

Abstract
The trauma of a severe burn injury induces a hypermetabolic response that increases morbidity and mortality. Previously, our group showed that insulin resistance post-burn injury is associated with endoplasmic reticulum (ER) stress. Evidence suggests that c-jun N-terminal kinase (JNK) -2 may be involved in ER stress-induced apoptosis. Here, we hypothesized that JNK2 contributes to the apoptotic response after burn injury downstream of ER stress. To test this, we compared JNK2 knockout mice (−/−) to wildtype mice after inducing a 30% total body surface area thermal injury. Animals were sacrificed after 1, 3 and 5 days. Inflammatory cytokines in the blood were measured by multiplex analysis. Hepatic ER stress and insulin signaling were assessed by Western Blotting and insulin resistance was measured by a peritoneal glucose tolerance test. Apoptosis in the liver was quantified by TUNEL staining. Liver function was quantified by AST and ALT activity assays. ER stress increased after burn in both JNK2−/− and wildtype mice, indicating that JNK2 activation is downstream of ER stress. Knockout of JNK2 did not affect serum inflammatory cytokines; however, the increase in IL-6 mRNA expression was prevented in the knockouts. Serum insulin did not significantly increase in the JNK2−/− group. On the other hand, insulin signaling (PI3K/Akt pathway) and glucose tolerance tests did not improve in JNK2−/−. As expected, apoptosis in the liver increased after burn injury in wildtype mice but not in JNK2−/−. AST/ALT activity revealed that liver function recovered more quickly in JNK2−/−. This study indicates that JNK2 is a central mediator of hepatic apoptosis after a severe burn.
Keywords: Severe burn injury, endoplasmic reticulum stress, inflammation, insulin resistance, JNK-2
INTRODUCTION
Despite remarkable advances in wound healing, mortality and morbidity of severe burn patients (30–40% total body surface area) remain poor due to the inflammatory and hypermetabolic responses (1–4). Burn-induced inflammation and hypermetabolism are characterized by increased protein turnover and degradation to meet metabolic demands, loss of lean body mass, compromised function of essential organs, hyperglycemia and insulin resistance (5).
Burn-induced hepatic damage and dysfunction worsen morbidity and delay recovery (6). Previous research has shown that severe burn impairs hepatocyte structure and function and leads to cell death, by both apoptosis and necrosis (7–9). In other disease models, liver damage has been linked to insulin resistance and inflammation (10). The liver may be influencing survival and recovery by modulating the systemic responses to thermal injury; therefore, it is crucial that we decipher the hepatic molecular mechanisms.
The source of hepatic damage post-burn is likely the endoplasmic reticulum (ER)1. ER stress is induced when there is an excess of unfolded proteins and is elevated in rodent models of severe burn injury (8). Hepatic ER stress is an important mediator of insulin resistance, inflammation and apoptosis (11–13). In vitro and in vivo studies by Ozcan et al demonstrated that ER stress mediates peripheral insulin resistance and type 2 diabetes at the molecular, cellular and organismal levels (11). ER stress leads to apoptosis through either IRE1α activation and/or calcium release (14, 15).
JNK2 proteins are activated downstream of ER stress (16). After phosphorylation of IRE-1 and activation of TRAF2 (16), activated JNK leads to serine phosphorylation of IRS-1, which reduces insulin receptor signaling (11). Increased JNK activation by treatment with palmitic acid, a saturated fatty acid, lead to insulin resistance in primary mouse hepatocytes (17). JNK inhibition 1 hr after smoke inhalation improved mouse survival by preventing inflammatory cell infiltration, cytokine release and airway apoptosis (18). Specifically, JNK activates pro-apoptotic Bim and inactivates anti-apoptotic Bcl-2 proteins (19). In the context of burn injury, rapid JNK activation has been observed in the liver and cardiac tissue (13, 20, 21). Activation in the cardiac tissue could be replicated by treatment with alpha1-adrenergic agonism but the consequences of JNK phosphorylation were not explored (20). In summary, JNK proteins have been linked to insulin resistance, inflammation and apoptosis in other disease settings; however, JNK’s role in burn has not been thoroughly defined.
The functions of JNK1, 2 and 3 differ, thus it is critical to study them separately. JNK1 phosphorylates cJun and has been associated with increasing insulin sensitivity (22, 23). Inhibitor studies in models of obesity and non-alcoholic fatty liver disease (NAFLD)3 have revealed that JNK1 mediates development of obesity, insulin resistance, steatosis, hepatitis, inflammation, apoptosis and liver injury (23, 24). Hepatic-specific knockdown of JNK1 reduces serum insulin and glucose in obese mice but increases glucose intolerance and insulin resistance in a model of NAFLD (25, 26). Less information is available regarding JNK2’s role. JNK2 may actually block cJun phosphorylation (22). JNK2−/− NAFLD studies suggest a role in mediating insulin resistance and steatohepatitis (23). JNK3 is primarily found in the brain and is involved in ischemic apoptosis (27).
The objective of this study was to examine JNK2’s role in mediating hypermetabolism, inflammation and apoptosis post-burn.
MATERIALS AND METHODS
Animals
The study protocol was approved by the Institutional Animal Care and Use Committee of Texas Medical Branch at Galveston. The National Academy Press Guide for the Care and Use of Laboratory Animals (1996) were met. C57BL/6 wildtype mice and JNK2 knockout mice (20–30 g) were purchased from Harlan (Houston, TX, USA) and housed for 1 wk prior to experiments.
Genotyping
Reverse transcription PCR was performed to confirm the genotype of the animals. Total RNA was isolated from liver samples, quantified and reverse transcribed. Wildtype JNK2 was identified with primers 5′-GGA GCC CGA TAG TAT CGA GTT ACC-3′ and 5′-GTT AGA CAA TCC CAG AGG TTG TGT G -3′. Mutant (knockout) JNK2 was identified with primers 5′-GGA GCC CGA TAG TAT CGA GTT ACC -3′ and 5′-CCA GCT CAT TCC TCC ACT CAT G-′ (Jackson Laboratory). Cycling parameters included one cycle of 94°C for 5 minutes then 35 cycles of 94°C for 30 seconds, 52°C for 1 minute, 72°C for 1 minute followed by one cycle of 72°C for 5 minutes.
Burn Injury
A 30% total body surface area burn was induced as previously described (8). Briefly, animals were anesthetized (80 mg/kg ketamine, 7.5 mg/kg xylazine), shaved along the dorsum and administered saline subcutaneously along the spinal cord (0.9%, 1mL). Mice were placed in a mold exposing a defined area of skin on their back and then lowered into 96 to 98°C water for 10 seconds to induce a full-thickness scald burn. Lactated Ringer’s solution (2mL) was administered for resuscitation and buprenorphine (0.1 mg/kg) for analgesia. Similar to burned mice, sham mice were treated with anesthesia, saline, Ringer’s and analgesia but were not lowered into a water bath.
Experimental Design
Thermal injury was induced in wildtype and JNK2−/− mice at day 0. Animals were sacrificed 1, 3 and 5 days post-burn and liver and serum samples were collected. There were 5–6 mice per group per time point.
Serum Protein Analysis: Multiplex
Serum concentrations of 11 cytokines and hormones were measured using a Linco multiplex bead array system (St. Charles, MO, USA) and MiraiBio software package (Hitachi, San Francisco, CA, USA). These included insulin, leptin, glucagon, GLP-1, MCP-1, IL-6, total PAI-1, resistin, amylin, peptide YY, and pancreatic polypeptide.
Hepatic Protein Analysis: Western Blotting
Immediately after sacrifice, liver tissue was snap frozen in liquid nitrogen and stored at −80°C. To isolate protein, approximately 100 mg of tissue was homogenized in a RIPA lysis buffer (containing protease and phosphatase inhibitors) and centrifuged. Protein concentration was quantified using a Pierce BCA assay kit (Thermo Scientific). SDS-PAGE and Western blotting were used to analyze 30 μg of each protein sample. Band intensities were analyzed with ImageJ software, with β-actin as the loading control.
The antibody against mouse ATF6 was purchased from Imgenex. Anti-GRP78/Bip antibody was purchased from Abcam. β-actin, Bcl-2, FoxO1 and phosphorylated (p) FoxO1 were purchased from Cell Signaling.
Gene Expression: Quantitative Real Time PCR
Total RNA was isolated from liver tissue following manufacturers instructions (Qiagen RNeasy Mini Kit), quantified and reverse transcribed (Applied Biosystems). Real-time PCR for XBP1spliced, Pdia3, Dnajb9, IL-6, and albumin expression was performed on cDNA with the housekeeping gene rRNA 18S.
Hepatic Apoptosis: TUNEL
Hepatic apoptosis was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)4. To prepare samples, liver tissue was isolated, formalin-fixed, paraffin-embedded and sectioned at 5μm each. We used a DeadEnd TUNEL fluorometric system from Promega. Briefly, the DeadEnd fluorometric TUNEL system detects fragmented DNA of apoptotic cells by incorporating fluorescein-12-dUTP (green) at the 3′-OH ends of the DNA strands using the enzyme terminal deoxynucleotidyl transferase (TdT, recombinant), which forms a polymeric tail. We used propidium iodide (red) as a nuclear counterstain. We then imaged the staining with a confocal microscope (Zeiss) and quantified apoptotic cells per cm2 of tissue.
Liver Injury: AST and ALT Activity Assays
Liver injury was assessed by quantifying serum levels of alanine aminotransferase (ALT)5 and aspartate aminotransferase (AST)6. BioVision AST and ALT activity assay kits were used.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM)7. Comparisons between groups and time points were made with a two-way analysis of variance and Bonferroni correction was performed when differences between means were found (GraphPad Prism 5, version 5.0a). Differences were considered significant when p < 0.05.
RESULTS
Figure 1 confirms the genotype of JNK2−/− and wildtype mice. No mortality was observed in wildtype or JNK2−/− mice. In the majority of parameters measured, no significant differences were found between sham wildtype and sham JNK2−/− mice. Trends that were observed are described below.
Figure 1.

JNK2 knockout genotype confirmed.
ER chaperones were activated after burn injury in the liver of wildtype and JNK2−/− (Fig 2A,B). Western blots of Bip showed a slight increase and blots of ATF6 demonstrated a significant increase at day 1 in both strains (p < 0.001 wildtype burn and p < 0.05 JNK2−/− burn vs. sham). Downstream targets of ER stress, spliced XBP1 and Dnajb9, were significantly elevated 5 days post-burn in wildtype mice (p< 0.05 vs. sham) but not in JNK2−/− mice (Fig 2C,E). Pdia3 transcription was moderately–but not significantly–elevated in wildtype mice but not in JNK2−/− mice (Fig 2D). Interestingly, the basel levels of downstream ER stress markers appeared slightly reduced in sham JNK2−/− mice compared to sham wildtype mice.
Figure 2. Hepatic ER stress.

was initiated post-burn in both wildtype and JNK2 knock out (JNK2 −/−) mice. Bip (A) and activated ATF6 (B) levels were quantified 1, 3 and 5 days post-burn relative to β-actin. Representative blots from sham and burn groups at day 1 are shown above each graph. Downstream effectors of ER stress were transcribed in wildtype mice but not JNK2 knockout mice, as indicated by mRNA expression of XBP1s, Pdia3, and Dnajb9 (C–E) measured by real time PCR of day 5 liver samples. Data presented are mean ± SEM. * p < 0.05 and *** p < 0.001 vs. Sham.
We performed a peritoneal glucose tolerance test but there were no differences evident between groups (data not shown). Serum insulin was significantly elevated 5 days post burn in wildtype mice (p < 0.05, day 5 vs. sham) but not in JNK2−/− (Fig 3A). Sham JNK2−/− mice displayed a slight (non-significant) increase in insulin compared to sham wildtype mice. Serum leptin decreased in both strains at 3 and 5 days post-burn (p < 0.01, Fig 3B). Interestingly, resistin did decrease in both strains but returned to sham levels later in JNK2−/− than in wildtype mice, at day 5 instead of day 3 (Fig 3C). The other hormones measured were not significantly different between groups (data not shown).
Figure 3. Serum insulin, leptin and resistin.

(A) Serum insulin was measured by ELISA before and 1, 3, 5 days after burn injury. * p < 0.05 vs. Day 0. # p < 0.05 vs. Day 3. Serum leptin (B) and resistin (C) were measured by multiplex analysis before and 1, 3, 5 days post-burn. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. Day 0. # p < 0.05, ## p < 0.01, ### p < 0.001 vs. Day 1. ΨΨΨ p < 0.001 vs. Day 3. γ p < 0.05 vs. Wildtype. Mean ± SEM.
Inflammatory cytokine IL-6 increased (non-significantly) in the serum of both wildtype and JNK2−/− (Fig 4A). On the other hand, IL-6 gene expression in the liver peaked only in wildtype mice (p < 0.01, day 5 wildtype vs. JNK2−/− and vs. sham); IL-6 mRNA levels did not increase in JNK2−/− (Fig 4B). The other cytokines and adipokines measured did not display significant differences between groups (data not shown).
Figure 4. Inflammatory cytokines.

in the serum were measured by ELISA (A). IL-6 mRNA expression in the liver was measured by qPCR relative to 18S rRNA (B). Mean ± SEM. ** p < 0.01 vs. all other means.
TUNEL staining indicated that hepatic apoptosis was prevented in JNK2−/− (Fig 5A,B). In hepatic tissue of wildtype mice collected 5 days after burn, TUNEL positive staining increased significantly (p < 0.01 vs. sham). No TUNEL staining was detected in hepatic tissue of JNK2−/− 5 days post-burn. Consistent with this, pro-survival (anti-apoptotic) signaling in the liver was higher when JNK2 is knocked out. In wildtype mice, FoxO1 phosphorylation significantly decreased 3 days post-burn (Fig 5C). In JNK2−/−, there was no significant change in FoxO1 phosphorylation after burn injury; however, lower FoxO1 levels were observed in sham JNK2−/− than sham wildtype mice. In JNK2 knockout mice, Bcl-2 levels were increased at day 1 (p < 0.05) compared to wildtype mice (Fig 5D).
Figure 5. Hepatic apoptosis.

was reduced in JNK2 −/− mice. Apoptosis was measured by TUNEL staining (A,B). Red (propidium iodide) staining indicates all nuclei and green staining indicates TUNEL positive cells (highlighted by white arrows). ** p < 0.01 vs. Sham. * p < 0.05 vs. Wildtype at day 5. Pro-survival (anti-apoptotic signaling in the liver was measured by Western blotting (C,D). Representative blots from day 1 post-burn are shown above each graph. Mean ± SEM. ** p < 0.01 and *** p < 0.001 vs. Sham. # p < 0.05 vs. Wildtype.
Liver injury was examined by measuring AST and ALT activities in the serum before and 1 and 3 days after burn (Fig 6). Both AST and ALT activities peaked on day 1 in both strains. Knockout of JNK2 lowers day 3 AST activity and day 1 ALT activity.
Figure 6. Liver injury and function.
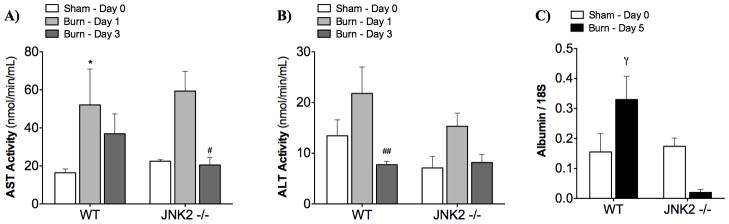
A) AST activity indicated that liver function improved more quickly in the JNK2 −/− mice. B) The peak in ALT activity was lower in JNK2 −/− mice (although not significant). C) Albumin mRNA levels were significantly lower in JNK2 −/− mice at 5 days post-burn. Mean ± SEM. * p < 0.05 vs. Day 0. # p < 0.05, ## p < 0.01 vs. Day 1. γ p < 0.05 vs. JNK2 −/−.
Liver function was evaluated by measuring albumin production in the liver. At 5 days post-burn, albumin synthesis in the liver (indicated by mRNA expression, Fig 6C) was reduced in JNK2−/− compared to wildtype mice (p < 0.05).
DISCUSSION
The overall objective of this study was to examine the role of JNK2 activation in the hypermetabolic, inflammatory and apoptotic response after a severe thermal injury. We hypothesized that JNK2 contributes predominantly to the apoptotic response after burn injury downstream of ER stress. This study indicates that JNK2 may influence hypermetabolism, does contribute to inflammation and is a central mediator of hepatic apoptosis after a severe burn.
First, we confirmed the genotype of the JNK2−/− and confirmed that JNK2 activation is downstream of chaperone activation during the hepatic unfolded protein response. Figure 1 confirms the knock out genotype, similar to that used in a study by Singh et al who were studying NAFLD (23). Figure 2 indicates increased hepatic ER stress (upstream chaperone activation and downstream effector transcription) after thermal injury in wildtype mice. The burned JNK2−/− mice also demonstrated ER chaperone activation compared to shams. Consistent with this, ER stress has previously been described upstream of JNK2 activation in obesity and ER stress studies (11, 16). In contrast, transcription of downstream effectors of ER stress (XBP1s, Pdia3, and Dnajb9) was unaffected by burn injury in the JNK2−/− mice. Previous models of the ER stress response illustrate XBP1s and JNK2 activation in parallel (28); however, our results strongly indicate that JNK2 activation is upstream of XBP1 splicing. These data suggest that the ER stress pathway is interrupted by JNK2 knockout.
Our second aim was to compare the metabolic response to a severe burn in wildtype and JNK2−/−(Figure 3). Primary mouse hepatocytes treated with palmitic acid exhibit sustained JNK activation and insulin resistance (17). In JNK2 knock out mice fed a high fat diet, obesity and insulin resistance remained (23). On the other hand, knockdown of JNK2 using antisense oligonucleotides prevented insulin resistance (23). As shown in Figure 3, burned JNK2 knockout mice fail to exhibit elevated insulin after burn injury relative to sham JNK2 knockouts, indicating that JNK2 may contribute to hyperinsulinemia in the post-burn stress response. On the other hand, insulin levels increased slightly (but not significantly) in JNK2 knockout shams compared to wildtype shams. An alternative interpretation of these data is that the JNK2 knockouts do not exhibit post-burn hyperinsulinemia because they start with higher insulin levels; this is difficult to be certain of, however, given that the elevation with JNK2−/− was not significant.
Next, we evaluated the influence of JNK2 on inflammation. After smoke inhalation, administration of a JNK inhibitor prevented the influx of inflammatory cells and cytokine release and significantly prolonged animal survival (18). In our analysis, the serum cytokine levels did not differ between wildtype and JNK2−/−; however; the peak in synthesis of IL-6 in the liver was entirely prevented after JNK2 knockout (Figure 4). These data suggest that JNK2 has a strong influence on post-burn IL-6 production in the liver but not on IL-6 production in other tissues, given that serum IL-6 does not differ between strains. This study focused on the liver post-burn but we are eager to study IL-6 production in adipose and muscle tissue in future research.
Finally, we examined hepatic apoptosis, injury, and function (Figures 5 and 6). Apoptosis has been noted after severe burn injury in several studies in the liver, heart and gastrointestinal tract (7, 8, 29). After JNK2 knock out, we observed a remarkable elimination of TUNEL-positive hepatocytes (Figure 5A,B). JNK induces apoptosis when the PI3K pathway is inhibited, either by stress or inhibitor (30). Knock out of JNK2 in a mouse model of LPS-induced toxic liver injury also resulted in significant reduction of apoptosis (31). JNK protein kinases have been implicated in deactivation of Bcl2, a mitochondrial anti-apoptotic protein (19). After burn injury, Bcl2 activation is elevated in wildtype mice but even more so in JNK2 knock out mice (Figure 5D).
The transcription factor forkhead box O 1 (FoxO1)8 is a key regulator of cell survival and mediator of insulin action. Dephosphorylation of FoxO1 activates and shuttles it from the cytoplasm to the nucleus where it up-regulates pro-apoptotic and gluconeogenic genes (32). Our data indicate that after burn injury in wildtype mice, phosphorylated FoxO1 decreased significantly compared to sham mice; however, phosphorylated FoxO1 did not change significantly after burn in JNK2 knockouts (Figure 5C). This suggests that JNK2 may be mediating apoptosis through FoxO1 dephosphorylation.
Caspase-3 activity was not affected by knockout of JNK2 in this model (data not shown). Instead, JNK2 may contribute to apoptosis through a mitochondrial pathway. We have previously described severe mitochondrial swelling and damage post-burn (33). Specifically, JNK2 may contribute to hepatocellular injury and death via mitochondrial permeability transition, as observed in an ischemia/reperfusion study by Theruvath et al (34). This would be interesting to confirm in a follow up study.
Liver structure and function is damaged after burn-induced hepatic apoptosis (7, 8). Hepatic damage has been linked to insulin resistance in the setting of streptozotocin-induced diabetes and to inflammation in the setting of acute phase inflammation (10). In our model of burn injury, both AST and ALT were elevated in serum of burned wildtype mice (Figure 6A,B). Knockout of JNK2 lowers day 3 AST activity and day 1 ALT activity, indicating reduced liver damage. JNK2 knock out in the toxic liver injury model produced the same reduction in ALT activity (31). In a mouse model of burn, we previously demonstrated that liver-secreted albumin decreased, indicating liver dysfunction (8). In this study, albumin synthesis in the liver (indicated by mRNA expression, Fig 6C) is reduced in JNK2−/− compared to wildtype mice.
A natural follow up study to this one will investigate the role of JNK1 in severe burn injury. Interestingly, Singh’s obesity study, knockout or knock down of JNK1 indicated that the two JNK isoforms have distinct effects on hepatocyte cell death and inflammation (23). Another interesting follow-up study would be to investigate the relationship between hepatic ER stress and mitochondrial dysfunction and to test a novel mitochondria-targeted antioxidant peptide, SS-31, which has been shown to reduce post-burn ER stress and apoptosis in skeletal muscle (35).
To summarize, JNK2 may influence hypermetabolism, does contribute to inflammation and is a central mediator of hepatic apoptosis after a severe burn. JNK2 may be an important therapeutic target to interrupt and prevent hepatic apoptosis and damage. If we alleviate hepatic damage, we may be able to maintain proper hepatic function, which could improve insulin sensitivity and prevent inflammation. Any effort to reduce hypermetabolism and inflammation will no doubt improve morbidity and mortality of severely burned patients.
Acknowledgments
This research was supported by an RO1 from the National Institutes of Health (GM087285-01), the CFI’s Leader’s Opportunity Fund (25407) and the Health Research Grant Program from the Physicians’ Services Incorporated Foundation. We are grateful to the SRI Genomics Core Facility for genotyping the samples.
Footnotes
ER - endoplasmic reticulum
JNK - c-jun N-terminal kinase
NAFLD – non-alcoholic fatty liver disease
TUNEL - terminal deoxyuridine nick end labeling
ALT - alanine aminotransferase
AST - aspartate aminotransferase
SEM - standard error of the mean
FoxO1 - forkhead box O 1
References
- 1.Jeschke MG, Chinkes DL, Finnerty CC, Kulp G, Suman OE, Norbury WB, Branski LK, Gauglitz GG, Mlcak RP, Herndon DN. Pathophysiologic response to severe burn injury. Ann Surg. 2008;248:387–401. doi: 10.1097/SLA.0b013e3181856241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finnerty CC, Herndon DN, Przkora R, Pereira CT, Oliveira HM, Queiroz DM, Rocha AM, Jeschke MG. Cytokine expression profile over time in severely burned pediatric patients. Shock. 2006;26:13–19. doi: 10.1097/01.shk.0000223120.26394.7d. [DOI] [PubMed] [Google Scholar]
- 3.Przkora R, Barrow RE, Jeschke MG, Suman OE, Celis M, Sanford AP, Chinkes DL, Mlcak RP, Herndon DN. Body composition changes with time in pediatric burn patients. J Trauma. 2006;60:968–971. doi: 10.1097/01.ta.0000214580.27501.19. discussion 971. [DOI] [PubMed] [Google Scholar]
- 4.Przkora R, Jeschke MG, Barrow RE, Suman OE, Meyer WJ, Finnerty CC, Sanford AP, Lee J, Chinkes DL, Mlcak RP, Herndon DN. Metabolic and hormonal changes of severely burned children receiving long-term oxandrolone treatment. Ann Surg. 2005;242:384–389. doi: 10.1097/01.sla.0000180398.70103.24. discussion 390–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biolo G, Fleming RY, Maggi SP, Wolfe RR. Transmembrane transport and intracellular kinetics of amino acids in human skeletal muscle. Am J Physiol. 1995;268:E75–84. doi: 10.1152/ajpendo.1995.268.1.E75. [DOI] [PubMed] [Google Scholar]
- 6.Jeschke MG, Micak RP, Finnerty CC, Herndon DN. Changes in liver function and size after a severe thermal injury. Shock. 2007;28:172–177. doi: 10.1097/shk.0b013e318047b9e2. [DOI] [PubMed] [Google Scholar]
- 7.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 8.Song J, Finnerty CC, Herndon DN, Boehning D, Jeschke MG. Severe burn-induced endoplasmic reticulum stress and hepatic damage in mice. Mol Med. 2009;15:316–320. doi: 10.2119/molmed.2009.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrow RE, Hawkins HK, Aarsland A, Cox R, Rosenblatt J, Barrow LN, Jeschke MG, Herndon DN. Identification of factors contributing to hepatomegaly in severely burned children. Shock. 2005;24:523–528. doi: 10.1097/01.shk.0000187981.78901.ee. [DOI] [PubMed] [Google Scholar]
- 10.Zafar M, Naeem-Ul-Hassan Naqvi S, Ahmed M, Kaimkhani ZA. Altered liver morphology and enzymes in streptozotocin induced diabetic rats. Int J Morphol. 2009;27:719–725. [Google Scholar]
- 11.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 12.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54:795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gauglitz GG, Halder S, Boehning DF, Kulp GA, Herndon DN, Barral JM, Jeschke MG. Post-Burn Hepatic Insulin Resistance Is Associated with Er Stress. Shock. 2009 doi: 10.1097/SHK.0b013e3181b2f439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 15.Deniaud A, Sharaf El Dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 16.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 17.Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci U S A. 2006;103:16454–16459. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Syrkina OL, Quinn DA, Jung W, Ouyang B, Hales CA. Inhibition of JNK activation prolongs survival after smoke inhalation from fires. Am J Physiol Lung Cell Mol Physiol. 2007;292:L984–991. doi: 10.1152/ajplung.00248.2006. [DOI] [PubMed] [Google Scholar]
- 19.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballard-Croft C, Maass DL, Sikes P, White J, Horton J. Activation of stress-responsive pathways by the sympathetic nervous system in burn trauma. Shock. 2002;18:38–45. doi: 10.1097/00024382-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Cho K, Zipkin RI, Adamson LK, Mcmurtry AL, Griffey SM, Greenhalgh DG. Differential regulation of c-jun expression in liver and lung of mice after thermal injury. Shock. 2000;14:182–186. doi: 10.1097/00024382-200014020-00018. [DOI] [PubMed] [Google Scholar]
- 22.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 25.Yang R, Wilcox DM, Haasch DL, Jung PM, Nguyen PT, Voorbach MJ, Doktor S, Brodjian S, Bush EN, Lin E, Jacobson PB, Collins CA, Landschulz KT, Trevillyan JM, Rondinone CM, Surowy TK. Liver-specific knockdown of JNK1 up-regulates proliferator-activated receptor gamma coactivator 1 beta and increases plasma triglyceride despite reduced glucose and insulin levels in diet-induced obese mice. J Biol Chem. 2007;282:22765–22774. doi: 10.1074/jbc.M700790200. [DOI] [PubMed] [Google Scholar]
- 26.Sabio G, Cavanagh-Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, Barrett T, Mora A, Kim JK, Davis RJ. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009;10:491–498. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka TA, Matsuhisa M, Yamasaki Y. Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and insulin resistance. Int J Biochem Cell Biol. 2005;37:1595–1608. doi: 10.1016/j.biocel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Lightfoot E, Jr, Horton JW, Maass DL, White DJ, Mcfarland RD, Lipsky PE. Major burn trauma in rats promotes cardiac and gastrointestinal apoptosis. Shock. 1999;11:29–34. doi: 10.1097/00024382-199901000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Molton SA, Todd DE, Cook SJ. Selective activation of the c-Jun N-terminal kinase (JNK) pathway fails to elicit Bax activation or apoptosis unless the phosphoinositide 3′-kinase (PI3K) pathway is inhibited. Oncogene. 2003;22:4690–4701. doi: 10.1038/sj.onc.1206692. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Jeschke MG, Gauglitz GG, Song J, Kulp GA, Finnerty CC, Cox RA, Barral JM, Herndon DN, Boehning D. Calcium and ER stress mediate hepatic apoptosis after burn injury. J Cell Mol Med. 2009;13:1857–1865. doi: 10.1111/j.1582-4934.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Theruvath TP, Snoddy MC, Zhong Z, Lemasters JJ. Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-Jun N-terminal kinase 2. Transplantation. 2008;85:1500–1504. doi: 10.1097/TP.0b013e31816fefb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HY, Kaneki M, Andreas J, Tompkins RG, Martyn JA. Novel mitochondria-targeted antioxidant peptide ameliorates burn-induced apoptosis and endoplasmic reticulum stress in the skeletal muscle of mice. Shock. 2011;36:580–585. doi: 10.1097/SHK.0b013e3182366872. [DOI] [PMC free article] [PubMed] [Google Scholar]