

Abstract
Mutations in GBA, the gene encoding glucocerebrosidase, the lysosomal enzyme deficient in Gaucher disease increase the risk for developing Parkinson disease. Recent research suggests a relationship between glucocerebrosidase and the Parkinson disease-related amyloid-forming protein, α-synuclein; however, the specific molecular mechanisms responsible for association remain elusive. Previously, we showed that α-synuclein and glucocerebrosidase interact selectively under lysosomal conditions, and proposed that this newly identified interaction might influence cellular levels of α-synuclein by either promoting protein degradation and/or preventing aggregation. Here, we demonstrate that membrane-bound α-synuclein interacts with glucocerebrosidase, and that this complex formation inhibits enzyme function. Using site-specific fluorescence and Förster energy transfer probes, we mapped the protein-enzyme interacting regions on unilamellar vesicles. Our data suggest that on the membrane surface, the glucocerebrosidase-α-synuclein interaction involves a larger α-synuclein region compared to that found in solution. In addition, α-synuclein acts as a mixed inhibitor with an apparent IC50 in the submicromolar range. Importantly, the membrane-bound, α-helical form of α-synuclein is necessary for inhibition. This glucocerebrosidase interaction and inhibition likely contribute to the mechanism underlying GBA-associated parkinsonism.
Keywords: Gaucher disease, Parkinson disease, fluorescence, protein-lipid interactions
1. Introduction
α-Synuclein (α-syn), the protein encoded by the SNCA gene, has been the topic of intense research since the discovery of its genetic connection to early-onset autosomal dominant Parkinson disease (PD) [1–5]. Tremendous efforts are specifically aimed at elucidating the mechanism of amyloid formation as deposits of fibrillar α-syn constitute the major proteinacious aggregates in Lewy bodies (LBs) [6, 7], the classical pathological hallmarks of PD and related synucleinopathies including dementia with LB and multiple systems atrophy. Despite the substantial work, it is still not known whether the amyloid aggregates themselves or intermediates formed during fibril formation are the cytotoxic agents. Moreover, it is unclear if alterations in normal physiological function of α-syn [8–11] also participate in pathogenesis.
As a consequence of the genetic association between Gaucher disease (GD) and PD [12–15], increasing attention is now focused on glucocerebrosidase (GCase, Fig. 1A), the lysosomal enzyme deficient in GD, and its involvement in PD. In fact, mutations in GCase are the most common genetic risk factor for parkinsonism [16]. Patients with PD carrying GCase mutations overall have an earlier onset of symptoms and more associated cognitive deficits [16]. Importantly, neuropathological studies of GD patients with synucleinopathies demonstrate the presence of mutant GCase in α-syn positive LBs [17], suggesting a potential relationship between the two proteins.
Fig 1.

α-Syn GCase interaction in the presence of vesicles. A, GCase crystal structure (PDB code 2NSX [60]). The intrinsic 12 Trp residues are shown in gray and were used as Förster energy transfer donors. Active site residues (E235 and E340) are in yellow. Loops 1–3, regions proposed to be important for substrate recognition and activity [60, 61], are highlighted in blue. For clarity, residues 286–290 were omitted. B, Schematic representation of α-syn primary sequence. The seven amphipathic imperfect repeat regions (blue) in the membrane binding domain (gray) and the acidic C-terminal region (red) are denoted. The location of Dns-labeling sites, residues 7, 40 and 57 (membrane binding domain) and residues 100 and 136 (C-terminal domain) are indicated. The same color representation is used in panels C and D. Location of peptide C23 (residue 118–140) is also indicated. C, Normalized change in mean residue ellipticity Δ[θ]) as a function of lipid-to-protein ratio (50 mM MES, 25 mM NaCl, pH 5.5, [WT α-syn] = 5 μM). Error bars indicate standard deviations from two independent measurements (top left). POPC/POPS vesicles induced secondary structure in α-syn (top left, inset). GCase titration curves for Dns136-α-syn (top right), Dns100-α-syn (bottom left), and Dns136-C23 peptide (bottom right). The titration curves were obtained from mean spectral wavelength (<λ>) in 50 mM MES, 25 mM NaCl at pH 5.5 in the absence (●) or presence of 0.9 mM POPC/POPS vesicles (■). Error bars indicate standard deviations from two replicate samples. Fits are shown as lines. D, Förster energy transfer from GCase to DnsX-α-syn. Measurements were performed in the presence of 2 μM GCase (Trp donor, excited at 295 nm) and 5 μM DnsX-α-syn (Dns acceptor), either in the absence or presence of vesicles at pH 5.5 (colored according to panel B). Fluorescence spectra of GCase and DnsX-α-syn alone are shown in gray and green, respectively. WT syn was used as a negative control.
Furthermore, results from mouse studies indicate that GCase deficiency can affect α-syn clearance and cause abnormal protein accumulation [18–20]. Neuronal overexpression of wild-type (WT) GCase appears to ameliorate α-syn pathology, suggesting GCase plays a role in α-syn degradation [19]. These data support the hypothesis that α-syn turnover is related to GCase levels and/or activity.
A recently proposed molecular mechanism by Mazzulli et al. entails a positive feedback loop [21]. Enzyme inactivity leads to the buildup of glucosylceramide, which in turn stabilizes the formation of α-syn oligomeric species, and the resulting abnormal α-syn accumulation further reduces GCase trafficking, leading to more glucosylceramide accumulation and α-syn aggregation.
In our own work, we identified a specific physical interaction between α-syn and GCase under lysosomal solution conditions, mapping the interaction site to the C-terminal residues of α-syn [22]. Notably, this complex formation is modulated by a common GD-related mutation, N370S. As lysosomes play an important role in α-syn degradation [23–25], we propose an alternate mechanism to explain the connection between the two proteins. We hypothesize that the interaction of α-syn with WT enzyme could have a beneficial effect by promoting lysosomal degradation of α-syn, or by inhibiting adverse α-syn aggregation. Thus, mutations that decrease the amount of enzyme reaching the lysosome or weaken the interaction, as seen with the N370S mutant, could reduce this benefit and increase the probability of α-syn pathology.
Although we suggested a hypothetical interaction model of the α-syn–GCase complex on the membrane surface of intralysosomal vesicles, experimental evidence was needed. Moreover, since the membrane is important for both GCase activity [26–31] and α-syn conformation [32–37], studies on protein-lipid interactions are particularly pertinent. While the effects of GCase on α-syn homeostasis are the subject of considerable work, a role for α-syn in enzyme function has not been established. Such information could have further implications and indicate other mechanisms responsible for the increased PD risk. Towards these goals, we examined the effect of membranes on the α-syn-GCase interaction and assessed how α-syn influences GCase activity.
2. Material and methods
2.1 Protein expression and sample preparation
The WT human α-syn plasmid (pRK172) was provided by M. Goedert (Medical Council Research Laboratory of Molecular Biology, Cambridge, UK) [38]. Single cysteine mutants (G7C, V40C, E57C, L100C and Y136C) were generated using the Quick-Change site-directed mutagenesis kit (Stratagene) and verified by DNA sequencing. α-Syn variants were prepared as previously described [39]. Taliglucerase alfa (recombinant GCase) was obtained from Protalix Biotherapeutics Corp. (Carmiel, Israel). Myozyme (recombinant acid α-glucosidase (α-glu A), Genzyme Corp. Cambridge, MA), was collected from remnants from patient infusions. β- and γ-synucleins were purchased and used as received from rPeptide (Bogart, GA). Recombinant apolipoprotein C-III was expressed and purified as described [40]. The plasmid was provided by Philippa Talmud (University College London Medical School, London, UK) [41]. Synthetic α-syn peptides (residues 1–15 containing W4 (N15) and residues 118–140 containing C136 (C23)) were purchased from AnaSpec, Inc. (Fremont, CA) and Bio-Synthesis, Inc. (Lewisville, TX), respectively. All protein samples were prepared in appropriate buffer (50 mM 2-(N-morpholino)ethanesulfonic acid (MES), 25 mM NaCl, pH 5.5 or 50 mM sodium phosphate, 25 mM NaCl, pH 7.4), buffer exchanged, and concentrated to desired concentration using Amicon Ultra YM3 and YM30 centrifugal filters (Millipore) for α-syn and GCase, respectively.
2.2 Protein and peptide labeling
Dansyl (Dns)-labeled proteins were prepared and purified as previously described [22]. In addition, C23 was labeled using a similar procedure with minor modifications. Peptide concentration was determined using a molar absorptivity (280 nm) = 2680 M−1 cm−1. The labeling reaction time was 2 h. Excess Dns was removed with a Hi Trap desalting column (GE Healthcare). Molecular weights of Dns-labeled proteins and peptides were confirmed by ESI-MS (Biochemistry Core Facility, NHLBI). Concentrations of labeled-protein and C23 peptide were determined using a molar absorptivity (336 nm) = 5700 M−1 cm−1 (Dns).
2.3 Lipid vesicles
Phospholipids: 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and bis(monooleoylglycero)phosphate, (BMP) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Glucocerebroside (GluCer) was purchased from Matreya, LLC (Pleasant Gap, PA). Phospholipid vesicles were made with the following composition: POPC/BMP (1:1 molar ratio), POPC/POPS (1:1 molar ratio), and POPC/POPS/GluCer (0.8:1:0.2 molar ratio). Chloroform solutions of lipid mixtures were dried under a nitrogen stream for 5–10 min followed by vacuum desiccation for 45 min. Dried samples were resuspended and solubilized in 50 mM MES, 25 mM NaCl, pH 5.5 (fluorescence experiments) or in 50 mM sodium acetate, 142 mM K2HPO4, pH 5.5 (activity assays) via bath sonication for 5 min. Vesicles were spontaneously formed by ultrasonication in a water bath (45 minutes, 50% duty cycle, microtip limit, Branson 450 Sonifier). Vesicle solutions were equilibrated overnight (14–24 h) at 37 °C. In the case of POPS/POPC/GluCer vesicles, lipid mixtures were sonicated and equilibrated overnight at 65–70 °C. Vesicles were freshly prepared for each experiment. An average vesicle hydrodynamic radius (POPC/POPS) was determined to be 45 nm by dynamic light scattering (Wyatt DynaPro NanoStar; fifty 5 s acquisitions, 25 °C).
2.4 Fluorescence spectroscopy
Fluorescence spectra were measured using a Fluorolog-3 spectrofluorimeter (HORIBA Jobin Yvon, Inc.) and conducted at 25 °C using a temperature controlled cuvette holder. GCase titrations were performed in buffer (pH 5.5) in a fluorometer cell with inner dimension of 3 × 3 × 25 mm. Concentrated GCase stocks were serially diluted (from 25 to 0.5 μM) while maintaining the concentration of DnsX-α-syn at 2 μM. A lipid-to-protein (L/P) of 450 (0.9 mM) was used. Dns-α-syn was excited at 340 nm and emission was monitored from 405 to 650 nm. Intrinsic GCase Trp fluorescence was excited at 295 nm and monitored from 300 to 570 nm. Energy transfer is considered to be significant when Trp was quenched at least 10%, with a concomitant increase in Dns quantum yield (QY).
2.5 Circular dichroism spectroscopy
Measurements were collected on a Jasco J-715 spectropolarimeter (1 nm steps, 1 nm bandwidth, 0.5 sec integration time, 50 nm/min, 25°C).
2.6 Activity assays
GCase activity was performed in 384-well flat dark bottom black polystyrene microplates (Product #3712, Corning, NY). Fluorogenic substrate, 4-methylumbelliferyl β-D-glucopyranoside (4MU-β-glu, Sigma Aldrich, St. Louis, MO) and lipid vesicles (POPC/POPS) were mixed in assay buffer (50 mM sodium acetate, 142 mM K2HPO4, pH 5.5) with GCase added last to a final volume of 15 μl. The concentration of each component varied in different experiments as specified.
Typically, the concentration of GCase was 50 nM (except when otherwise noted) in the presence of 350 μM POPS/POPC and 1 mM 4MU-β-Glu. The plates were incubated for 10–15 min at 37 °C, shaking at 550 rpm. To stop the reaction and increase the pH, 15 μl of stop solution (1 M NaOH, 1 M glycine) was added to each well. Emission was recorded (excitation and emission wavelengths were 355 and 460 nm, respectively) using an EnVision Multilabel Plate Reader (PerkinElmer, Waltham, MA). GCase activity was also measured in the presence of 3 mM sodium taurocholate (NaTc). Similarly, α-glu A (50 nM) activity was measured in the presence of 350 μM POPC/POPS and 1 mM 4MU-α-Glu.
GCase activity was assayed using its natural substrate and the Amplex Red Glucose assay kit (Catalog #A22189, Invitrogen, Carlsbad, CA) as previously described [42]. This kit detects the release of glucose from GluCer incorporated into POPC/POPS vesicles. All activity data represent average results from two independent experiments each with two replicates.
2.7 Data analysis
Total helical content was calculated as previously described [37]. The mean spectral wavelength, <λ>, was calculated and fit to a simple two-state model to obtain apparent dissociation constant (Kd(App)) as previously described using IGOR Pro 6.01 (Wavemetrics) [22]. Equations for sigmoidal dose-response and nonlinear regression fitting were used to obtain values for IC50 (GraphPad Prism 5.0). To determine the mode of inhibition by α-syn, enzyme velocity vs. substrate concentration were fit using nonlinear regression to different equations describing competitive, uncompetitive, noncompetitive and mixed inhibition (GraphPad Prism 5.0). These models were compared based on P values and goodness of fit (R2). In addition, Lineweaver-Burk plots were analyzed.
3. Results
3.1 Fluorescent probes of α–syn GCase interaction on the membrane
Based on our prior work [22], we postulated that N-terminal α-syn is membrane associated on the surface of intralysosomal vesicles, forming an α-helical structure via its seven imperfect amphipathic 11 aa repeats (Fig. 1B), while its C-terminal tail would interact with GCase. To substantiate this hypothesis, we employed Dns-labeled α-syn at two C-terminal sites, distal (Dns100) and internal (Dns136) to the previously identified region of interaction (residues 118–137). Dns is an excellent probe of protein-protein interaction because the fluorophore is exquisitely responsive to its local environment [39]. Since anionic lipids are important for both the α-syn membrane interaction [43] and GCase activity [27, 29–31], and PS specifically enhances activity, we chose POPC/POPS vesicles as a membrane model.
Vesicle binding and α-syn secondary structural transformation were verified using circular dichroism (CD) spectroscopy. As expected [37], the spectral characteristics exhibited typical negative maxima for α-helical conformation upon the addition of vesicles (Fig. 1C top left inset). A midpoint transition was observed at a lipid-to-protein ratio (L/P) ~ 65 ± 5 approaching saturation at L/P ~ 200 (Fig. 1C top left) with a maximal total helical content of ~ 66%.
We examined the interaction between membrane-bound α-syn (L/P = 450) and GCase. For both Dns sites (Dns100 and Dns136), the fluorescence spectra are similar in the absence and presence of vesicles consistent with an aqueous environment, confirming that the C-terminal tail remains in solution. However, upon the addition of GCase, Dns emissive properties become site- and lipid-dependent (Fig. 1C). In general, the Dns spectrum shifts to a higher energy and increases in intensity upon enzyme interaction, indicating a change to a more hydrophobic surrounding and a reduction in fluorophore mobility.
The titration curve obtained for Dns136 in buffer is nearly indistinguishable from our previous data despite the use of different sources of recombinantly produced human GCase (Fig. 1C top right). In the presence of vesicles, spectral changes are moderately attenuated. At binding saturation, the mean wavelength change Δ<λ>) is smaller than that found in buffer, indicating that the local environment of Dns136 is slightly more exposed to solvent in the complex on the membrane surface. A larger apparent dissociation constant (Kd(app)) is seen with membrane-bound compared to free α-syn in solution (8(1) vs. 1.0(1) μM).
The trend for Dns100 is opposite to that seen with Dns136, where binding is stronger and spectral changes are larger for the membrane-bound form (Fig. 1C bottom left). In accord with our previous nuclear magnetic resonance (NMR) data, residue 100 exhibits minimal interaction with GCase in solution (Kd(app) ~ 40(1) μM). However, in the presence of vesicles, a strong affinity (Kd(app) ~ 1.0(2) μM) is obtained, indicating that residue 100 has become closer to the interacting site.
3.2 Binding of α-syn to the membrane impacts its interaction with GCase
To elucidate the role of the α-syn membrane binding domain on complex formation, we used a synthetic peptide containing residues 118–140 (C23) with a cysteine at residue 136 derivatized with Dns (DnsC23). In buffer, DnsC23 shows a binding affinity similar to the full length protein (Kd(app) ~ 2.0(2) μM, Fig. 1C bottom right) demonstrating that this region alone is sufficient to bind GCase and that the first 117 residues are minimally involved, in agreement with our Dns7 fluorescence and NMR data [22]. In contrast, binding of DnsC23 to GCase is greatly perturbed in the presence of vesicles (Fig. 1C bottom right). Despite its affinity for GCase in solution, the C-terminal peptide alone is unable to interact with the enzyme on the membrane surface, and clearly the remainder of the polypeptide is needed.
3.3 Förster energy transfer from GCase Trp to Dns-α-syn
To map complex formation from the perspective of GCase, we exploited its 12 intrinsic Trp residues as Förster energy transfer donors (Fig. 1A) and Dns-labeled α-syn as the acceptor (Fig. 1B). We prepared several Dns-variants at different sites spanning the α-syn sequence including residues in the membrane-binding (7, 40, and 57) and putative GCase-interacting (100 and 136) domains. Each Dns-variant was confirmed to have vesicle binding properties similar to the WT protein by CD spectroscopy (Fig. S1).
The Förster distance for the Trp-Dns pair is 22 Å. Energy transfer should occur when the Dns fluorophore is within a distance of 11–33 Å from any of the 12 Trp residues resulting in Trp fluorescence quenching and Dns fluorescence enhancement. To validate our method, we reexamined the protein-enzyme interaction in solution (Fig. 1D). WT α-syn was used as a negative control. The contribution from direct excitation (295 nm) of Dns is also shown for all variants in the absence of GCase (Fig. 1D and S2).
As anticipated, significant energy transfer is only observed for Dns100 and Dns136 with Dns136 exhibiting the higher QY increase (Fig. 1D and S2A). We established that the enhancement observed is due to energy transfer, as it is greater than would be expected from changes due to interaction with GCase alone (Fig. S2A). These data clearly show that the C-terminal region is the site of interaction, as it is in close proximity to GCase. Consistent with this, we observed the highest <λ> shift for Dns136 (~6 nm) followed by Dns100 (~2 nm), whereas negligible changes were found for Dns7, Dns40, and Dns57 (Table S1).
Having established the utility of energy transfer to map the protein-enzyme interaction in solution, we carried out similar measurements in the presence of vesicles. In accordance with the fluorescence data (Fig. 1C), we observed greater enhancement for Dns100 than for Dns136 (Fig. 1D and S2B). Interestingly, energy transfer also occurred for Dns7, Dns40, and Dns57 (Fig. 1D and S2B). Unlike in solution, these residues now are in closer proximity to GCase on the vesicle. By comparing changes due to selective Dns excitation (340 vs. 295 nm), we ascertained that the Dns fluorescence enhancement observed is due to energy transfer from GCase, and not from changes due to enzyme interaction. The QY changes caused by Dns excitation (340 nm) are significantly smaller than the observed intensity increases (Fig. S2B).
We note that Dns57 undergoes a spectral shift, while Dns7 and Dns40 are essentially unchanged, indicating that Dns57 directly interacts with GCase (Table S1). Though residues in the membrane binding domain are lipid sensitive (Fig. S3), the observed spectral changes can be directly attributed to inter-protein interactions or conformational changes upon enzyme binding, as spectra comparisons are made from membrane-bound samples. Similar to the behavior found in solution, this complex formation in the presence of vesicles is pH dependent and selective for conditions typical of the lysosome (Fig. S4).
3.4 α-Syn inhibits GCase activity
To determine the effect of α-syn on GCase catalytic activity, we adapted the widely used 4MU-β-Glu assay to include phospholipid vesicles as mimics of intralysosomal vesicles [31]. First, we obtained Km values for 4MU-β-Glu (0.6(1) mM) and POPC/POPS vesicles (0.1(1) mM) to optimize the assay (Fig. S5). Based on these results, GCase activity was measured using 1 mM 4MU-β-Glu and 350 μM POPC/POPS vesicles.
Upon addition of α-syn, inhibition was observed with a maximal inhibition of 76 ± 10% and an estimated IC50 of 215 nM (95% C.I. = 140 – 320 nM, Fig. 2A top). In contrast, GCase activity was unperturbed by the presence of C23 up to 10 μM (Fig. S6). Since this peptide only interacts with GCase in solution and not on the membrane surface (vide supra), inhibition is attributable to direct protein-protein interaction on the membrane. As a negative control, we used a synthetic peptide containing the first 15 N-terminal residues derived from α-syn (N15) that binds to the membrane [44], but does not interact with GCase (Fig. S6). As an additional control, we used another membrane binding protein, apolipoprotein C-III and also found no changes to GCase activity (Fig. S6).
Fig 2.

α-Syn inhibits GCase activity. A, Relative GCase activity as a function of added α-, β-, and γ-synuclein in the presence of 350 μM POPC/POPS vesicles ([GCase] = 50 nM and [4MU-β-glu] = 1 mM, IC50 ~215, 850, and 685 nM, respectively). Top Inset, Effect of α-syn on GluCer turnover by GCase ([GCase] = 50 nM and 350 μM POPC/POPS/GluCer vesicles). Bottom, Relative GCase activity as a function of added α-syn in the presence of 350 μM POPC/BMP vesicles ([GCase] = 50 nM and [4MU-β-glu] = 1 mM, IC50 ~230 nM). B, Comparison of amino acid sequences of α-, β-, and γ-synuclein. N-terminal residues (1–44) were omitted as the sequences are nearly identical (95.5 and 84% for β and γ-syn, respectively). Differences are colored in red and the residue is colored yellow if the side chain change retains similar charge. Alignment was made using the program ALIGN available at www.uniprot.org/align/
To examine inhibition specificity, we investigated the other two members of the synuclein family [45], β- and γ-syn (Fig. 2A top). All synucleins have a highly conserved N-terminal domain (residues 1–44: 95.5 and 84% sequence identity for β-syn and γ-syn, respectively) with variable central portion and C-terminus (overall 59 and 50% sequence identity for β-syn and γ-syn, respectively, Fig. 2B). Both β-syn and γ-syn exhibit reduced inhibition with comparable IC50 of 850 and 685 nM, respectively. Lastly, we verified that GCase inhibition occurs with its natural substrate by incorporating glucosylceramide (10% mol ratio) into lipid vesicles. It is clear that the addition of α-syn greatly diminishes GCase activity on its natural substrate (Fig. 2A, top inset).
3.4.1 Effect of phospholipid composition
To test the effect of phospholipid composition, BMP, another negatively charged lipid, was chosen because it constitutes a major phospholipid component found in intralysosomal vesicles [27, 46, 47]. Nearly identical behavior was observed (Fig. 2A bottom). Finally, we demonstrated enzyme specificity using another lysosomal hydrolase, α-glu A (Figure S6). As expected, α-syn had no effect on α-glu A activity since they do not interact (Figure S7). These results clearly indicate that α-syn selectively inhibits GCase activity, independent of substrate and lipid composition.
3.4.2 Role of α-syn structure in GCase inhibition
We substituted POPC/POPS vesicles with NaTc, a detergent used to emulsify the substrate 4MU-β-Glu and shown to enhance GCase catalytic activity [48, 49]. Interestingly, the inhibitory effect of α-syn is completely abolished in the presence of NaTc as compared to POPC/POPS (Fig. 3A). While α-syn still binds to GCase (Kd (app) ~11(1) μM, Fig. 3B) in the presence of NaTc, α-syn does not develop any α-helical secondary structure and remains disordered (Fig. 3B inset). These results suggest that the specific conformational changes on the vesicle surface and α-syn–GCase interaction are important for inhibition (Fig. 3C).
Fig 3.

Effect of α-syn secondary structure on GCase activity. A, Comparison of GCase activity in the presence of NaTc (black) and POPC/POPS vesicles (gray) upon the addition of α-syn. B, GCase titration curve obtained from mean spectral wavelength (<λ>) of Dns136-α-Syn at pH 5.5 in the presence of 3 mM NaTc. Fit is shown as a line. Inset, Circular dichroism spectra of α-syn in the presence of NaTc (black) and POPC/POPS vesicles (gray). C, A schematic diagram depicting that while α-syn interacts with GCase in the presence of both NaTc and POPC/POPS vesicles, only the α-helical vesicle-bound α-syn inhibits enzyme activity.
3.4.3 Mode of inhibition
We characterized the inhibitory effect of α-syn in detail by measuring the GCase reaction velocity as a function of substrate concentration (0 – 4 mM) at several protein concentrations (0, 0.15, 0.3, and 1.25 μM, Fig. 4A). By fitting the data, we find that α-syn acts as a mixed inhibitor, increasing and decreasing apparent Km and Vmax, respectively (Fig. 4B). Similar results were obtained by Lineweaver-Burk analysis (Fig. S8).
Fig 4.
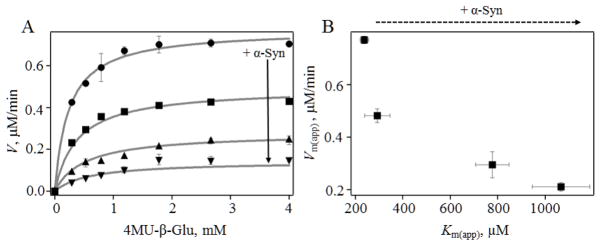
Mechanism of inhibition by α-syn. A, GCase (10 nM) reaction velocity as a function of added 4MU-β-glu in the presence of 0 μM (●), 0.15 μM (■), 0.3 μM (▲) and 1.25 μM (▼) α-syn. Fits to Michaelis-Menten equation for mixed inhibition are shown as lines. B, Plot of apparent Vm(app) vs. Km(app) extracted from fits.
4. Discussion
4.1 α–Syn GCase interaction on the membrane
In prior work, we showed that the interaction between α-syn and GCase is site-specific and occurs only in acidic solutions. We now extend our study to consider how membranes would modulate this complex formation. Lipids function not only as the substrate for GCase in intralysosomal vesicles, but also a surface for α-syn conformation alteration. For the first time, we demonstrate that the two proteins associate both on and off the membrane, with comparable affinity, with Kd(app) in the micromolar range.
Binding of α-syn to the membrane is critical for complex formation. In the absence of the N-terminal membrane binding domain, α-syn and GCase do not associate on the membrane, whereas in solution, this complex stays intact. Due to the likelihood of electrostatic repulsion between the negatively charged membrane surface and the acidic C-terminal tail, we suggest that at least some of the N-terminal membrane-binding residues, 1–95, are required to anchor α-syn to the membrane, thereby promoting and/or stabilizing its binding to GCase at the C-terminus.
In the presence of vesicles, the GCase interaction site on α-syn appears to shift to include residues N-terminal to the buffer binding location, supported by the observed differences between Dns100 and Dns136. Moreover, we interpret the fluorescence enhancement exhibited by N-terminal Dns probes (7, 40, and 57) upon GCase binding as an expansion of the interaction interface on α-syn. These fluorescence results clearly suggest that the structure of the membrane-bound protein-enzyme complex is altered from that previously characterized in buffer solution alone.
Although explicit distances could not be obtained due to the presence of multiple donors, the Förster energy transfer data (i.e. intensity enhancement) can be used to glean information regarding proximity between different Dns sites and GCase. Taking our previous data into consideration together with other factors including the orientation of α-syn and GCase at the membrane interface, we propose a new schematic model for the membrane-bound α–syn GCase complex (Fig. 5).
Fig 5.
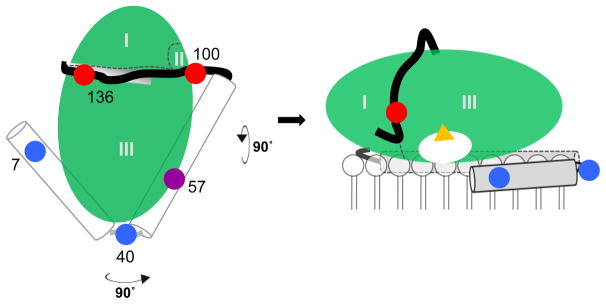
Schematic model of the α-syn–GCase complex at the membrane interface. Left, View from the top. Membrane binding and α-helical domains of α-syn (residues 1–37 and 51–92) are represented as cylinders and connected by a flexible linker. The disordered C-terminal tail is drawn black. This open horseshoe structure is adapted from the sodium dodecyl sulfate micelle-bound structure (PDB code 1XQ8 [53]). The five fluorescent probes are colored coded to indicate membrane-binding (blue: Dns7 and Dns40), GCase-binding (red: Dns100 and Dns136) and membrane-and-GCase binding (purple: Dns57). In this model, α-syn residues 118–140 reside in the groove between domains I and III of GCase (See Fig. S9 for more detailed renderings). Right, View from the side. The GCase active site is represented by yellow triangle. The relative orientations (tilt) of the helices on the bilayer need not be identical since there is a flexible hinge connecting them.
GCase is depicted as an ellipsoid (Fig. 5 right) with the active site pocket (shown as a white oval) facing the membrane. The concave surface near loop 1, the proposed location of enzyme-associating C-terminal α-syn residues [22], is also proximal to the membrane surface (Fig. S9). Because there would be strong electrostatic repulsion between the phospholipid headgroups and carboxyl sidechains on α-syn, we suggest that the C-terminal tail is pulled away from the lipid surface, moving towards the aqueous interface along the groove (white rectangle, Fig. 5 left). Given the disordered nature of the C-terminal tail, this region can adopt a flexible conformation and wrap around GCase.
To orient the rest of the α-syn polypeptide, we considered the two structurally characterized membrane conformations adopted by the first 100 residues, the bent or horseshoe [50–53] and the extended helix [54–56]. The two structures are not mutually exclusive and it is thought that they can interchange [57, 58]. Based on our data, we suggest that α-syn is more likely to adopt an open horseshoe helix (Fig. 5). In the horseshoe helix model, there are two, anti-parallel helices comprised of residues 3–37 (N-helix) and 45–92 (C-helix), with a short, 7-residue linker in between [53]. In accord with this model, probes in the helical regions, Dns7, Dns40, and Dns57, are all membrane sensitive Δ<λ>), whereas the C-terminal probes are unperturbed by the presence of phospholipids.
In the presence of GCase, Dns7 and Dns40 exhibit little to no changes, Dns57 is moderately affected, and Dns100 and Dns136 are dramatically altered. From this, we infer that the N-terminal membrane interaction is maintained, and that the GCase binding site has become larger, potentially involving residue 57. The observed energy transfer between GCase and N-terminal residues 7 and 40 indicates that these sites are within distances of 11 to 33 Å, but not directly interacting with GCase. If α-syn is in a fully extended helix, then we would not observe energy transfer from GCase to α-syn upon binding, as the distance for Dns7 would be at least 55 Å from any nearest Trp. However, since we do not have explicit distance constraints, many other models are possible. Nevertheless, this simplified and low resolution view can be used to begin to understand why membrane bound α-helical α-syn inhibits GCase activity.
4.2 α-Syn is a mixed inhibitor
Despite the current interest in defining the role of GCase in PD, our study is the first to investigate whether α-syn has a direct effect on GCase function. We show that α-syn is a potent GCase inhibitor only when it adopts an α-helical conformation. The observed mixed mode of inhibition suggests that at the membrane interface, α-syn binding influences both substrate accessibility and turnover.
It is plausible that on the one hand, direct association by the C-terminus can alter enzyme conformation and lead to inhibition by α-syn. However, given that in the NaTc assay activity is unchanged by α-syn, we suggest that other regions of α-syn maybe hindering or altering either GCase-membrane interaction and/or conformational changes necessary for function. Inhibition may also be as a result of steric hindrance by α-syn, occluding substrate binding and/or product release. Alternatively, α-syn-lipid interaction can cause structural perturbation of the bilayer [44], which could change the local lipid organization and influence enzyme activity. As residue 57 is the only site found thus far to interact with both the membrane and GCase, we postulate that the C-helix is involved. The importance of central and C-terminal domains is also highlighted by the stronger inhibitory effect by α-syn compared to both β-syn and γ-syn. The only significant differences in amino acid sequences are found within residues 59–140, particularly in the deletion of residues 74–84 in β-syn and differences observed in the C-terminus in γ-syn. Nevertheless, future studies are necessary to pinpoint the specific residues responsible for inhibition.
4.3 Implications for disease association
This study supports the notion that GCase deficiency can contribute to the pathogenesis of the synucleinopathies. This newly identified inhibition of GCase activity by the membrane-bound protein-enzyme complex offers a parallel explanation to the bidirectional model [21]: mutations that reduce GCase levels or weaken α-syn interaction lead to reduced lysosomal degradation, and result in an accumulation of α-syn and substrate, which then mediates the interaction of the protein-enzyme complex on vesicles, leading to further loss of activity (Fig. 6). Since the helical content and binding to vesicles of α-syn are dependent on lipid-to-protein stoichiometry [43], we postulate that the accumulation of intralysosomal vesicles can be another important link that affects α-syn GCase vesicle interaction and loss of activity. Thus, the observed interplay between the α-syn GCase association and activity can be another potential self-perpetuating mechanism connecting enzyme deficiency and the accumulation of α-syn and substrate. A recent study found evidence that GCase activity is significantly reduced in brain autopsy tissue from patients with sporadic PD [59], which supports the hypothesis that increased α-syn levels can cause GCase deficiency directly, even in the absence of mutant enzyme.
Fig 6.
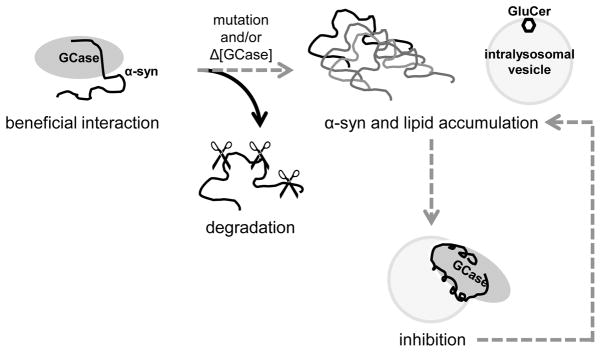
Molecular links between glucocerebrosidase and α-synuclein. In normal functional lysosomes, wild-type GCase interacts with α-syn, facilitating degradation. In some cases, mutant and/or decreased levels of wild-type GCase increase the probability of α-syn accumulation, as well as the buildup of glucocerebroside (GluCer). The inhibitory effect of α-syn may lead to a secondary loss of enzyme activity, which would further compound the problem.
This link between membrane-bound α-syn and GCase could be one of many possible mechanisms that connect GD and PD. However, since only a minority of GD patients or carriers develops PD, other pathological factors are likely involved. Nevertheless, it is now possible to search for proteins and other molecules that could modulate this α-syn GCase interaction and to evaluate their effects on enzyme activity. Once identified, they will be tested in cellular models to explore their impact on GCase activity, α-syn aggregation, and cell health.
Supplementary Material
Highlights.
α-Synuclein and glucocerebrosidase interact at the membrane interface
Complex formation is selective at a lysosomal pH
Membrane-bound, α-helical α-synuclein inhibits glucocerebrosidase function
A new molecular connection between Parkinson and Gaucher diseases is proposed
Acknowledgments
This work was supported by the Intramural Research Program at the National Institutes of Health, National Heart, Lung, and Blood Institute and National Human Genome Research Institute. The recombinant glucocerebrosidase used was a gift from Protalix Biotherapeutics, Carmiel, Israel. We thank Duck-Yeon Lee (Biochemistry Core Facility, NHBLI) for technical assistance with mass spectrometry and Grzegorz Piszczek (Biophysics Core Facility, NHLBI) for use of CD spectrometer and DLS instrument. We also are grateful to Ehud Goldin and Jim Gruschus for many helpful discussions.
Abbreviations
- α-syn
α-synuclein
- PD
Parkinson disease
- LBs
Lewy bodies
- GD
Gaucher disease
- GCase
glucocerebrosidase
- WT
wild-type
- α-glu A
acid α-glucosidase
- N15
α-syn peptide residues 1–15
- C23
α-syn peptide residues 118–140
- MES
2-(N-morpholino)ethanesulfonic acid
- Dns
dansyl
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- BMP
bis(monooleoylglycero)phosphate
- GluCer
glucocerebroside
- QY
quantum yield
- 4MU-β-glu
4-methylumbelliferyl β-D-glucopyranoside
- <λ>
mean spectral wavelength
- Kd(App)
apparent dissociation constant
- CD
circular dichroism
- NMR
nuclear magnetic resonance
- NaTc
sodium taurocholate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, DiIorio G, Golbe LI, Nussbaum RL. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 2.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nature Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 3.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Tortosa EG, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 4.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. α-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841–841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 5.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 6.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 7.Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett. 1997;239:45–48. doi: 10.1016/s0304-3940(97)00891-4. [DOI] [PubMed] [Google Scholar]
- 8.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes snare-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng FR, Vivacqua G, Yu S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J Chem Neuroanat. 2011;42:242–248. doi: 10.1016/j.jchemneu.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Norris EH, Giasson BI, Lee VMY. α-Synuclein: Normal function and role in neurodegenerative diseases. Curr Top Dev Biol. 2004;60:17–54. doi: 10.1016/S0070-2153(04)60002-0. [DOI] [PubMed] [Google Scholar]
- 11.Uversky VN, Eliezer D. Biophysics of parkinson’s disease: Structure and aggregation of α-synuclein. Curr Protein Pept Sci. 2009;10:483–499. doi: 10.2174/138920309789351921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bras J, Singleton A, Cookson MR, Hardy J. Emerging pathways in genetic Parkinson’s disease: Potential role of ceramide metabolism in Lewy body disease. FEBS J. 2008;275:5767–5773. doi: 10.1111/j.1742-4658.2008.06709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sidransky E. Gaucher disease and parkinsonism. Mol Genet Metab. 2005;84:302–304. doi: 10.1016/j.ymgme.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Velayati A, Yu WH, Sidransky E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep. 2010;10:190–198. doi: 10.1007/s11910-010-0102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med. 2011;17:485–493. doi: 10.1016/j.molmed.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010;120:641–649. doi: 10.1007/s00401-010-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, Kolodziej P, Kahn I, Saftig P, Woulfe J, Rochet JC, Glicksman MA, Cheng SH, Grabowski GA, Shihabuddin LS, Schlossmacher MG. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol. 2011;69:940–953. doi: 10.1002/ana.22400. [DOI] [PubMed] [Google Scholar]
- 19.Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, Passini MA, Grabowski GA, Schlossmacher MG, Sidman RL, Cheng SH, Shihabuddin LS. CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A. 2011;108:12101–12106. doi: 10.1073/pnas.1108197108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu YH, Sun Y, Ran H, Quinn B, Witte D, Grabowski GA. Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol Genet Metab. 2011;102:436–447. doi: 10.1016/j.ymgme.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yap TL, Gruschus JM, Velayati A, Westbroek W, Goldin E, Moaven N, Sidransky E, Lee JC. α-Synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem. 2011;286:28080–28088. doi: 10.1074/jbc.M111.237859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of α-synuclein in vivo. J Biol Chem. 2010;285:13621–13629. doi: 10.1074/jbc.M109.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 25.Pan TH, Kondo S, Le WD, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- 26.Kolter T, Sandhoff K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010;584:1700–1712. doi: 10.1016/j.febslet.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 27.Sandhoff K, Kolter T. Biochemistry of glycosphingolipid degradation. Clin Chim Acta. 1997;266:51–61. doi: 10.1016/s0009-8981(97)00166-6. [DOI] [PubMed] [Google Scholar]
- 28.Vaccaro AM, Tatti M, Ciaffoni F, Salvioli R, Barca A, Scerch C. Effect of saposins a and c on the enzymatic hydrolysis of liposomal glucosylceramide. J Biol Chem. 1997;272:16862–16867. doi: 10.1074/jbc.272.27.16862. [DOI] [PubMed] [Google Scholar]
- 29.Morimoto S, Kishimoto Y, Tomich J, Weiler S, Ohashi T, Barranger JA, Kretz KA, O’Brien JS. Interaction of saposins, acidic lipids, and glucosylceramidase. J Biol Chem. 1990;265:1933–1937. [PubMed] [Google Scholar]
- 30.Vaccaro AM, Tatti M, Ciaffoni F, Salvioli R, Maras B, Barca A. Function of saposin c in the reconstitution of glucosylceramidase by phosphatidylserine liposomes. FEBS Lett. 1993;336:159–162. doi: 10.1016/0014-5793(93)81631-9. [DOI] [PubMed] [Google Scholar]
- 31.Wilkening G, Linke T, Sandhoff K. Lysosomal degradation on vesicular membrane surfaces - enhanced glucosylceramide degradation by lysosomal anionic lipids and activators. J Biol Chem. 1998;273:30271–30278. doi: 10.1074/jbc.273.46.30271. [DOI] [PubMed] [Google Scholar]
- 32.Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. α-Synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 33.Bodner CR, Dobson CM, Bax A. Multiple tight phospholipid-binding modes of α-synuclein revealed by solution NMR spectroscopy. J Mol Biol. 2009;390:775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci U S A. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JC, Langen R, Hummel PA, Gray HB, Winkler JR. α-Synuclein structures from fluorescence energy-transfer kinetics: Implications for the role of the protein in Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:16466–16471. doi: 10.1073/pnas.0407307101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound α-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci U S A. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfefferkorn CM, Lee JC. Tryptophan probes at the α-synuclein and membrane interface. J Phys Chem B. 2010;114:4615–4622. doi: 10.1021/jp908092e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 39.Yap TL, Pfefferkorn CM, Lee JC. Residue-specific fluorescent probes of α-synuclein: Detection of early events at the N- and C-termini during fibril assembly. Biochemistry. 2011;50:1963–1965. doi: 10.1021/bi2000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gangabadage CS, Zdunek J, Tessari M, Nilsson S, Olivecrona G, Wijmenga SS. Structure and dynamics of human apolipoprotein cIII. J Biol Chem. 2008;283:17416–17427. doi: 10.1074/jbc.M800756200. [DOI] [PubMed] [Google Scholar]
- 41.Liu HQ, Talmud PJ, Lins L, Brasseur R, Olivecrona G, Peelman F, Vandekerckhove J, Rosseneu M, Labeur C. Characterization of recombinant wild type and site-directed mutations of apolipoprotein c-III: Lipid binding, displacement of apoE, and inhibition of lipoprotein lipase. Biochemistry. 2000;39:9201–9212. doi: 10.1021/bi0009441. [DOI] [PubMed] [Google Scholar]
- 42.Motabar O, Goldin E, Leister W, Liu K, Southall N, Huang WW, Marugan JJ, Sidransky E, Zheng W. A high throughput glucocerebrosidase assay using the natural substrate glucosylceramide. Anal Bioanal Chem. 2012;402:731–739. doi: 10.1007/s00216-011-5496-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfefferkorn CM, Jiang Z, Lee JC. Biophysics of α-synuclein membrane interactions. Biochim Biophys Acta. 2012;1818:162–171. doi: 10.1016/j.bbamem.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfefferkorn CM, Heinrich F, Sodt AJ, Maltsev AS, Pastor RW, Lee JC. Depth of α-synuclein in a bilayer determined by fluorescence, neutron reflectometry, and computation. Biophys J. 2012;102:613–621. doi: 10.1016/j.bpj.2011.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clayton DF, George JM. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 46.Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392:193–197. doi: 10.1038/32440. [DOI] [PubMed] [Google Scholar]
- 47.Kobayashi T, Beuchat MH, Chevallier J, Makino A, Mayran N, Escola JM, Lebrand C, Cosson P, Kobayashi T, Gruenberg J. Separation and characterization of late endosomal membrane domains. J Biol Chem. 2002;277:32157–32164. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 48.Peters SP, Coyle P, Glew RH. Differentiation of β-glucocerebrosidase from β-glucosidase in human tissues using sodium taurocholate. Arch Biochem Biophys. 1976;175:569–582. doi: 10.1016/0003-9861(76)90547-6. [DOI] [PubMed] [Google Scholar]
- 49.Urban DJ, Zheng W, Goker-Alpan O, Jadhav A, Lamarca ME, Inglese J, Sidransky E, Austin CP. Optimization and validation of two miniaturized glucocerebrosidase enzyme assays for high throughput screening. Comb Chem High Throughput Screen. 2008;11:817–824. doi: 10.2174/138620708786734244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC. A broken alpha -helix in folded alpha -synuclein. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 51.Bortolus M, Tombolato F, Tessari I, Bisaglia M, Mammi S, Bubacco L, Ferrarini A, Maniero AL. Broken helix in vesicle and micelle-bound α-synuclein: Insights from site-directed spin labeling-epr experiments and md simulations. J Am Chem Soc. 2008;130:6690–6691. doi: 10.1021/ja8010429. [DOI] [PubMed] [Google Scholar]
- 52.Drescher M, Veldhuis G, van Rooijen BD, Milikisyants S, Subramaniam V, Huber M. Antiparallel arrangement of the helices of vesicle-bound α-synuclein. J Am Chem Soc. 2008;130:7796–7797. doi: 10.1021/ja801594s. [DOI] [PubMed] [Google Scholar]
- 53.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human α-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 54.Bussell R, Jr, Eliezer D. A structural and functional role for 11-mer repeats in α-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329:763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- 55.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci U S A. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trexler AJ, Rhoades E. α-Synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lokappa SB, Ulmer TS. α-Synuclein populates both elongated and broken helix states on small unilamellar vesicles. J Biol Chem. 2011;286:21450–21457. doi: 10.1074/jbc.M111.224055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robotta M, Braun P, vanRooijen B, Subramaniam V, Huber M, Drescher M. Direct evidence of coexisting horseshoe and extended helix conformations of membrane-bound α-synuclein. ChemPhysChem. 2011;12:267–269. doi: 10.1002/cphc.201000815. [DOI] [PubMed] [Google Scholar]
- 59.Gegg ME, Burke D, Heales SJR, Cooper JM, Hardy J, Wood NW, Schapira AHV. Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol. 2012;72:455–463. doi: 10.1002/ana.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lieberman RL, Wustman BA, Huertas P, Powe AC, Pine CW, Khanna R, Schlossmacher MG, Ringe D, Petsko GA. Structure of acid β-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat Chem Biol. 2007;3:101–107. doi: 10.1038/nchembio850. [DOI] [PubMed] [Google Scholar]
- 61.Dvir H, Harel M, McCarthy AA, Toker L, Silman I, Futerman AH, Sussman JL. X-ray structure of human acid-β-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003;4:704–709. doi: 10.1038/sj.embor.embor873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.