the pHR-azide complex
Previous structural analyses of pHR (6,7) have shown that the primary binding site (site I) of chloride ion exists in the vicinity of the Schiff base (Fig. 3 d). In this study, a diffraction dataset at 1.9 Å resolution was collected from a C2 crystal that was soaked in a postcrystallization solution containing 0.2 M sodium azide at pH 7.0 and then flash-cooled in dim light. It was found that the chloride ion in site I and the nearby water molecule (Wat502) were replaced with an azide ion (Fig. 3 b). This replacement was not accompanied by any significant conformational changes in the surrounding residues. The crystal structure of the pHR-azide complex at pH 9 was nearly identical to that observed at neutral pH.
Figure 3.
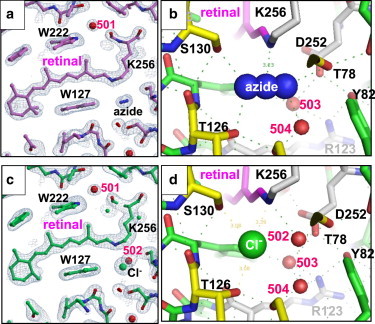
(a–d) The structure of the azide-bound purple form of pHR (a and b) is compared with that of the chloride-bound purple form (c and d). The 2Fo − Fc maps in panels a and c are contoured at 1.5 σ.
Light-induced structural changes in the pHR-azide complex
Because the lifetime of Malk increases significantly with increasing pH, one would expect a larger amount of Malk to accumulate when a C2 crystal that has been soaked at a higher pH is illuminated. Indeed, the magnitude of light-induced structural change increased significantly when the pH of the soaking solution was increased. (When a thin crystal soaked at pH 9 was exposed to orange light, a large fraction of the protein was converted into a reaction state(s) with λmax at 410 nm; Fig. S3). Fig. 4 b shows the light-induced structural change observed when the pHR-azide complex in the C2 crystal at pH 9 was exposed to orange light. It is clear that the three subunits in the asymmetric unit undergo different structural changes under illumination, namely, subunit B undergoes a very large structural change (e.g., a profound outward movement of the cytoplasmic half of helix F), whereas no significant movement of helix F is induced in the other subunits (Fig. S4).
Figure 4.
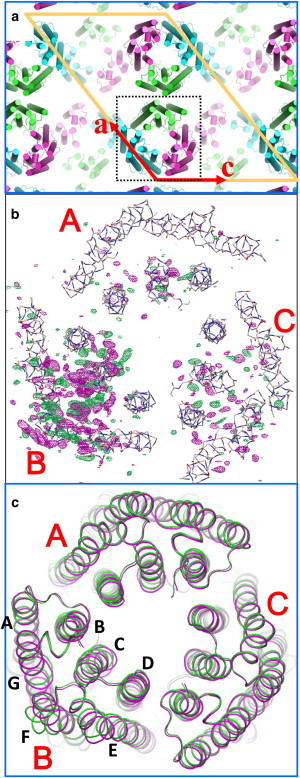
(a) Protein packing in the C2 crystal. Subunits A, B, and C are shown in green, blue, and magenta, respectively. (b) Light-induced structural change in the pHR-azide complex. The |Flight| − |Fdark| difference map (positive densities in magenta, negative in green) is contoured at 3.5 σ. (c) The structural model of the reaction state (green) is compared with that of the unphotolyzed state (magenta).
Fig. 5 a shows the |Flight|– |Fdark| difference map in the cytoplasmic side of subunit B. It shows that retinal’s isomerization into the 13-cis/15-anti configuration is accompanied by a large upward movement of the C13 methyl, pushing the indole ring of Trp-222 upward, which in turn causes large movements of most residues in the cytoplasmic half of helix F. Simultaneously, the side chain of Ile-134, which contacts the C13 methyl of retinal in the unphotolyzed state, rotates so as to enlarge a cavity in the cytoplasmic vicinity of the Schiff base (Fig. 6). Whereas the unphotolyzed state has one water molecule (Wat-501) in the cytoplasmic vicinity of the Schiff base, two additional water molecules (Wat-701 and Wat-702) are incorporated in this region. In addition to these water molecules, three water molecules (Wat-704 through Wat-706) are incorporated into a long cavity that is created by the outward movement of the cytoplasmic half of helix F.
Figure 5.
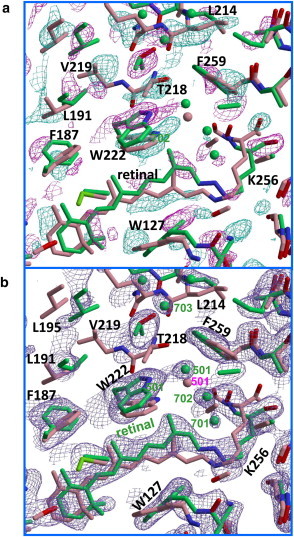
Light-induced structural change in subunit B. (a) |Flight| − |Fdark| difference map (positive densities in purple, negative in cyan), contoured at 3.0 σ and overlaid on the structural models of the reaction state (green) and the unphotolyzed state (pink). Green and pink spheres represent water molecules in the reaction state and the unphotolyzed state, respectively. (b) 2Fo – Fc map, contoured at 1.2 σ.
Figure 6.
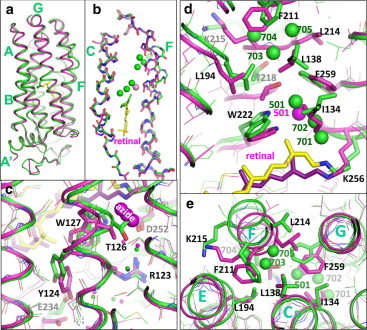
The structural model of the reaction state trapped in subunit B (green) is compared with the unphotolyzed state of the azide-bound purple form (magenta). (a) Side view of subunit B. (b) Deformation of helices C and F. (c) Light-induced structural change in the extracellular region. (d and e) Structural change in the cytoplasmic region. Carbon atoms and water molecules (spheres) in the reaction state and the unphotolyzed state are shown in green and magenta, respectively.
The largest movement (∼4 Å) in the main chain of helix F is observed at Lys-215 (Fig. 6 e). It is noteworthy that the outward movement of the cytoplasmic half of helix F is accompanied by a large swing of the benzene ring of F259 in helix G. This swing, together with a movement of F211 near the protein surface, contributes to the creation of a long cavity that extends from the Schiff base to the side chain of Lys-215. Meanwhile, the cytoplasmic half of helix E undergoes a noticeable inward movement, partly filling the open space between helices E and F.
A large light-induced structural change is also seen on the extracellular side of subunit B. First, the middle moiety of helix C deforms in such a manner that the side chain of Thr-126 moves toward Asp-252, leading to a remarkable shrinkage of site I (Fig. 6 c). As a result of this shrinkage, the azide ion in site I is squeezed out to the external medium. Second, the structure of the FG loop is largely distorted. This distortion is coupled with a flip-flop motion of the side chain of Glu234.
In subunit A, the light-induced conformational change is confined to the extracellular half of helix C (Fig. S4 a). As observed with subunit B, the middle moiety of helix C deforms so greatly that the azide ion is expelled from site I. However, retinal takes on an all-trans configuration and the cytoplasmic half of helix F barely deforms in the reaction state trapped in subunit A. In many aspects, its protein structure is similar to that of the anion-depleted blue or yellow form that is generated in the absence of small anions (7).
The light-induced structural change in subunit C is larger than that observed in subunit A (Fig. S4 c and Fig. S5). The structural analysis that was performed using the approximation that only one reaction state was trapped in subunit C suggested that retinal takes on the 13-cis/15-anti configuration in the major reaction state trapped in subunit C. In this reaction state, the middle moiety of helix C deforms greatly, just as observed in subunit B, but the main-chain structure of helix F is unaltered. The structural change in subunit C is characterized by a noticeable movement of helix G toward the extracellular side and large movements of several residues in the cytoplasmic half (Fig. S4). These movements create a large cavity between Val-74 and Phe-259, in which a water molecule (Wat-701) is hydrogen-bonded to the main chain of Thr-71 (Fig. 7 b and Fig. S5 c).
Figure 7.
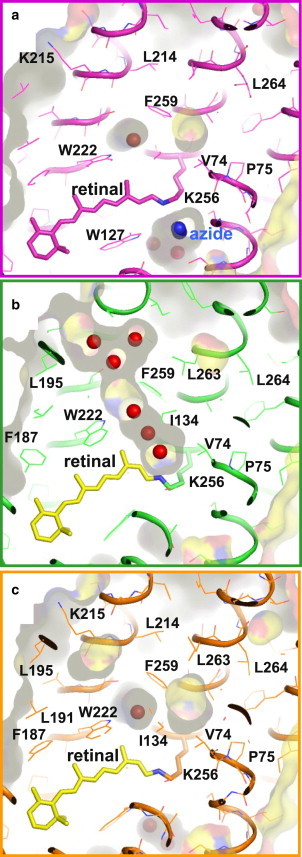
(a–c) Internal cavities in the unphotolyzed state (a) and the reaction state trapped in subunit B (b) and subunit C (c).
Discussion
Various crystallographic analyses of photoactive proteins have shown that the formation/decay kinetics of a reaction state are greatly affected by the crystal lattice force (36,37). In this study, we found that the three subunits in the asymmetric unit of the C2 crystal of the pHR-azide complex underwent different structural changes upon exposure to visible light (Fig. 4). This observation is understandable when the crystal lattice force is taken into account. With respect to subunit A, several residues in the cytoplasmic half of helix F contact residues in helix G of subunit A in a neighboring trimer related by a 2-fold axis. With respect to subunit C, residues in the EF loop interact with the BC loop of subunit C in a neighboring trimer related by a 2-fold screw axis. In these subunits, the motional freedom of helices F and/or G are reduced by the protein-protein contacts. In subunit B, on the other hand, the EF loop faces toward an open space in the unit cell and there is no major obstacle against the outward movement of helix F.
Our major concern is, which reaction state was trapped in subunit B? It should be noted that no large deformation of helix F was detected when the C2 crystal was exposed to strong visible light at neutral pH. Because the lifetime of Malk increases significantly with increasing pH, it is reasonable to consider that the outward movement of the cytoplasmic half of helix F in subunit B occurs when Malk is generated. This statement does not necessarily exclude the possibility that an equilibrium state between Malk and Nalk was trapped in subunit B. Meanwhile, no deformation of the cytoplasmic half of helix F was observed in another reaction state with 13-cis retinal that was generated in subunit C. This reaction state is characterized by a sliding movement of helix G toward the extracellular side and a large deformation of the middle moiety of helix C. It seems likely that the branching reaction leading to Malk was inhibited in subunit C so that another reaction state (most likely Mcis) accumulated under illumination at room temperature. In subunit A, on the other hand, a reaction state with all-trans retinal was trapped. Because its protein structure is similar to that of the anion-depleted yellow form generated in the absence of small anions, it seems possible that the protein conformation of the unphotolyzed state in subunit A is altered in a passive way when subunit B undergoes a large structural change. (Note that when a thin crystal was exposed to orange light, a large fraction of the protein was converted into reactions states with λmax at 410 nm; Fig. S3). An alternative interpretation is that a side reaction leading to the yellow form with all-trans retinal becomes significant in subunit A, where the motional freedom of the transmembrane helices is very restricted (Fig. S6).
A peculiar structural feature of the reaction state trapped in subunit B is the formation of a long cavity in the interhelical space between the retinal Schiff base and the cytoplasmic surface (Fig. 7). This cavity is filled with a linear cluster of water molecules, which would allow a rapid proton exchange between the Schiff base and the cytoplasmic medium. This structural feature is not shared by the unphotolyzed state of the pHR-azide complex, in which the interhelical space in the cytoplasmic half is completely closed. In contrast to the opening of the interhelical space in the cytoplasmic half, the cavity in the extracellular vicinity of the Schiff base is shrunk in Malk. This shrinkage is caused by a large deformation of the middle moiety of helix C. The structure of the reaction state trapped in subunit C suggests that the shrinkage of site I occurs before the formation of Malk. Thus, one can postulate that the middle moiety of helix C functions as another type of valve by which proton transport between the Schiff base and the extracellular medium is regulated.
Absorption kinetics data of the pHR-azide complex at pH 6 showed that in the presence of 1 M sodium azide, the M state decayed very rapidly into the O state (with a time constant of 0.2 ms at 24°C). At a lower concentration of sodium azide, however, a nonnegligible fraction of M was found to have a much longer lifetime (>20 ms at 24°C) even at pH 6. This observation is in line with a previous report that the decay of the M state of the pHR-azide complex was accelerated by an increasing concentration of sodium azide (13). A possible explanation for this acceleration is that an azide molecule is inserted into the interhelical space of the cytoplasmic half, and it shuttles a proton between the Schiff base and the cytoplasmic surface. When the pKa value (∼4.8) of azide is taken into account, it seems possible that this insertion often occurs at neutral or acidic pH, whereas it is rare at high pH. Thus, one can explain the absorption kinetics data observed under various solvent conditions by supposing that the pHR-azide complex undergoes a bypass reaction when the azide molecule (in the protonated form) is inserted into the cytoplasmic region before or during the M state.
It is noteworthy that the reaction state trapped in subunit C has a large cavity in the vicinity of the main chain of Lys-256 (Fig. 7 c). This cavity, which is surrounded by the side chains of Thr-71, Val-74, Ile-134, L138, and F259, is large enough to contain at least one water molecule. If this same cavity is created upon formation of Mcis at acidic pH, an azide molecule in its neutralized form could be inserted into this hydrophobic cavity. In this case, one would expect the inserted azide molecule to function as a proton donor to the deprotonated Schiff base, and, as a consequence, reprotonation of the Schiff base (i.e., formation of the O state) to occur so rapidly that the branching reaction into Malk would be avoided. Owing to the low pKa-value of azide, this azide-mediated proton transfer cannot occur at pH 9, where reprotonation of the Schiff base is suggested to occur after a linear cluster of water is formed between the Schiff base and the cytoplasmic medium.
It has been reported that the outward movement of the cytoplasmic half of helix F occurs when the N state of BR is generated in an alkaline suspension of purple membrane (22,24). A similar deformation of helix F was also suggested to occur upon formation of the MN state of the D96N mutant of BR (23,25,26), whose protein structure is reported to be similar to the N state of wild-type BR. Interestingly, the light-induced structural change observed in subunit B is not much different from what was previously observed upon formation of the N state of BR (24). However, no large deformation of helix F was detected when the M state of wild-type BR was generated in 3D crystals (37–39). It appears that in wild-type BR, the deformation of helix F, which is driven by a repellent force between the C13 methyl of retinal and Trp-182, is accompanied by proton transfer from Asp-96 to the deprotonated Schiff base. It is noteworthy that all of the residues (L138, L194, F211, L214, and F259) that contact the inserted water molecules (Wat-704 through Wat-706) in Malk are highly conserved among archaeal rhodopsins (L97, L152, F172, L175, and F219 in BR) (40). In the unphotolyzed state of any archaeal rhodopsin with known structure, these residues come together to close the interhelical space in the cytoplasmic half (41–43). Thus, our proposal that the cytoplasmic half of helix F acts as a sort of valve against intraprotein proton transport seems to be applicable to BR and other archaeal retinylidene proteins with proton-pumping activity. A similar idea was recently proposed by another group (44). Our recent crystallographic study of the P63 crystal of archaerhodopsin-2, which contains four subunits per asymmetric unit, showed that a large light-induced deformation of helix F (due to formation of the N state) is induced in a subunit that has the EF loop facing an open space (R. Fujii and T. Kouyama, unpublished data).
Previous crystallographic analyses of photoreaction intermediates of BR showed that the extracellular half of helix C is largely distorted in the M and O states generated in the P622 crystal (37,45). (This distortion is less significant in the P63 crystal, in which the three glycolipids in the central region of the trimer are replaced with other lipid components (38).) A similar deformation of helix C is observed in the proton-pumping cycle of the pHR-azide complex. Thus, it is possible that in many archaeal rhodopsins, the extracellular half of helix C functions as another type of valve by which backward movement of proton is prevented. (This statement does not necessarily exclude the possibility that BR possesses an additional valve, as proposed by Wolf et al. (46).)
Recent structural analyses of sensory rhodopsin-2 from N. pharaonis (NpSR2) showed that the EF loop undergoes a large movement upon formation of the M state (47,48). Although a significant deformation of helix F was reported to occur in the M state of NpSR2 (47), the magnitude of its deformation is much smaller than that observed in Malk of the pHR-azide complex. Before discussing whether there is an essential difference in intrinsic properties between NpSR2 and pHR, it is important to investigate light-induced structural changes in NpSR2 using a crystal that ensures that there is no obstacle against the outward movement of helix F.
Our recent crystallographic analyses of the pHR-bromide complex suggested that the following conformational changes are induced under illumination: 1), the bromide ion in site I is translocated across the Schiff base upon formation of the L state, in which the bromide ion interacts with the Schiff base with the NH bond directing to the cytoplasmic side; and 2), in a reaction state accumulating under illumination at 240 K, helix F in subunit B is largely distorted, as observed in the pHR-azide complex under illumination at pH 9 (unpublished data). Together with these results, our observation of a large deformation of helix F in Malk provides a clue for understanding the anion pumping mechanism of pHR. It is noteworthy that the deformation of helix F is accompanied by the formation of a small hole between helices E and F (Fig. 7 a). This hole is exposed to the cytoplasmic medium and is also connected to the long cavity created in the interhelical region of the cytoplasmic half. Interestingly, the ε amino group of Lys-215 comes close to this hole. It is also interesting to note that Thr-218 OH participates in a long hydrogen-bond network formed between the Schiff base and Lys-215 in Malk (Fig. 6 d). An intriguing possibility is that Lys-215 and Thr-218 mediate a proton transfer from the cytoplasmic medium to the halide ion that has been translocated across the Schiff base, and the resultant HCl escapes from the protonated Schiff base and diffuses toward the cytoplasmic side along the long cavity created in the interhelical region of the cytoplasmic half. This scenario is compatible with a previous mutagenesis study of HR that showed that the reaction kinetics in the late half of the anion-pumping cycle is perturbed by replacements of Lys-215 and Thr-218 (49). Furthermore, Váró et al. (50) reported that the N-to-O reaction is strongly electrogenic, and the corresponding reaction is only chloride dependent. A more ingenious experimental setup is necessary to clarify the details of the chloride release process.
Acknowledgments
We thank Drs. N. Shimidzu and K. Baba for helping with data collection at the SPring-8 beamlines BL26B2 and BL38B1.
This work was supported by a grant-in-aid from the Ministry of Education, Science, and Culture of Japan.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Accession Numbers
The crystallographic coordinates of the azide-bound purple form of pHR at pH 7 and the reaction states (together with the unphotolyzed state) of the pHR-azide complex at pH 9 have been deposited in the Protein Data Bank with accession codes 3ABW and 3VVK.
Supporting Material
References
- 1.Mukohata Y., Kaji Y. Light-induced ATP synthesis dependent on halorhodopsin-pH regulation. Arch. Biochem. Biophys. 1981;208:615–617. doi: 10.1016/0003-9861(81)90551-8. [DOI] [PubMed] [Google Scholar]
- 2.Bivin D.B., Stoeckenius W. Photoactive retinal pigments in haloalkaliphilic bacteria. J. Gen. Microbiol. 1986;132:2167–2177. doi: 10.1099/00221287-132-8-2167. [DOI] [PubMed] [Google Scholar]
- 3.Schobert B., Lanyi J.K. Halorhodopsin is a light-driven chloride pump. J. Biol. Chem. 1982;257:10306–10313. [PubMed] [Google Scholar]
- 4.Essen L.O. Halorhodopsin: light-driven ion pumping made simple? Curr. Opin. Struct. Biol. 2002;12:516–522. doi: 10.1016/s0959-440x(02)00356-1. [DOI] [PubMed] [Google Scholar]
- 5.Kolbe M., Besir H., Oesterhelt D. Structure of the light-driven chloride pump halorhodopsin at 1.8 A resolution. Science. 2000;288:1390–1396. doi: 10.1126/science.288.5470.1390. [DOI] [PubMed] [Google Scholar]
- 6.Kouyama T., Kanada S., Ihara K. Crystal structure of the light-driven chloride pump halorhodopsin from Natronomonas pharaonis. J. Mol. Biol. 2010;396:564–579. doi: 10.1016/j.jmb.2009.11.061. [DOI] [PubMed] [Google Scholar]
- 7.Kanada S., Takeguchi Y., Kouyama T. Crystal structures of an O-like blue form and an anion-free yellow form of pharaonis halorhodopsin. J. Mol. Biol. 2011;413:162–176. doi: 10.1016/j.jmb.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Váró G., Zimányi L., Lanyi J.K. Photocycle of halorhodopsin from Halobacterium salinarium. Biophys. J. 1995;68:2062–2072. doi: 10.1016/S0006-3495(95)80385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guijarro J., Engelhard M., Siebert F. Anion uptake in halorhodopsin from Natromonas pharaonis studied by FTIR spectroscopy: consequences for the anion transport mechanism. Biochemistry. 2006;45:11578–11588. doi: 10.1021/bi060753j. [DOI] [PubMed] [Google Scholar]
- 10.Hegemann P., Oesterbelt D., Steiner M. The photocycle of the chloride pump halorhodopsin. I: Azide-catalyzed deprotonation of the chromophore is a side reaction of photocycle intermediates inactivating the pump. EMBO J. 1985;4:2347–2350. doi: 10.1002/j.1460-2075.1985.tb03937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bamberg E., Tittor J., Oesterhelt D. Light-driven proton or chloride pumping by halorhodopsin. Proc. Natl. Acad. Sci. USA. 1993;90:639–643. doi: 10.1073/pnas.90.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Váró G., Brown L.S., Lanyi J.K. Proton transport by halorhodopsin. Biochemistry. 1996;35:6604–6611. doi: 10.1021/bi9601159. [DOI] [PubMed] [Google Scholar]
- 13.Kulcsár A., Groma G.I., Váró G. Characterization of the proton-transporting photocycle of pharaonis halorhodopsin. Biophys. J. 2000;79:2705–2713. doi: 10.1016/S0006-3495(00)76508-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lakatos M., Groma G.I., Váró G. Characterization of the azide-dependent bacteriorhodopsin-like photocycle of salinarum halorhodopsin. Biophys. J. 2002;82:1687–1695. doi: 10.1016/S0006-3495(02)75521-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mevorat-Kaplan K., Brumfeld V., Sheves M. Effect of anions on the photocycle of halorhodopsin. Substitution of chloride with formate anion. Biochemistry. 2005;44:14231–14237. doi: 10.1021/bi051323g. [DOI] [PubMed] [Google Scholar]
- 16.Váró G. Analogies between halorhodopsin and bacteriorhodopsin. Biochim. Biophys. Acta. 2000;1460:220–229. doi: 10.1016/s0005-2728(00)00141-9. [DOI] [PubMed] [Google Scholar]
- 17.Brown L.S., Lanyi J.K. Determination of the transiently lowered pKa of the retinal Schiff base during the photocycle of bacteriorhodopsin. Proc. Natl. Acad. Sci. USA. 1996;93:1731–1734. doi: 10.1073/pnas.93.4.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metz G., Siebert F., Engelhard M. Asp85 is the only internal aspartic acid that gets protonated in the M intermediate and the purple-to-blue transition of bacteriorhodopsin. A solid-state 13C CP-MAS NMR investigation. FEBS Lett. 1992;303:237–241. doi: 10.1016/0014-5793(92)80528-o. [DOI] [PubMed] [Google Scholar]
- 19.Sasaki J., Brown L.S., Lanyi J.K. Conversion of bacteriorhodopsin into a chloride ion pump. Science. 1995;269:73–75. doi: 10.1126/science.7604281. [DOI] [PubMed] [Google Scholar]
- 20.Facciotti M.T., Rouhani S., Glaeser R.M. Crystal structures of bR(D85S) favor a model of bacteriorhodopsin as a hydroxyl-ion pump. FEBS Lett. 2004;564:301–306. doi: 10.1016/S0014-5793(04)00208-X. [DOI] [PubMed] [Google Scholar]
- 21.Kouyama T., Murakami M. Structural divergence and functional versatility of the rhodopsin superfamily. Photochem. Photobiol. Sci. 2010;9:1458–1465. doi: 10.1039/c0pp00236d. [DOI] [PubMed] [Google Scholar]
- 22.Kamikubo H., Kataoka M., Lanyi J.K. Structure of the N intermediate of bacteriorhodopsin revealed by x-ray diffraction. Proc. Natl. Acad. Sci. USA. 1996;93:1386–1390. doi: 10.1073/pnas.93.4.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramaniam S., Lindahl M., Henderson R. Protein conformational changes in the bacteriorhodopsin photocycle. J. Mol. Biol. 1999;287:145–161. doi: 10.1006/jmbi.1999.2589. [DOI] [PubMed] [Google Scholar]
- 24.Vonck J. Structure of the bacteriorhodopsin mutant F219L N intermediate revealed by electron crystallography. EMBO J. 2000;19:2152–2160. doi: 10.1093/emboj/19.10.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dencher N.A., Sass H.J., Büldt G. Water and bacteriorhodopsin: structure, dynamics, and function. Biochim. Biophys. Acta. 2000;1460:192–203. doi: 10.1016/s0005-2728(00)00139-0. [DOI] [PubMed] [Google Scholar]
- 26.Hirai T., Subramaniam S. Protein conformational changes in the bacteriorhodopsin photocycle: comparison of findings from electron and X-ray crystallographic analyses. PLoS ONE. 2009;4:e5769. doi: 10.1371/journal.pone.0005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ihara K., Narusawa A., Kouyama T. A halorhodopsin-overproducing mutant isolated from an extremely haloalkaliphilic archaeon Natronomonas pharaonis. FEBS Lett. 2008;582:2931–2936. doi: 10.1016/j.febslet.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 28.Hayakawa N., Kasahara T., Kouyama T. Effect of xenon binding to a hydrophobic cavity on the proton pumping cycle in bacteriorhodopsin. J. Mol. Biol. 2008;384:812–823. doi: 10.1016/j.jmb.2008.09.075. [DOI] [PubMed] [Google Scholar]
- 29.Chan T.F. An improved algorithm for computing the singular value decomposition. ACM Trans. Math. Softw. 1982;8:72–83. [Google Scholar]
- 30.Steller I., Bolotovsky B., Rossmann M.G. An algorithm for automatic indexing of oscillation images using Fourier analysis. J. Appl. Cryst. 1997;D30:1036–1040. [Google Scholar]
- 31.Dodson E.J., Winn M., Ralph A. Collaborative Computational Project, number 4: providing programs for protein crystallography. Methods Enzymol. 1997;277:620–633. doi: 10.1016/s0076-6879(97)77034-4. [DOI] [PubMed] [Google Scholar]
- 32.Brünger A.T., Adams P.D., Warren G.L. Crystallography & NMR system. CNS: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.McRee D.E. Academic Press; San Diego: 1993. Practical Protein Crystallography. [Google Scholar]
- 34.Scharf B., Engelhard M. Blue halorhodopsin from Natronobacterium pharaonis: wavelength regulation by anions. Biochemistry. 1994;33:6387–6393. doi: 10.1021/bi00187a002. [DOI] [PubMed] [Google Scholar]
- 35.Reference deleted in proof.
- 36.Moukhametzianov R., Klare J.P., Gordeliy V.I. Development of the signal in sensory rhodopsin and its transfer to the cognate transducer. Nature. 2006;440:115–119. doi: 10.1038/nature04520. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto M., Hayakawa N., Kouyama T. Crystal structures of different substrates of bacteriorhodopsin’s M intermediate at various pH levels. J. Mol. Biol. 2009;393:559–573. doi: 10.1016/j.jmb.2009.08.047. [DOI] [PubMed] [Google Scholar]
- 38.Luecke H., Schobert B., Lanyi J.K. Structural changes in bacteriorhodopsin during ion transport at 2 angstrom resolution. Science. 1999;286:255–261. doi: 10.1126/science.286.5438.255. [DOI] [PubMed] [Google Scholar]
- 39.Sass H.J., Büldt G., Ormos P. Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature. 2000;406:649–653. doi: 10.1038/35020607. [DOI] [PubMed] [Google Scholar]
- 40.Ihara K., Umemura T., Mukohata Y. Evolution of the archaeal rhodopsins: evolution rate changes by gene duplication and functional differentiation. J. Mol. Biol. 1999;285:163–174. doi: 10.1006/jmbi.1998.2286. [DOI] [PubMed] [Google Scholar]
- 41.Enami N., Yoshimura K., Kouyama T. Crystal structures of archaerhodopsin-1 and -2: Common structural motif in archaeal light-driven proton pumps. J. Mol. Biol. 2006;358:675–685. doi: 10.1016/j.jmb.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 42.Royant A., Nollert P., Navarro J. X-ray structure of sensory rhodopsin II at 2.1-A resolution. Proc. Natl. Acad. Sci. USA. 2001;98:10131–10136. doi: 10.1073/pnas.181203898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luecke H., Schobert B., Spudich J.L. Crystal structure of sensory rhodopsin II at 2.4 angstroms: insights into color tuning and transducer interaction. Science. 2001;293:1499–1503. doi: 10.1126/science.1062977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freier E., Wolf S., Gerwert K. Proton transfer via a transient linear water-molecule chain in a membrane protein. Proc. Natl. Acad. Sci. USA. 2011;108:11435–11439. doi: 10.1073/pnas.1104735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J., Yamazaki Y., Kouyama T. Crystal structure of the O intermediate of the Leu93→Ala mutant of bacteriorhodopsin. Proteins. 2012;80:2384–2396. doi: 10.1002/prot.24124. [DOI] [PubMed] [Google Scholar]
- 46.Wolf S., Freier E., Gerwert K. How does a membrane protein achieve a vectorial proton transfer via water molecules? ChemPhysChem. 2008;9:2772–2778. doi: 10.1002/cphc.200800703. [DOI] [PubMed] [Google Scholar]
- 47.Gushchin I., Reshetnyak A., Gordeliy V. Active state of sensory rhodopsin II: structural determinants for signal transfer and proton pumping. J. Mol. Biol. 2011;412:591–600. doi: 10.1016/j.jmb.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 48.Klare J.P., Bordignon E., Steinhoff H.J. Transmembrane signal transduction in archaeal phototaxis: the sensory rhodopsin II-transducer complex studied by electron paramagnetic resonance spectroscopy. Eur. J. Cell Biol. 2011;90:731–739. doi: 10.1016/j.ejcb.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 49.Sato M., Kubo M., Demura M. Role of putative anion-binding sites in cytoplasmic and extracellular channels of Natronomonas pharaonis halorhodopsin. Biochemistry. 2005;44:4775–4784. doi: 10.1021/bi047500f. [DOI] [PubMed] [Google Scholar]
- 50.Váró G., Needleman R., Lanyi J.K. Light-driven chloride ion transport by halorhodopsin from Natronobacterium pharaonis. 2. Chloride release and uptake, protein conformation change, and thermodynamics. Biochemistry. 1995;34:14500–14507. doi: 10.1021/bi00044a028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.