

In response to a stimulus that makes neurons fire repetitively, many neurons, such as CA1 and CA3 pyramidal cells, develop a slow, long-lasting hyperpolarization after the firing of a number of action potentials (Fig. 1 A) (1). This is called a slow-after-hyperpolarization, or sAHP. The sAHP was first described in neurons more than 30 years ago (2–4). The sAHP develops with a time course of hundreds of milliseconds and decays with a time course of seconds (Fig. 1 A). The slow AHP is called “slow” to distinguish it from the fast and medium AHPs that have faster time-courses than the sAHP (5–8).
Figure 1.
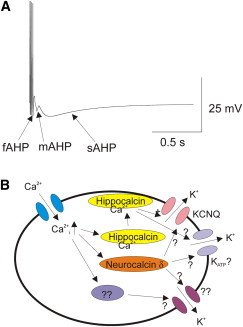
Time course and signaling cascade of an sAHP. (A) Simulation of a train of action potentials in a neuron with a fast AHP (fAHP), medium AHP (mAHP), and a slow AHP (sAHP). (B) Calcium influx through voltage-gated calcium channels (blue) in response to the train of action potentials raises the intracellular calcium. Calcium binds to intracellular calcium sensor proteins: for example, hippocalcin (yellow) and neurocalcin (orange). Upon calcium binding, hippocalcin exposes a myristoylated residue and translates to the plasma membrane, where it activates potassium channels underlying the sAHP. Both KCNQ (pink) and KATP (gray) channels have been implicated as the potassium channel underlying the sAHP. Other calcium sensor proteins (purple) and other potassium channels (magenta) might also contribute to the sAHP.
The negative membrane potential and increased membrane conductance during an sAHP prevents further action potentials from being generated as long as the sAHP lasts. This prevents overexcitation of the neuron in response to repetitive or long-lasting stimulus inputs (1). Indeed, malfunction of the sAHPs have been implicated in epilepsy (9). The sAHP endows the cell with an activity-dependent excitation that has been suggested to be important for synaptic plasticity and memory (10). The sAHP is also very strongly modulated by neurotransmitters, allowing neurotransmitters to alter the firing properties of the neuron (11–13). Here in a study in a recent issue of Biophysical Journal, Tzingounis and co-workers tackle the question: What determines the slow time course of the sAHP?
It was early on determined that calcium inflow during the train of action potentials was necessary for the development of the sAHP (2–4). It was proposed that it was the accumulation of intracellular calcium during the train of action potentials that cause the sAHP. It was also early on shown that the hyperpolarization of the sAHP is due to the opening of potassium channels (2). However, the identity of the potassium channels that open during the sAHP remained unknown for many years. The mechanism of how calcium opens these potassium channels was also a mystery. It was just recently that some of these questions started to be answered.
The initial idea of how a rise in intracellular calcium would activate the sAHP was that intracellular calcium directly binds to and activates calcium-activated potassium channels. The search for these calcium-activated channels has gone on for a long time. Many different potassium channels have been suggested as the ones underlying the sAHP. For example, potassium channels that directly are modulated by calcium, such as the BK (14) and SK channels (15), have been proposed as the elusive sAHP potassium channel. However, subsequent studies have shown that these channels do not underlie the sAHP, but that instead these channels contribute to the fast (6,16) and medium AHPs (15,17), respectively (which have a much faster time course than the sAHP (Fig. 1 A)).
It was not until 2007 that Tzingounis et al. (18) showed that a diffusible, cytosolic calcium-binding protein, hippocalcin, contributes to the sAHP in CA1 pyramidal neurons in the hippocampus. Hippocalcin is a member of the visinin-like neuronal calcium-sensor protein family. Tzingounis et al. (18) showed that a knockout of hippocalcin drastically reduced the sAHP and that upregulation of hippocalcin increased the sAHP. They further show that, upon calcium binding, hippocalcin exposes a myristoylated group and translates to the plasma membrane, presumably to activate the sAHP potassium channels in the plasma membrane (Fig. 1 B) (18). Even if the knockout of hippocalcin drastically reduced the sAHP, it did not completely remove the sAHP (18). This suggests that there are several calcium sensors contributing to the sAHP in these cells.
Interestingly, hippocalcin is highly expressed in hippocampal neurons, but it is not highly expressed in all parts of the brain that exhibit an sAHP (19). More recently, another member of the visinin-like neuronal calcium sensor proteins, neurocalcin δ, has been shown to contribute to the sAHP in cortical neurons (19)—cells which do not express hippocalcin at high levels, but still have a robust sAHP. That the sAHP is activated by cytosolic calcium sensors is consistent with earlier findings that the sAHP seems to be sensing the bulk calcium in the cytosol and not the calcium inflow in micro domains around the plasma membrane, as is the case for the fAHP and the mAHP.
So which channels are involved in the sAHP?
Tzingounis et al. (18) showed that potassium channels in the KCNQ family contribute to the potassium currents during an sAHP in CA3 pyramidal cells (Fig. 1 B) (20,21). Knockouts of KCNQ channels, or expression of dominant negative KCNQ subunits, decreased the amplitude of the sAHP (20,21). Later on, Soh and Tzingounis (22) showed that UCL2077, a blocker of the sAHP, blocks KCNQ channels—further strengthening the idea that KCNQ channels underlie the sAHP. However, knockout of specific members of the KCNQ channel family did not completely abolish the sAHP, just reduce the sAHP amplitude (20,21). This suggests that there are several channels contributing to the sAHP, maybe even different channels in different cell types. For example, KATP channels have been suggested to contribute to the sAHP in some cell types, because glibenclamide, a specific KATP blocker, reduces the amplitude of the sAHP in these cells (23).
So what determines the slow time course of the sAHP?
The time course of the sAHP has been shown to poorly track the time course of the intracellular calcium concentration (24). Tzingounis and co-workers here study the effects of modulating the kinetics of KCNQ channels on the kinetics of the sAHP in both wild-type mice and hippocalcin knockout mice.
First, Tzingounis and co-workers show that hippocalcin knockout reduces the sAHP in CA3 pyramidal cells in the hippocampus, as previously shown for CA1 and L5 pyramidal cells (18,19). The sAHP is reduced by >50% in the hippocalcin knockout animals, suggesting that hippocalcin is the major calcium sensor for the sAHP in CA3 cells. However, there is still an sAHP in the knockout animals, showing that there are most likely several calcium sensors, possibly Neurocalcin δ, that contribute to the sAHP in CA3 cells.
Second, using Retigabine, a compound that alters the kinetics of the KCNQ currents (25,26), the authors further show that the rise phase of the sAHP is determined by the kinetics of KCNQ channel opening in both wild-type and hippocalcin knockout animals. By clever use of different calcium buffers, such as BAPTA, to alter the calcium handling in the cells, the authors could show that Retigabine is not slowing the rise phase of the sAHP by acting on the calcium sensor itself. In the presence of intracellular BAPTA, the calcium binding to the calcium sensor is slowed down, as seen by a drastically slowed rise phase of the sAHP. This suggests that, in the presence of BAPTA, calcium-binding to the calcium sensor is now the rate-limiting step during the rise phase of the sAHP. In the presence of BAPTA, Ritigabine has no effect on the rise phase of the sAHP, suggesting that Ritigabine is not acting on the calcium sensor. Instead, this strongly suggests that Ritigabine is acting on the KCNQ channels to slow the rise phase of the sAHP.
In wild-type mice, Ritigabine does not affect the decay phase of the sAHP, suggesting that the decay is not dependent on the KCNQ channel kinetics. Instead, this result suggests that the decay is dependent on the slow decay kinetics of another process in the sAHP cascade. Surprisingly, in hippocalcin knockout animals, the decay of the sAHP is accelerated by Ritigabine, presumably by accelerating the KCNQ channel kinetics. The mechanism for this acceleration of the sAHP decay by Retigabine in the knockout animals is not obvious, because Ritigabine accelerates the opening, and not the closing, of KCNQ channels. One would expect that the closing kinetics, and not the opening kinetics, to be the most important for determining the decay kinetics of the sAHP.
These data further strengthen the proposed role of hippocalcin and KCNQ channels in the generation of the sAHP. However, more players are probably involved, especially more types of calcium sensors, because an sAHP, albeit of a smaller amplitude, is still present in hippocalcin knockout animals. There is always the possibility that the sAHP calcium sensors, and sAHP potassium channels that generate the sAHP, are different in different cell types. It is also possible that the different calcium sensors activate different types of potassium channels (Fig. 1 B). In addition, the molecular mechanism of how the different calcium sensors, such as hippocalcin, activate the sAHP channels is not completely understood.
Recently, it has been proposed that PIP2 production is down-stream of calcium, because overexpressing the enzyme PIP5K that produces PIP2 greatly sensitizes the sAHP to calcium inflow (27). PIP2 has earlier been shown to increase the current in many channels, including KCNQ (28–30) and KATP (31–33) channels, so it is easy to see that PIP2 could be a part of the sAHP cascade. Clearly, more studies are necessary on both the molecular and cellular levels, to enable understanding of the mechanism underlying the sAHP.
Acknowledgments
The work in the author’s laboratory is funded by the National Institutes of Health’s Heart, Lung, and Blood Institute (grant No. R01-HL095920) and the James & Esther King Biomedical Research Program, Florida Department of Health (grant No. 3KB02).
References
- 1.Hille B. Sinauer Associates; Sunderland, MA: 2001. Ion Channels of Excitable Membranes. [Google Scholar]
- 2.Alger B.E., Nicoll R.A. Epileptiform burst afterhyperpolarization: calcium-dependent potassium potential in hippocampal CA1 pyramidal cells. Science. 1980;210:1122–1124. doi: 10.1126/science.7444438. [DOI] [PubMed] [Google Scholar]
- 3.Hotson J.R., Prince D.A. A calcium-activated hyperpolarization follows repetitive firing in hippocampal neurons. J. Neurophysiol. 1980;43:409–419. doi: 10.1152/jn.1980.43.2.409. [DOI] [PubMed] [Google Scholar]
- 4.Schwartzkroin P.A., Stafstrom C.E. Effects of EGTA on the calcium-activated afterhyperpolarization in hippocampal CA3 pyramidal cells. Science. 1980;210:1125–1126. doi: 10.1126/science.6777871. [DOI] [PubMed] [Google Scholar]
- 5.Meech R.W. Calcium-dependent potassium activation in nervous tissues. Annu. Rev. Biophys. Bioeng. 1978;7:1–18. doi: 10.1146/annurev.bb.07.060178.000245. [DOI] [PubMed] [Google Scholar]
- 6.Storm J.F. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J. Physiol. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Storm J.F. An after-hyperpolarization of medium duration in rat hippocampal pyramidal cells. J. Physiol. 1989;409:171–190. doi: 10.1113/jphysiol.1989.sp017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Storm J.F. Potassium currents in hippocampal pyramidal cells. Prog. Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- 9.McCormick D.A., Contreras D. On the cellular and network bases of epileptic seizures. Annu. Rev. Physiol. 2001;63:815–846. doi: 10.1146/annurev.physiol.63.1.815. [DOI] [PubMed] [Google Scholar]
- 10.Disterhoft J.F., Wu W.W., Ohno M. Biophysical alterations of hippocampal pyramidal neurons in learning, aging and Alzheimer’s disease. Aging Res. Rev. 2004;3:383–406. doi: 10.1016/j.arr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Madison D.V., Nicoll R.A. Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature. 1982;299:636–638. doi: 10.1038/299636a0. [DOI] [PubMed] [Google Scholar]
- 12.Andrade R., Nicoll R.A. Pharmacologically distinct actions of serotonin on single pyramidal neurones of the rat hippocampus recorded in vitro. J. Physiol. 1987;394:99–124. doi: 10.1113/jphysiol.1987.sp016862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haas H.L., Konnerth A. Histamine and noradrenaline decrease calcium-activated potassium conductance in hippocampal pyramidal cells. Nature. 1983;302:432–434. doi: 10.1038/302432a0. [DOI] [PubMed] [Google Scholar]
- 14.Barrett J.N., Magleby K.L., Pallotta B.S. Properties of single calcium-activated potassium channels in cultured rat muscle. J. Physiol. 1982;331:211–230. doi: 10.1113/jphysiol.1982.sp014370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bond C.T., Herson P.S., Adelman J.P. Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J. Neurosci. 2004;24:5301–5306. doi: 10.1523/JNEUROSCI.0182-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lancaster B., Nicoll R.A. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J. Physiol. 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villalobos C., Shakkottai V.G., Andrade R. SKCa channels mediate the medium but not the slow calcium-activated afterhyperpolarization in cortical neurons. J. Neurosci. 2004;24:3537–3542. doi: 10.1523/JNEUROSCI.0380-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzingounis A.V., Kobayashi M., Nicoll R.A. Hippocalcin gates the calcium activation of the slow afterhyperpolarization in hippocampal pyramidal cells. Neuron. 2007;53:487–493. doi: 10.1016/j.neuron.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villalobos C., Andrade R. Visinin-like neuronal calcium sensor proteins regulate the slow calcium-activated afterhyperpolarizing current in the rat cerebral cortex. J. Neurosci. 2010;30:14361–14365. doi: 10.1523/JNEUROSCI.3440-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tzingounis A.V., Heidenreich M., Jentsch T.J. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc. Natl. Acad. Sci. USA. 2010;107:10232–10237. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tzingounis A.V., Nicoll R.A. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc. Natl. Acad. Sci. USA. 2008;105:19974–19979. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soh H., Tzingounis A.V. The specific slow afterhyperpolarization inhibitor UCL2077 is a subtype-selective blocker of the epilepsy associated KCNQ channels. Mol. Pharmacol. 2010;78:1088–1095. doi: 10.1124/mol.110.066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanner G.R., Lutas A., Yellen G. Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J. Neurosci. 2011;31:8689–8696. doi: 10.1523/JNEUROSCI.5951-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lasser-Ross N., Ross W.N., Yarom Y. Activity-dependent [Ca2+]i changes in guinea pig vagal motoneurons: relationship to the slow afterhyperpolarization. J. Neurophysiol. 1997;78:825–834. doi: 10.1152/jn.1997.78.2.825. [DOI] [PubMed] [Google Scholar]
- 25.Tatulian L., Delmas P., Brown D.A. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J. Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tatulian L., Brown D.A. Effect of the KCNQ potassium channel opener retigabine on single KCNQ2/3 channels expressed in CHO cells. J. Physiol. 2003;549:57–63. doi: 10.1113/jphysiol.2003.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villalobos C., Foehring R.C., Andrade R. Essential role for phosphatidylinositol 4,5-bisphosphate in the expression, regulation, and gating of the slow afterhyperpolarization current in the cerebral cortex. J. Neurosci. 2011;31:18303–18312. doi: 10.1523/JNEUROSCI.3203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loussouarn G., Park K.H., Escande D. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 2003;22:5412–5421. doi: 10.1093/emboj/cdg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H., Craciun L.C., Logothetis D.E. PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 30.Suh B.C., Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- 31.Baukrowitz T., Schulte U., Fakler B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- 32.Enkvetchakul D., Loussouarn G., Nichols C.G. The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys. J. 2000;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shyng S.L., Nichols C.G. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]