

Summary
After replicating in epithelial cells, alphaherpesviruses such as Pseudorabies Virus (PRV) invade axons of peripheral nervous system neurons and undergo retrograde transport toward the distant cell bodies. Although several viral proteins engage molecular motors to facilitate transport, the initial steps and neuronal responses to infection are poorly understood. Using compartmented neuron cultures to physically separate axon infection from cell bodies, we found that PRV infection induces local protein synthesis in axons, including proteins involved in cytoskeletal remodeling, intracellular trafficking, signaling, and metabolism. This rapid translation of axonal mRNAs is required for efficient PRV retrograde transport and infection of cell bodies. Furthermore, induction of axonal damage, which also induces local protein synthesis, prior to infection reduces virion trafficking, suggesting that host damage signals and virus particles compete for retrograde transport. Thus, similar to axonal damage, virus infection induces local protein translation in axons and viruses likely exploit this response for invasion.
Introduction
Alphaherpesviruses such as human pathogen herpes simplex virus 1 (HSV-1), and animal pathogen PRV cause mild to severe neuronal pathologies after entry into the peripheral nervous system (PNS). Alphaherpesvirus particles are capable of moving both toward the cell body (retrograde) and away from the cell body (anterograde) (Smith et al., 2001). In natural hosts, the infection usually stops in the PNS where a quiescent infection is established, but the infection may continue on to the central nervous system (CNS), particularly in non-natural hosts, causing encephalitis, paralysis and death (Brittle et al., 2004; Pomeranz et al., 2005). To move efficiently within axons to and from cell bodies and also from neuron to neuron, viral gene products engage molecular motors and move virion complexes on microtubules (Enquist et al., 2002; Smith et al., 2004). However most of the initial interactions between infecting virions and axons, particularly how viral particles engage the transport machinery for efficient long distance movement to the nucleus remain largely unknown.
The first contact of alphaherpesvirus virions with the host cell occurs via the interactions of viral membrane glycoproteins (gC, gB, gD, gH/gL) with cellular heparan sulfate receptors and herpesvirus entry mediators (e.g. HVEM, nectin1) leading to the fusion of viral envelope with the host cell plasma membrane (Krummenacher et al., 2003; Spear et al., 2000; Turner et al., 1998). These initial interactions and subsequent internalization of the virions trigger tyrosine phosphorylation of cellular proteins together with calcium signaling (Cheshenko et al., 2003; Qie et al., 1999). By unknown mechanisms, this signaling cascade enables the efficient microtubule dependent transport and nuclear translocation of incoming viral genomes in a Beta-importin dependent manner (Ojala et al., 2000). Successful nuclear import of the viral genomes is a key step in viral infection. How this crucial step is carried out efficiently in neurons is of our particular interest since the axons of mammalian neurons can be separated from the cell body by centimeters to meters.
The axons of developing and mature mammalian neurons contain endoplasmic reticulum, Golgi, RNA silencing machinery components, and the complete protein synthesis machinery including several localized mRNAs (Merianda et al., 2009; Murashov et al., 2007). These axonal mRNAs are transported from the cell body as messenger ribonucleoprotein (mRNP) granules and their local translation in axons enables spatial control of homeostasis and production of rapid responses (reviewed in (Lin and Holt, 2008; Wang et al., 2010)). Local translation in axons regulates growth cone navigation and integrity during development and regeneration (Piper and Holt, 2004). Mature mammalian neurons also contain axonal translation machinery that produce new proteins after external stimuli which are utilized to inform neuronal soma in response to injury (Hanz et al., 2003; Taylor et al., 2009; Verma et al., 2005; Yudin et al., 2008; Zheng et al., 2001).
Here we demonstrate that efficient retrograde transport of PRV particles after axonal infection requires new protein synthesis. We used the tri-chamber neuronal culture system (Ch’ng and Enquist, 2005) to physically separate axons from cell bodies. We found that local inhibition of protein synthesis in isolated axons significantly and rapidly reduced the number of virus particles moving towards the cell body. We confirmed the induction of new protein synthesis in axons after infection using synthetic amino acid labeling. We further purified labeled proteins and identified them using nano-scale ultra high performance liquid chromatography coupled to high-resolution mass spectrometry (nano-uHPLC-MS). This analysis revealed a subset of nascent proteins produced after axonal infection including those involved in cytoskeleton remodeling, intracellular trafficking, signal transduction and metabolism. Our findings demonstrate a striking similarity between the local events caused by axonal entry of virions and the retrograde injury signaling mechanism in axons (Hanz et al., 2003).
Experimental Procedures
Cell lines and virus strains
The porcine kidney epithelial cell line (PK15) was purchased from the American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin and streptomycin. Becker is a wild-type laboratory strain (Platt et al., 1979). PRV GS443 expresses VP26–green fluorescent protein (GFP) in a PRV Becker background (Smith et al., 2001). PRV 180 encodes mRFP1-VP26 in a PRV-Becker background (del Rio et al., 2005). PRV 233 is a gB-null mutant of PRV-Becker expressing diffusible GFP from the gG locus under the control of a CMV promoter (Curanovic and Enquist, 2009). The gB-null viral strains were propagated, and their titers were determined in PK15 cells stably transfected with LP64e3, a plasmid expressing PRV gB under the control of the cytomegalovirus (CMV) promoter (Lisa Pomeranz, personal communication). Neuronal infections were done using 106 pfu of PRV unless otherwise noted.
Neuronal cultures
Superior cervical ganglia (SCG) were isolated from day-17 Sprague- Dawley rat embryos (Hilltop Labs, Inc., Scottsdale, PA) and neurons were cultured in tri- chambers as described previously (Ch’ng and Enquist, 2005). Protocol is detailed in Extended Experimental Procedures.
Viral infection of compartmented neuronal cultures
For the analysis of retrograde spread in the soma compartment (S), 1% methocel including neuronal media was placed in the middle/methocel (M) compartment 30 min. before the infection to block possible diffusion of viral particles through the grooves. PRV inoculum was then added in the N-chamber. Depending on the assay infected axons or soma were imaged either immediately, or at indicated hours post infection (h p.i.). For titer assays, contents of S, M, or N compartments were collected by scraping the dish with a pipette tip. All titers were determined on PK15 cells and are expressed in pfu/ml.
Antibodies and Reagents
All antibodies and reagents are detailed in Extended Experimental Procedures.
Immunofluorescence (IF) staining and imaging
Dissociated SCG neurons were plated onto glass-bottom MatTek dishes or chambered optical plastic dishes (Ibidi). Each dish or chamber compartment was rinsed once with phosphate-buffered saline (PBS) and fixed with ice cold methanol for 15 min at −20°C. Imaging was done using a Ti-Eclipse inverted epifluorescence microscope (Nikon). Images were acquired and processed using the NIS-Elements imaging software suite (version 3.2; Nikon). For comparative analysis, fluorescence intensity, exposure time and other parameters were kept the same for each antibody or RNA probe. Detailed staining protocol was given in Extended Experimental Procedures.
Live-cell imaging
Live-cell imaging was performed on a Ti-Eclipse inverted epifluorescence microscope (Nikon) equipped with a Cool Snap ES2 camera (Photometrics). Neuron cultures were kept in a humidified stage-top incubator (Live Cell Instrument) at 37°C with 5% CO2 during imaging. The percentage of moving virus particles was calculated manually using NIS-Elements imaging software. Representative kymographs were generated using Image J ‘make kymograph’ plug-in.
Western blot analysis
For Total cell extracts were prepared in RIPA-light buffer supplemented with 1 mM DTT and protease inhibitor cocktail (Roche). Lysates were kept 30 minutes on ice and sonicated to shear genomic DNA. Further details can be found in Extended Experimental Procedures.
siRNA transfections
Delivery of silencing RNAs into neurons and axons were done using cationic magnetic nanoparticles (Magnetofection). The reagents and protocols are detailed in Extended Experimental Procedures.
Quantitative RT-PCR
Q-RT-PCR was performed with Eppendorf Realplex Mastercycler. Reaction mixture was prepared using Kapa Syber Fast qPCR kit and samples were prepared as triplicates. Each experiment was done in duplicates. Please refer to Extended Experimental Procedures for details.
Detection and identification of L-AHA containing proteins
Bio-orthogonal noncanonical amino acid tagging (BONCAT) was performed by using click chemistry reagents (Invitrogen). L-azidohomoalanine (L-AHA) was used as a methionine surrogate. The azide moiety of this amino acid was used for the copper catalyzed azide-alkyne cycloaddition reaction either with Alexa Fluor488-alkyne (Invitrogen) for fluorescent staining or alkyne-agarose resin (Invitrogen) for proteomic analysis according to manufacturers protocol. For the detailed protocol please refer to Extended Experimental Procedures.
RNA fluorescent in situ hybridization (RNA-FISH)
For the detection of specific mRNA molecules in neurons, we used Panomics QuantiGene ViewRNA ISH Cell Assay kit (Affymetrix). Rat Anxa2, Prph and LIS1 probes were ordered as type I probe sets whereas human/rat/mouse 18S rRNA probe was ordered as type 4 probe set to be able to do dual labeling. The protocol is detailed in Extended Experimental Procedures.
Statistical analysis
One-way ANOVA with Tukey’s post-test or Student-t test were performed using GraphPad Prism 5.0d for Max OS X (GraphPad Software). Numbers included in the text, bar graphs, and figure legends throughout the manuscript indicate the mean ± standard error of the mean (SEM).
Mass Spectrometry Data Acquisition and Analysis
Peptides were prepared from the captured proteins using on-bead trypsin digestion (Trypsin Gold, Promega) overnight at 37 °C. Digest solution with beads were subjected to 10 KDa cut-off membrane ultrafiltration (Microcon YM-10, Millipore), the filters were washed with 50% acetoniltrile (ACN), 0.1% formic acid (FA), and the peptide liquor was concentrated in a speedvac until near dryness. Peptides were desalted using StageTip micro-scale reversed-phase chromatography (Rappsilber et al., 2003), and then subjected to reversed-phase nano-LC-MS and MS/MS performed on a nano-flow capillary high pressure HPLC system (Nano Ultra 2D, Eksigent, Dublin, CA) coupled to an LTQ-Orbitrap XL hybrid mass spectrometer (ThermoFisher Scientific, San Jose, CA) outfitted with a NanoMate ion source robot (Advion, Ithaca, NY). Aggregate search results were imported into Scaffold software (Proteome Software, Portland, OR), consolidated according to the PeptideProphet and ProteinProphet parsimony algorithms (ISB, Seattle WA), subjected to spectral counting quantitation analysis, and filtered to contain 95% protein/80% peptide confidence, requiring 2 peptides per protein assignment. For further details please refer to Extended Experimental Procedures.
Results
Inhibition of protein synthesis in axons reduces virus yield in the cell bodies
To test whether axonal protein translation affects retrograde PRV infection, primary rat superior cervical ganglia (SCG) neurons were cultured in tri-chambers (Ch’ng and Enquist, 2005). These neurons are preferred because of their infection efficiency with PRV, homogeneity and robust axonal growth in vitro providing an excellent tool to study directional virus infection. We first verified that we could inhibit local protein translation in axons with little effect on the cell bodies (Figure 1A) by adding the protein synthesis inhibitor cycloheximide (CHX) in the N-chamber media of uninfected neurons. We assayed phosphorylation state of eIF2 alpha (Kimball, 1999) at various times after CHX addition to verify translation inhibition. Notably, phosphorylated eIF2 alpha increased in the N-chamber axons after two hours of treatment. In contrast, there was no significant effect on eIF2 alpha phosphorylation in neuron cell bodies for at least 24 hours (Figure 1B) indicating that a CHX mediated block in canonical protein translation could be achieved specifically in separate compartments of the tri-chamber.
Figure 1. The effect of axonal protein synthesis inhibition on retrograde PRV infection.

(A) Tri-chamber neuron culture is schematically represented (S: soma, M: middle/methocel, N: neurite). (B) Steady state levels of phosphorylated eIF2-alpha after CHX treatment of N-chamber axons. At indicated time points after CHX addition, both S- and N-chambers were lysed in dish and 25 μl of each sample were run on a 12% SDS gel and the membranes were stained with phosphorylated eIF2-alpha and beta-actin antibodies after Western blotting. (C) PRV GS443 retrograde infection in tri-chambers. CHX was added to N-chambers 1 h before infection. Images of S-chambers were taken 20 hpi (4x magnification). (D) S-chamber virus yields were calculated as pfu, 20 hpi in the absence or presence of CHX either in the N-/or S-chambers. Data are the mean ± SEM with**P<0.01 using one sample t-test. ND: not detected, n≥3.
Next, we tested the effect of axonal protein synthesis inhibition on retrograde PRV infection. N-chamber axons were infected with PRV expressing GFP-tagged capsid proteins (PRV GS443) 1 hour after CHX treatment (Smith et al., 2001). GFP-capsid accumulation in the neuron cell bodies (S-chamber) indicated productive infection and allowed us to monitor the efficiency of infection 20 hours post infection (hpi). Neurons that project axons to the N-chamber were labeled by adding a lipophilic dye (DiI) because only a fraction of total cell bodies project axons to the N-chamber (Ch’ng and Enquist, 2005). Addition of CHX to the S-chamber completely blocked retrograde infection as expected since virus replication requires extensive protein synthesis in the cell body. Interestingly, when CHX was applied only to the distal N-chamber, we also saw a significant reduction in retrograde infection of cell bodies (Figure 1C). To validate this finding, we quantified the virus yield in the S-chambers with and without CHX treatment using a standard plaque assay. In vehicle treated chambers retrograde infection yielded 2.13× ×105 ± 2.7×104 plaque forming units (pfu)/ml. Adding CHX to axons reduced the virus yield in cell bodies by approximately 400-fold (5.33×102 ± 2.46×102 pfu/ml) compared to the controls (Figure 1D). This result suggested that efficient retrograde alphaherpesvirus infection requires local protein synthesis in axons.
Axonal transport of virus particles is regulated by local protein synthesis
We used Q- PCR to quantitate viral genomes and live-cell microscopy to monitor particle movement in axons. We also compared the effects of cytosolic protein synthesis inhibitor CHX to global (cytosolic and mitochondrial) protein synthesis inhibitor emetine hydrochloride (Lietman, 1971). To focus on local events in axons soon after infection and to minimize the duration of inhibitor treatment, we limited our observations to 2 hours. We first determined the extent of retrograde particle movement in N, M, and S compartments by monitoring viral DNA in capsids by Q-PCR (Figure 2A). We converted the DNA quantity in each chamber, including the infection supernatant (SN), to pfu by determining the genome/infectious unit ratio of titered stocks. By 2 hours after wild type PRV (Becker) infection, 50% of the input inoculum had not yet entered axons and remained in the SN (6.3×105 pfu). About 10% of the input inoculum had entered N- chamber axons (1×105 pfu) at this time. Even this early after infection, some viral particles (5.05×103 pfu) had moved to the middle axonal compartment, but none were detectable in the S-chamber (Figure 2A). Addition of protein synthesis inhibitors to N-chambers resulted in a significant reduction in viral genomes in the M-chambers. CHX inhibition reduced the amount of viral genomic DNA in the M-chamber by 76.3 ± 6.1 % and emetine treatment reduced viral genomic DNA by 86.5 ± 11.3 % compared to the untreated controls (Figure 2B). Because both inhibitors had comparable effects, it is likely that cytoplasmic protein synthesis and not mitochondrial protein synthesis is required for efficient retrograde transport.
Figure 2. Determining the effect of axonal protein synthesis on retrograde PRV transport (see also figure S1 and movie S1).

(A) PRV Becker inoculum was added to the N-chamber for 2h, followed by the removal of the infection supernatant and the lysis of the axons (M and N-chamber) and cell bodies (S-chamber). Viral DNA in the infection supernatant and all three chambers was quantified by Q-PCR (number of samples=3, ND=not detected). (B) Q-PCR quantification of virus particles in the M-chamber in the absence or presence of CHX and emetine. Inhibitors were added to the N-chamber 1 h before PRV 443 infection. 2 hpi M-chamber axons were lysed in the dish and the amount of viral DNA was determined. (C) Motile and still virus particles in axons in the absence or presence of CHX and emetine were quantified by live-cell imaging. Virus movement was recorded for 2 min. as 1 frame/second. The ratio of moving to total number of capsids was calculated. Minimum 3 different areas in an N-chamber were chosen for each condition (n=number of experiments, see also supplemental figure 1C). Results are normalized to the control condition. Kymographs of the movies were also shown. Vertical lines indicate stationary virus particles whereas diagonal lines indicate motile virus. Data are the mean ± SEM with *P<0.05 using one sample t-test.
Next, we determined how inhibition of protein synthesis affected the early retrograde particle motion in axons by live cell microscopy. N-chamber axons were infected with PRV GS443 following 1 hour treatment of CHX, emetine, or vehicle. Even in the presence of protein synthesis inhibitors, GFP capsids were moving as early as 10–15 minutes after addition of the inoculum, as in the control samples, demonstrating that inhibition of local protein synthesis had little effect on viral entry. Moreover, the dynamics of those particles that were moving in any condition were not significantly different from the control infection. However, the ratio of moving particles to the total number of particles that entered was reduced by 65 ± 1.3 % in CHX treated axons and by 62 ± 2.9 % in emetine treated axons (Figure 2C and Movie S1). A comparable inhibitory effect was observed when CHX was added 1 hour after the PRV infection (Figure S1). These results demonstrate that inhibition of local translation in axons decreases the number of particles that undergo retrograde transport.
PRV infection stimulates local protein synthesis in axons
To further assess the effects of PRV infection on local protein synthesis, we labeled nascent proteins in axons using “click chemistry” strategy of bio-orthogonal noncanonical amino acid tagging (BONCAT) (Dieterich et al., 2010; Dieterich et al., 2007; Kiick et al., 2002). In this approach, methionine in the medium is replaced with an azide-bearing synthetic amino acid, L-azidohomoalanine (L-AHA). Initially we used dissociated neurons in standard cultures to image nascent protein synthesis after PRV infection. Neurons were infected with PRV Becker and fixed at 2 hpi. As a control, we labeled uninfected cultures with L-AHA for 2 hours (Figure 3A; a). Emetine added during this time markedly suppressed the appearance of L-AHA labeled proteins (Figure 3A; b). L-AHA containing proteins increased in PRV infected dissociated neurons, especially in axons, as highlighted by arrows (Figure 3A; c) and labeling was blocked by the addition of emetine (Figure 3A; d). Higher magnification images of L-AHA labeled proteins in axons after PRV infection are shown in figure 3A (panels i – k). To verify that the L-AHA labeled proteins are indeed in PRV infected neurons, we infected neurons with a PRV recombinant that carries VP26-mRFP in capsids (PRV 180). We saw increased L-AHA labeling in axons harboring mRFP capsids close to the neuronal soma (Figure 3A; o and p).
Figure 3. Detection of newly synthesized proteins in axons by click chemistry.

(A) Dissociated SCG neurons were incubated with 50 μM L-AHA and labeled with alkyne-488 in the absence or presence of emetine. Neurons were either mock infected or infected with Becker or PRV 180. Panels a-d and i-k and o show L-AHA label and e-h, l-n and r show phase contrast images. Panel p shows capsid label (a-h; scale bar is equal to 25 μm, i-r; scale bar is equal to 200 μm). (B) N- chamber axons were incubated and labeled with L-AHA and alkyne-488. Axons were either kept uninfected (a), cut (e) or infected with PRV 180 (b-d, f-h). Panels a, b and e, f show L-AHA label, c and g show capsids, and d and h are the merged images (scale bar is equal to 25 μm). Arrows denote axonal L-AHA labeling, asterix indicate growth cones and the dashed line shows the cut site.
Next, to verify that these newly made, infection specific proteins are made locally in axons, we introduced L-AHA only to the N-chamber of standard tri-chambers. As shown in Figure 3B, panel a, uninfected axons show limited labeling with L-AHA. Note the small speckles of new proteins throughout one axon (arrows). Because damage to axons increases protein synthesis (Yudin et al., 2008), we severed axons in the N-chamber from their cell bodies and monitored new protein synthesis. In contrast to the mock axons, the intensity of L-AHA labeling increased markedly when axons were damaged indicating new protein production as an injury response consistent with the published data (Figure 3B, e). Remarkably, a similar increase in L-AHA labeling was observed in undamaged axons 2 hpi (Figure 3B, c and g). We noted that the signal intensity was particularly intense in growth cones, which are known to utilize local protein translation machinery for development and navigation (Zivraj et al., 2010) (Figure 3B, g). Infecting viral particles also accumulated at these sites (panel h). Collectively, our data suggest that local protein synthesis occurs rapidly after axonal entry of PRV particles, and that when synthesis of these protein is blocked, transport of virus particles to distant neuronal cell bodies is markedly reduced.
Axonal damage reduces retrograde movement of PRV particles
Since both axonal damage and PRV infection induce local protein synthesis in axons, we tested whether these processes are related. In these studies we used an alternative tri-chamber design in which the neuron cell bodies are seeded in the middle chamber and project their axons to the left or right compartments (equivalent to the N-chamber). We cut axons in the right N-chamber with a scalpel prior to virus infection and the left N-chamber was kept uncut as a control (Figure 4A). We observed that cut axon ends still connected to their cell bodies (proximal) after axotomy were highly dynamic and developed vesiculations by two hours after the cut as noted by others (Figure 4B; b and Movie S2) (Kerschensteiner et al., 2005; Tuck and Cavalli, 2010). One hour after the cut, lesioned axons were infected with PRV GS443 and capsid movement in proximal and distal axon segments were immediately recorded. Surprisingly, axon segments disconnected from their cell bodies (distal) transported viral particles efficiently throughout the imaging time (up to 3 hours) comparable to what was observed in the uncut axons on the left compartment (Figure 4B; a and c, and Movie S2). Remarkably, PRV capsids were significantly less motile in the proximal segments of the cut axons (65.1 ± 5.3 % reduction) compared to particle motility in uncut axons, and distal axon fragments (Figure 4C; Movie S2). We conclude that axonal damage induced prior to virus infection does not increase but rather reduces viral particle trafficking significantly in axons proximal (but not distal) to the cut site. These observations suggest that there is competition between the retrograde transport of damage signals and virus particles.
Figure 4. Retrograde PRV infection in injured axons (see also movie S2).

(A) Experimental design is illustrated. Dotted line represents the cut site. (B) Corresponding phase contrast images of uncut (a) and cut (b and c) axons. Axons shown in b are still connected to cell bodies (proximal) whereas axons in c are disconnected (distal). Scale bar is equal to 25 μm.(C) Virus particle movement was recorded in a, b, and c and the results are shown in the graph. Data are the mean ± SEM with **P<0.01 using one sample t-test (n=number of movies). Kymographs are shown for the corresponding sections below the graph. Vertical lines indicate stationary virus particles whereas diagonal lines indicate motile virus.
Identification of newly synthesized proteins in axons upon PRV infection
To test whether PRV infection induces a global increase in protein synthesis, we labeled newly synthesized proteins with L-AHA and identified them using the BONCAT strategy together with high resolution mass spectrometry-based proteomic analysis (Figure 5A). Controls included uninfected axons incubated with virus-free stock media and infected axons not incubated with L-AHA. L-AHA containing proteins were then purified by using an alkyne-agarose resin and digested into peptides on beads with trypsin using a modified FASP methodology (Wisniewski et al., 2009). The results of the two independent experiments were compared and after eliminating all the proteins that were detected in both controls, we identified a group of newly made proteins unique to PRV infected axons. Pathway analysis revealed that these proteins mainly function in cytoskeleton remodeling, intracellular trafficking, energy metabolism, and signaling pathways (Figure 5B and Table S1). Five of these proteins: Peripherin, annexin A2, pafah1b1/LIS1, gapdh, and pkm2 were found to be conserved between both data sets even after the application of high stringency filters for peptide assignment and protein inference (95% protein/80% peptide confidence based on the PeptideProfit and ProteinProfit algorithms utilized by the Scaffold software, additionally requiring 2 peptides per protein) (Figure 5B). We further investigated peripherin, annexin A2, and LIS1 (Figure S2) because of their putative relevance to the retrograde transport phenotypes we observed.
Figure 5. Identification and classification of newly synthesized proteins in axons after PRV infection (see also figure S2 and table S1).
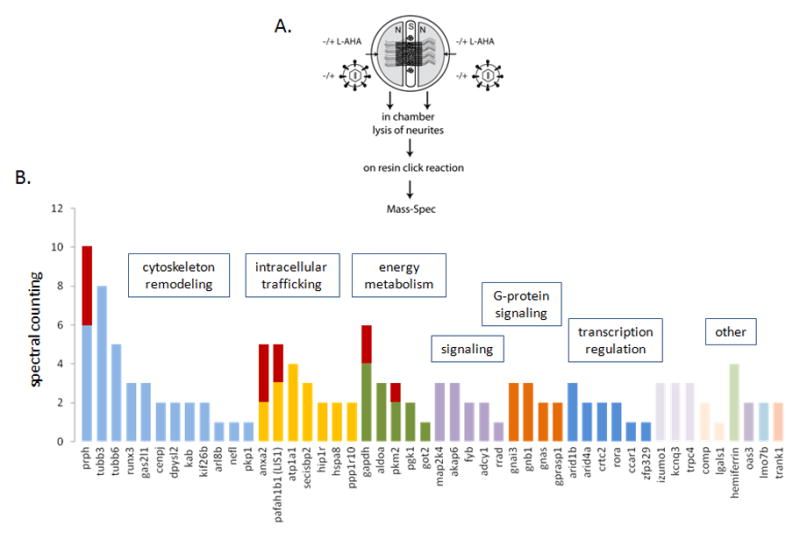
(A) Experimental design is illustrated. (B) Functional categorization of PRV specific proteins (Metacore, DAVID). The results of 2 independent, summed experiments were shown in the bar graph. Bars indicate the raw spectral counting values for each protein. Red bars denote that the corresponding proteins are identified with high confidence in both experiments. In this case, the spectral counting from each experiment is shown proportionally by two colors.
Peripherin (Prph) yielded the highest spectral counts and represented the most probable newly synthesized protein during retrograde PRV infection. The second most prominent candidate was Annexin A2 (Anxa2), followed by Pafah1b1 (LIS1). The mRNAs of these three proteins have previously been shown to be present in DRG axons (Gumy et al.; Willis et al., 2005), but their local synthesis in axons had not been described. We confirmed the presence of these transcripts in SCG axons by fluorescence labeling of RNAs using gene specific probes and an 18S rRNA probe as a control (Figure 6A). Anxa2, Prph and LIS1 mRNAs were sparsely distributed as small and large puncta in axons (panels a, b, and c), whereas 18S rRNA was abundant and non-uniformly distributed as small speckles along axons (panels d–f).
Figure 6. Localization of identified proteins and their mRNAs in SCG axons (see also figure S3).

(A) Axons in tri-chambers were stained with fluorescent RNA probes for Anxa2 (a), Prph (b) and LIS1 (c), 2 days after siNT transfection in axons. 18S rRNA staining is shown in panels d–f, and merged images are shown in g–i. (B) Mock (a–f) or PRV GS443 infected axons (g–s) were stained with anti-Anxa2 (a and g), anti-Prph (b–h) and anti-LIS1 (c and i) antibodies. Capsid localization (j–l), merged (m–o) and phase-contrast (p–s) images were shown. Scale bar is equal to 25 μm. (B) (C) Dissociated SCG neurons were transfected either with siNT or siRNAs against Anxa2, Prph, and LIS1, and cells were either used for IF staining or Western blotting. After staining with the corresponding antibodies, fluorescent densities were measured (n>10) and shown in the graph (C) and protein bands were shown in (D). Beta-actin was used as loading control. Data are the mean ± SEM with ***P<0.001 using one sample t-test.
We determined the axonal localization of these proteins by IF staining of mock and PRV infected axons, 2 hpi (Figure 6B). In mock infected axons, Anxa2 and LIS1 showed speckled and punctate distribution (panels a and c) whereas Prph was diffusively distributed throughout the axons (panel b). In PRV GS443 infected axons, Anxa2 accumulated in distinct areas (panel g) that are invaded by virus particles (panel j). Interestingly, Prph localization changed from diffuse (panel h) to patchy staining in infected axons (panel k). LIS1 staining in PRV infected axons was indistinguishable from mock condition (panel i).
LIS1, Anxa2, and Prph are involved in retrograde trafficking of PRV in axons
We used RNAi silencing to assess the roles of axonal Anxa2, Prph, or LIS1 translation on PRV retrograde infection. We first examined silencing efficiency on a single cell level by IF staining of dissociated neurons at 96 hours post transfection (Figure S3A). Fluorescence intensities were measured in control (siNT) and gene specific siRNA transfected cells and depicted in the graph after staining with corresponding antibodies (Figure 6C). Protein and mRNA levels in cells after gene silencing were also determined by western blot (Figure 6D) and RNA hybridization (Figure S3B) analyses, respectively. Collectively, these data confirmed that we significantly knockdown the expression of all three targets. We also ensured that these siRNAs were functional in axons by transfecting only the N-chambers. Target mRNAs were then stained with fluorescent probes proving the effect of siRNAs also in axons (Figure S3C).
Finally, to determine the effect of local silencing of the synthesis of these proteins on PRV retrograde infection, we transfected axons by siNT, siAnxa2, siPrph, or siLIS1 before infecting with PRV. For the retrograde spread assay, we used a recombinant PRV (PRV 233) that encodes a diffusible GFP protein, but lacks gB, which is required for membrane fusion and cell entry (Babic et al., 1993; Curanovic and Enquist, 2009; Liu et al., 2008). The PRV 233 stock was grown in gB complementing cells, so that gB was incorporated in progeny virions that were infectious. However, after replication in primary neurons, newly produced progeny, which lack gB, cannot spread to the neighboring cells for secondary infection (Curanovic and Enquist, 2009). As a result, we were able to assess unambiguously the observed effects of axon-to-cell body transport events on primary infected cells. We infected N-chamber axons with PRV 233–48 hours after siRNA transfection. At 24 hpi, we quantified the number of GFP-positive cell bodies in the S-chamber (Figure 7A). The lipohilic dye DiI was added to label cell bodies that projected axons to the N-chamber (panels a–d). The ratio of GFP positive (infected) cells to DiI positive cells was determined in control and gene specific siRNA transfected samples.
Figure 7. The effect of axonal gene knockdown on retrograde PRV infection.

(A) Images show representative areas in the S-compartments. N-chambers were transfected with either siNT or gene-specific siRNAs before PRV 233 infection. DiI stained cell bodies (a–d), GFP positive cell bodies (e–h), merged images (i–l), and phase contrast images (m–o) are shown. (B) Graph depicts the percentage of GFP positive to DiI positive cell bodies of either siNT or gene-specific siRNA transfected axons. Data shown are the mean of 3 independent experiments consisting of duplicates. Data are the mean ± SEM with *P<0.05,**P<0.01 and ***P<0.001 using one sample t-test. (C) Hypothesized cascade of events that take place in retrograde PRV infection of healthy and injured axons. 1. Virion attachment and entry, increase in cytosolic calcium and phosphorylation events 2. Translation of axonal mRNAs 3. Formation of dynein coupled transport complexes. 4. Beta-importin dependent nuclear localization and subsequent transcription and replication of viral genomes.
When the axons in the N-chamber were infected with PRV 233 (105 pfu) and the inoculum was removed one hour after infection, less than 10% of all S-chamber cell bodies (7.087 ± 1.15%) were infected at 24 hpi. Following siRNA knockdown of Anxa2, Prph, and LIS1, the percentage of GFP positive cells was significantly reduced to 1.1 ± 0.4%, 2.96 ± 1.56%, and 0.4 ± 0.1% respectively (Figure 7B). We conclude that reducing LIS1 and Anxa2 expression, and to a lesser degree Prph expression in axons significantly reduces the number of retrogradely infected cell bodies in the distant S-chamber. Because PRV 233 has no known replication deficiency, this defect must arise from inefficient retrograde transport of capsids from axons to cell bodies. Taken together, our results suggest that efficient long-distance axonal transport of PRV particles requires rapid local axonal synthesis of host cell trafficking proteins such as dynein regulator LIS1, the vesicle transport regulator Anxa2, and an intermediate filament, Prph after infection (Figure 7C).
Discussion
The PNS functions because of efficient long distance connections between axons and cell bodies. Distant events at axonal termini, such as tissue damage, inflammation, and infection must be recognized and transmitted to the distant neuronal nucleus to produce an appropriate response both at the site of the insult and in the central neuronal circuits. How PNS axon termini sense viral infection and inflammation in their peripheral targets is an important, but poorly understood problem. For alphaherpesvirus particles, PNS invasion appears to be inevitable once the infection occurs in the epithelial cells. The reasons for this efficient uptake and spread into the PNS are not well understood. Here we suggest a mechanism involving local protein synthesis in axons at the site of infection that increases the efficiency of long distance virus transport towards neuronal cell bodies. We also suggest that damage responses in the axons may interfere with PNS invasion. We identify several newly synthesized proteins that are necessary for efficient retrograde herpesvirus infection.
We first observed that efficient retrograde infection of neurons after axonal infection requires axonal protein synthesis. We used compartmented SCG neurons to isolate axons from cell bodies and test local effects of protein synthesis inhibitors. We found that inhibition of cytosolic protein synthesis in isolated axons reduced the subsequent virus yield in cell bodies up to 400 fold. Using direct imaging of virus particles in axons shortly after infection, we confirmed that inhibition of local protein synthesis reduced the number of motile capsids in axons. Since many locally synthesized proteins in axons are known to be mitochondrial (Hillefors et al., 2007), we compared the effect of cycloheximide, a cytosolic protein synthesis inhibitor, to emetine, a cytosolic and mitochondrial translation inhibitor. Both inhibitors significantly and comparably reduced the number of virions that reached mid-axons in two hours indicating that the synthesis of cytosolic proteins is induced in retrograde PRV infection.
The BONCAT methodology and click chemistry allowed us to detect infection-stimulated protein synthesis in axons. We know that herpesvirus particle entry into cells requires receptor attachment and subsequent membrane fusion (Krummenacher et al., 2003; Turner et al., 1998). These events are followed by an increase in cytosolic calcium and enhanced phosphorylation events (Cheshenko et al., 2003). Recently, Tcherkezian et. al. showed that the transmembrane receptor DCC (Deleted in Colorectal Carcinoma) links extracellular stimuli with the intracellular protein synthesis machinery in axons via tethering ribosomal subunits and the eukaryotic initiation factor 4E (eIF4E) complex (Tcherkezian et al., 2010). Similar transmembrane signaling might trigger protein synthesis in axons when PRV envelope glycoprotein, gD engages the host receptor nectin 1 (De Regge et al., 2006; Krummenacher et al., 2003).
Physical damage to axons or the presence of inflammatory cytokines has been shown to induce local protein synthesis in axons (Hanz et al., 2003; Melemedjian et al., 2010). Calcium dependent beta-importin translation in axons results in the formation of a complex of beta/alpha- importin heterodimers and dynein motors. These complexes have high affinity to nuclear localization signals on the signaling cargo which are directed to the neuronal nuclei (Hanz et al., 2003; Yudin et al., 2008). We know that retrograde transport of poliovirus particles from injected muscle to the central nervous system in mice is enhanced by a muscle damage at the site of infection (Kuss et al., 2008). In contrast, tissue damage blocks neural invasion by PRV in vivo (Duale et al., 2009; Duale et al., 2010). We tested whether such an injury-induced environment in axons would be beneficial to virus transport by inducing axonal damage in compartmented neurons before infecting the axons. We found that pre-induction of a lesion in axons resulted in a severe (65.1%) reduction in the efficiency of retrograde infection compared to controls supporting the in vivo observations. In contrast, virus particles that entered distal axon segments physically disconnected from their cell bodies moved efficiently until they reached the cut site. Proximal damaged axons connected to their cell bodies were inefficient in transporting viral particles presumably because the transport and/or mRNA translation machinery is already co-opted by the danger signaling molecules/complexes. On the other hand, since the distal cut axon segments do not have cell body targeted injury signaling complexes (because the signaling should start at the cut site and move towards the minus end of microtubules), new protein synthesis and retrograde transport machinery can be utilized by the infecting virions. Because the distal axons are predicted to be in the latency phase of degeneration (for 24–48 hours), it is not surprising that they are active for viral transport (reviewed in (Wang et al., 2012)) (Figure 7C).
The similarity between the axonal damage response and axonal response to PRV infection is striking: Both induce local calcium and cytokine influx which triggers translation of specific mRNAs and formation of nucleus targeted complexes (involving beta-importin) that use dynein based fast transport machinery. According to our hypothesis, since they both depend on the same rapid transport machinery to reach the neuronal nucleus, injury competitively inhibits virus transport in the same axon. Recently, Fainzilber and colleagues reported a phosphoproteomic study on the injured nerve axoplasm revealing a highly complex and extensive signaling network including Abl, Akt, p38 and protein kinase C (Michaelevski et al., 2010). Such diversity of signals and mass movement of large signaling complexes directed towards the nucleus was suggested to affect all other signaling and transport machinery along that axon which supports our competitive inhibition theory.
We further identified the newly synthesized proteins induced by axonal infection. Proteomic analyses revealed a small pool of nascent proteins including several actin cytoskeleton and intermediate filament remodeling proteins, intracellular trafficking and signaling mediators, as well as energy metabolism enzymes. While the total number of newly synthesized proteins detected in this study may represent only a fraction of all those induced upon infection, it represents a technical tour-de-force in the context of a physiologically relevant primary neuron culture system in which the axonal sub-compartment is subject to PRV infection.
Cytoskeleton remodeling proteins have been associated with herpesvirus attachment and entry in sensory neurons which were shown to be activated upon gD and nectin 1 interaction (De Regge et al., 2006). Nectins have been known to induce filopodia and lamellipodia formation through cdc42 and rac1 signaling (Kawakatsu et al., 2002). These findings support our hypothesis that new protein synthesis in axons might be triggered via virus induced transmembrane signaling upon receptor attachment.
The neuronal type III intermediate filament, peripherin, was a prominent candidate identified upon axonal infection. It is known to be involved in regeneration of neurites. Peripherin might act as a signaling molecule since another type III intermediate filament vimentin was shown to be produced locally in axons and cleaved by calpain upon injury (Perlson et al., 2005). In this process, newly synthesized and cleaved vimentin plays a central role in transporting kinase signaling complexes to the cell nucleus by binding to both phosphorylated extracellular signal regulated kinase (ERK) -in a calcium dependent manner- and Beta-importin (Perlson et al., 2006). It was striking that, peripherin localization changed dramatically in PRV infected axons. Such relocalization may mean that this protein is modified after infection. Peripherin mRNPs containing several copies of translationally silent mRNAs were detected on microtubules in rat pheochromocytoma cells (PC12) (Chang et al., 2006) consistent with our observation of Prph mRNA in large puncta in axons. Our data suggests that viral entry induces synthesis of new Prph molecules that might contribute to the formation of high efficiency retrograde trafficking complexes as in the case of injury signaling.
Annexin A2 synthesis was also specifically detected in axons upon PRV infection. Annexins are calcium regulated phospholipid binding proteins that organize membrane domains and regulate endocytic and exocytic transport. Annexin A2 particularly regulates lipid raft organizations at sites where plasma membrane contacts actin cytoskeleton (reviewed in (Hayes et al., 2004) and (Gerke et al., 2005)). Extracellular herpesvirus virions contain Anxa2 although its function in the virion is unknown (Kramer et al., 2011). We observed a significant defect in retrograde PRV infection when Anxa2 translation was silenced in the axons. Therefore, it may be that Anxa2 mediates the initial actin cytoskeleton interaction of capsids after membrane fusion and cell entry.
The local synthesis of LIS1, a multifunctional dynein regulator, was a key finding. LIS1 participates in organelle, membrane vesicle and microtubule transport (Faulkner et al., 2000; Smith et al., 2000). LIS1 contains several WD repeats that mediate protein-protein interactions (Reiner et al., 1993). When LIS1 synthesis was inhibited, large vesicle transport in axons was dramatically reduced (Yi et al., 2011). Similarly, we observed that efficient retrograde transport of infecting virus particles depends on the rapid synthesis of LIS1 in axons. We know that LIS1 and NudE form a complex and adapt dynein motors to carry heavy and large cargo by preventing dynein detachment from microtubules and by enhancing motor force (McKenney et al., 2010). This function of LIS1 might increase the transport efficiency of the relatively large PRV particles moving on microtubules.
It is surprising to find that new synthesis of proteins such as, peripherin, Anxa2, and LIS1 is stimulated after axonal herpesvirus infection when these proteins are already present in axons. Perhaps the nascent proteins are not modified, which distinguishes them from the pre-existing pool so that they can act as messengers. Another possibility might be that new protein synthesis is a strategy to localize all the necessary components of a signaling transport complex that can ensure the rapid nuclear transport of viral genomes of numerous incoming viruses. The existing pool of these components might not be immediately accessible to virus particles even if they are already present in the axons due to prior engagement with other complexes or spatial segregation from the incoming capsids. In such cases, new synthesis of the needed molecules provides an excellent mechanism to assemble new and numerous virus transport complexes.
We show here that PRV infection of axons induces a robust and rapid response at the site of infection involving local translation of host proteins. This effect occurs hours before anything happens in the cell body and nucleus. Such local monitoring of peripheral tissues by PNS neurons is essential for survival but at the same time can be cleverly exploited by alphaherpesviruses to efficiently invade the nervous systems of their hosts. We focused only on three proteins that are locally produced in axons in response to infection. Clearly many other proteins are locally synthesized after infection that bear further study.
Supplementary Material
Highlights.
Pseudorabies Virus (PRV) infection stimulates local protein synthesis in axons.
Infection induces the synthesis of LIS1, peripherin, and annexin A2 in axons.
Axonal inhibition of protein synthesis retards retrograde PRV capsid transport.
Prior damage in axons significantly reduces retrograde PRV capsid transport.
Acknowledgments
We thank Emre Koyuncu and the members of the Enquist lab especially Andrea Granstedt, Tal Kramer, Matthew Taylor and Ren Song for their comments and critical reading of the manuscript. L.W.E. is supported by the US National Institutes of Health grant R01NS033506-18.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Babic N, Mettenleiter TC, Flamand A, Ugolini G. Role of essential glycoproteins gII and gp50 in transneuronal transfer of pseudorabies virus from the hypoglossal nerves of mice. J Virol. 1993;67:4421–4426. doi: 10.1128/jvi.67.7.4421-4426.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittle EE, Reynolds AE, Enquist LW. Two modes of pseudorabies virus neuroinvasion and lethality in mice. J Virol. 2004;78:12951–12963. doi: 10.1128/JVI.78.23.12951-12963.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’ng TH, Enquist LW. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J Virol. 2005;79:10875–10889. doi: 10.1128/JVI.79.17.10875-10889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Shav-Tal Y, Trcek T, Singer RH, Goldman RD. Assembling an intermediate filament network by dynamic cotranslation. J Cell Biol. 2006;172:747–758. doi: 10.1083/jcb.200511033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshenko N, Del Rosario B, Woda C, Marcellino D, Satlin LM, Herold BC. Herpes simplex virus triggers activation of calcium-signaling pathways. J Cell Biol. 2003;163:283–293. doi: 10.1083/jcb.200301084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curanovic D, Enquist LW. Virion-incorporated glycoprotein B mediates transneuronal spread of pseudorabies virus. J Virol. 2009;83:7796–7804. doi: 10.1128/JVI.00745-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Regge N, Nauwynck HJ, Geenen K, Krummenacher C, Cohen GH, Eisenberg RJ, Mettenleiter TC, Favoreel HW. Alpha-herpesvirus glycoprotein D interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J Cell Biol. 2006;174:267–275. doi: 10.1083/jcb.200510156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rio T, Ch’ng TH, Flood EA, Gross SP, Enquist LW. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J Virol. 2005;79:3903–3919. doi: 10.1128/JVI.79.7.3903-3919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, Schuman EM. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Lee JJ, Link AJ, Graumann J, Tirrell DA, Schuman EM. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat Protoc. 2007;2:532–540. doi: 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- Duale H, Hou S, Derbenev AV, Smith BN, Rabchevsky AG. Spinal cord injury reduces the efficacy of pseudorabies virus labeling of sympathetic preganglionic neurons. J Neuropathol Exp Neurol. 2009;68:168–178. doi: 10.1097/NEN.0b013e3181967df7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duale H, Lyttle TS, Smith BN, Rabchevsky AG. Noxious colorectal distention in spinalized rats reduces pseudorabies virus labeling of sympathetic neurons. J Neurotrauma. 2010;27:1369–1378. doi: 10.1089/neu.2010.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist LW, Tomishima MJ, Gross S, Smith GA. Directional spread of an alpha-herpesvirus in the nervous system. Vet Microbiol. 2002;86:5–16. doi: 10.1016/s0378-1135(01)00486-2. [DOI] [PubMed] [Google Scholar]
- Faulkner NE, Dujardin DL, Tai CY, Vaughan KT, O’Connell CB, Wang Y, Vallee RB. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- Gumy LF, Yeo GS, Tung YC, Zivraj KH, Willis D, Coppola G, Lam BY, Twiss JL, Holt CE, Fawcett JW. Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA. 17:85–98. doi: 10.1261/rna.2386111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, et al. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40:1095–1104. doi: 10.1016/s0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- Hayes MJ, Rescher U, Gerke V, Moss SE. Annexin-actin interactions. Traffic. 2004;5:571–576. doi: 10.1111/j.1600-0854.2004.00210.x. [DOI] [PubMed] [Google Scholar]
- Hillefors M, Gioio AE, Mameza MG, Kaplan BB. Axon viability and mitochondrial function are dependent on local protein synthesis in sympathetic neurons. Cell Mol Neurobiol. 2007;27:701–716. doi: 10.1007/s10571-007-9148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakatsu T, Shimizu K, Honda T, Fukuhara T, Hoshino T, Takai Y. Trans-interactions of nectins induce formation of filopodia and Lamellipodia through the respective activation of Cdc42 and Rac small G proteins. J Biol Chem. 2002;277:50749–50755. doi: 10.1074/jbc.M209846200. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc Natl Acad Sci U S A. 2002;99:19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SR. Eukaryotic initiation factor eIF2. Int J Biochem Cell Biol. 1999;31:25–29. doi: 10.1016/s1357-2725(98)00128-9. [DOI] [PubMed] [Google Scholar]
- Kramer T, Greco TM, Enquist LW, Cristea IM. Proteomic characterization of pseudorabies virus extracellular virions. J Virol. 2011;85:6427–6441. doi: 10.1128/JVI.02253-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, Eisenberg RJ, Cohen GH. Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J Virol. 2003;77:8985–8999. doi: 10.1128/JVI.77.16.8985-8999.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuss SK, Etheredge CA, Pfeiffer JK. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog. 2008;4:e1000082. doi: 10.1371/journal.ppat.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietman PS. Mitochondrial protein synthesis: inhibition by emetine hydrochloride. Mol Pharmacol. 1971;7:122–128. [PubMed] [Google Scholar]
- Lin AC, Holt CE. Function and regulation of local axonal translation. Curr Opin Neurobiol. 2008;18:60–68. doi: 10.1016/j.conb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WW, Goodhouse J, Jeon NL, Enquist LW. A microfluidic chamber for analysis of neuron-to-cell spread and axonal transport of an alpha-herpesvirus. PLoS One. 2008;3:e2382. doi: 10.1371/journal.pone.0002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney RJ, Vershinin M, Kunwar A, Vallee RB, Gross SP. LIS1 and NudE induce a persistent dynein force-producing state. Cell. 2010;141:304–314. doi: 10.1016/j.cell.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, Dussor GO, Price TJ. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci. 2010;30:15113–15123. doi: 10.1523/JNEUROSCI.3947-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merianda TT, Lin AC, Lam JS, Vuppalanchi D, Willis DE, Karin N, Holt CE, Twiss JL. A functional equivalent of endoplasmic reticulum and Golgi in axons for secretion of locally synthesized proteins. Mol Cell Neurosci. 2009;40:128–142. doi: 10.1016/j.mcn.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelevski I, Segal-Ruder Y, Rozenbaum M, Medzihradszky KF, Shalem O, Coppola G, Horn-Saban S, Ben-Yaakov K, Dagan SY, Rishal I, et al. Signaling to transcription networks in the neuronal retrograde injury response. Sci Signal. 2010;3:ra53. doi: 10.1126/scisignal.2000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashov AK, Chintalgattu V, Islamov RR, Lever TE, Pak ES, Sierpinski PL, Katwa LC, Van Scott MR. RNAi pathway is functional in peripheral nerve axons. FASEB J. 2007;21:656–670. doi: 10.1096/fj.06-6155com. [DOI] [PubMed] [Google Scholar]
- Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol Cell Biol. 2000;20:4922–4931. doi: 10.1128/mcb.20.13.4922-4931.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, Fainzilber M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005;45:715–726. doi: 10.1016/j.neuron.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Perlson E, Michaelevski I, Kowalsman N, Ben-Yaakov K, Shaked M, Seger R, Eisenstein M, Fainzilber M. Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J Mol Biol. 2006;364:938–944. doi: 10.1016/j.jmb.2006.09.056. [DOI] [PubMed] [Google Scholar]
- Piper M, Holt C. RNA translation in axons. Annu Rev Cell Dev Biol. 2004;20:505–523. doi: 10.1146/annurev.cellbio.20.010403.111746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt KB, Mare CJ, Hinz PN. Differentiation of vaccine strains and field isolates of pseudorabies (Aujeszky’s disease) virus: thermal sensitivity and rabbit virulence markers. Arch Virol. 1979;60:13–23. doi: 10.1007/BF01318093. [DOI] [PubMed] [Google Scholar]
- Pomeranz LE, Reynolds AE, Hengartner CJ. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev. 2005;69:462–500. doi: 10.1128/MMBR.69.3.462-500.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qie L, Marcellino D, Herold BC. Herpes simplex virus entry is associated with tyrosine phosphorylation of cellular proteins. Virology. 1999;256:220–227. doi: 10.1006/viro.1999.9673. [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- Smith GA, Gross SP, Enquist LW. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A. 2001;98:3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Pomeranz L, Gross SP, Enquist LW. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc Natl Acad Sci U S A. 2004;101:16034–16039. doi: 10.1073/pnas.0404686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear PG, Eisenberg RJ, Cohen GH. Three classes of cell surface receptors for alphaherpesvirus entry. Virology. 2000;275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Berchtold NC, Perreau VM, Tu CH, Li Jeon N, Cotman CW. Axonal mRNA in uninjured and regenerating cortical mammalian axons. J Neurosci. 2009;29:4697–4707. doi: 10.1523/JNEUROSCI.6130-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherkezian J, Brittis PA, Thomas F, Roux PP, Flanagan JG. Transmembrane receptor DCC associates with protein synthesis machinery and regulates translation. Cell. 2010;141:632–644. doi: 10.1016/j.cell.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck E, Cavalli V. Roles of membrane trafficking in nerve repair and regeneration. Commun Integr Biol. 2010;3:209–214. doi: 10.4161/cib.3.3.11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner A, Bruun B, Minson T, Browne H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol. 1998;72:873–875. doi: 10.1128/jvi.72.1.873-875.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, Fawcett JW. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25:331–342. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DO, Martin KC, Zukin RS. Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 2010;33:173–182. doi: 10.1016/j.tins.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis D, Li KW, Zheng JQ, Chang JH, Smit A, Kelly T, Merianda TT, Sylvester J, van Minnen J, Twiss JL. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–791. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Yi JY, Ori-McKenney KM, McKenney RJ, Vershinin M, Gross SP, Vallee RB. High-resolution imaging reveals indirect coordination of opposite motors and a role for LIS1 in high-load axonal transport. J Cell Biol. 2011;195:193–201. doi: 10.1083/jcb.201104076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudin D, Hanz S, Yoo S, Iavnilovitch E, Willis D, Gradus T, Vuppalanchi D, Segal-Ruder Y, Ben- Yaakov K, Hieda M, et al. Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron. 2008;59:241–252. doi: 10.1016/j.neuron.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, Twiss JL. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J Neurosci. 2001;21:9291–9303. doi: 10.1523/JNEUROSCI.21-23-09291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivraj KH, Tung YC, Piper M, Gumy L, Fawcett JW, Yeo GS, Holt CE. Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J Neurosci. 2010;30:15464–15478. doi: 10.1523/JNEUROSCI.1800-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.