

Abstract
Over the years, study of the disorders of hemoglobin has served as a paradigm for gaining insights into the cellular and molecular biology, as well as the pathophysiology, of inherited genetic disorders. To date, more than 1000 disorders of hemoglobin synthesis and/or structure have been identified and characterized. Study of these disorders has established the principle of how a mutant genotype can alter the function of the encoded protein, which in turn can lead to a distinct clinical phenotype. Genotype/phenotype correlations have provided important understanding of pathophysiological mechanisms of disease. Before presenting a brief overview of these disorders, we provide a summary of the structure and function of hemoglobin, along with the mechanism of assembly of its subunits, as background for the rationale and basis of the different categories of disorders in the classification.
More than 1000 hemoglobin disorders are known. These can be broadly classified into two general categories: defects in globin expression (i.e., the thalassemias) and defects in globin structure (e.g., sickle cell disease).
An impressive degree of molecular “engineering” was necessary for the evolution of a multisubunit protein that higher organisms require for optimal oxygen homeostasis and buffering of acidic metabolic waste products. Each globin subunit must form a stable linkage with heme (ferroprotoporphyrin IX) situated on the external surface of the protein so that oxygen in the RBC cytosol can bind reversibly to the hemes’ iron atoms. Moreover, the hydrophobic cleft into which the heme is inserted must be able to protect the Fe2+ heme iron from oxidation to Fe3+, which is incapable of binding oxygen.3 Delicate noncovalent interactions between unlike globin subunits are required for the hemoglobin tetramer α2β2 to bind and unload oxygen in a cooperative manner, thereby assuring maximal transport to actively metabolizing cells. This phenomenon is reflected by a sigmoid oxygen-binding curve that depends on hemoglobin tetramer having two quaternary structures: the T or deoxy conformer that has low oxygen affinity and the R or oxy conformer that has high oxygen affinity. Further fine-tuning of hemoglobin function comes from its allosteric behavior triggered by the binding of two small effector molecules 2,3-BPG, and protons to specific sites on the T structure distant from the heme groups.4 To endow the blood with high oxygen carrying capacity hemoglobin must be stuffed into flexible circulating RBCs. A remarkably high degree of solubility is required for hemoglobin to achieve an intracellular concentration of ∼34 g/dl or 5 mm (tetramer).
To reach such a high corpuscular hemoglobin concentration it is essential that α-globin as well as β-globin (or γ-globin) mRNA be expressed at very high levels during erythroid differentiation. Moreover, α- and non-α-globin synthesis must be closely matched. As explained in Nienhuis and Nathan (2012), subunit imbalance is central to the pathophysiology of the thalassemias. Free α-globin subunits are particularly toxic to erythroid cells. This threat is alleviated by the presence of α-hemoglobin stabilizing protein (AHSP), a chaperone that is expressed at high levels in erythroid cells and binds specifically and tightly to heme-intact α-globin subunits (Kihm et al. 2002; Feng et al. 2004; Mollan et al. 2010, 2012). The AHSP protects the cell from potentially toxic oxidized (Fe3+) heme until it is reduced to functional Fe2+ heme by cytochrome b5 reductase. On encountering a free heme-intact β-globin subunit, the α-globin dissociates from AHSP to form the extremely stable αβ dimer. As discussed later in this work, this process is facilitated by electrostatic attraction between positively charged α-globin subunits and negatively charged β-globin subunits.
In view of the multiple molecular constraints that are required for high-level production of fully functional and highly soluble hemoglobin, it is no surprise that Murphy’s law is in full force: whatever can go wrong will. As discussed briefly here, and in much more detail in Thein (2013), Higgs (2013), Nienhuis and Nathan (2012), and Musallam et al. (2012), mutations of globin genes that impair synthesis give rise to thalassemia and anemia of varying degree. In addition, well-defined clinical and hematologic phenotypes are associated with mutations that alter the structure of globin subunits, discussed in more detail in Thom et al. (2013). Impairment of hemoglobin solubility can be caused either by the formation of intracellular polymers (sickle cell disease) or by the development of amorphous precipitates (congenital Heinz body hemolytic anemia). Abnormalities of oxygen binding can lead either to erythrocytosis (high O2 affinity mutants) or to cyanosis (low O2 affinity mutants). Some globin mutants have structural alterations within the heme pocket that result in oxidation of the heme iron and pseudocyanosis because of methemoglobinemia.
GENERAL CLASSIFICATION OF HEMOGLOBIN DISORDERS
Hemoglobin disorders can be broadly classified into two general categories (as listed in Table 1):
Those in which there is a quantitative defect in the production of one of the globin subunits, either total absence or marked reduction. These are called the thalassemia syndromes.
Those in which there is a structural defect in one of the globin subunits.
Table 1.
Classification of hemoglobin disorders
|
I. QUANTITATIVE DISORDERS OF GLOBIN CHAIN SYNTHESIS/ACCUMULATION The thalassemia syndromes A. β-Thalassemia Clinical classification: β-Thalassemia minor or trait β-Thalassemia major β-Thalassemia intermedia Biochemical/genetic classification: β0-Thalassemia β+-Thalassemia δ-Thalassemia γ-Thalassemia Lepore fusion gene δβ-Thalassemia εγδβ-Thalassemia HPFH “Dominant” β-thalassemia (structural variants with β-thalassemia phenotype) β-Thalassemia with other variants: HbS/β-thalassemia HbE/β-thalassemia Other B. α-Thalassemia Deletions of α-globin genes: One gene: α+-thalassemia Two genes in cis: α0-thalassemia Two genes in trans: homozygous α+-thalassemia (phenotype of α0-thalassemia) Three genes: HbH disease Four genes: Hydrops fetalis with Hb Bart's Nondeletion mutants: Hb Constant Spring Other C. De novo and acquired α-thalassemia α-Thalassemia with mental retardation syndrome (ATR): Due to large deletions on chromosome 16 involving the α-globin genes Due to mutations of the ATRX transcription factor gene on chromosome X α-Thalassemia associated with myelodysplastic syndromes (ATMDS): Due to mutations of the ATRX gene |
II. QUALITATIVE DISORDERS OF GLOBIN STRUCTURE: STRUCTURAL VARIANTS OF HEMOGLOBIN A. Sickle cell disorders SA, sickle cell trait SS, sickle cell anemia/disease SC, HbSC disease S/β thal, sickle β-thalassemia disease S with other Hb variants: D, O-Arab, other SF, Hb S/HPFH B. Hemoglobins with decreased stability (unstable hemoglobin variants) Mutants causing congenital Heinz body hemolytic anemia Acquired instability—oxidant hemolysis: Drug-induced, G6PD deficiency C. Hemoglobins with altered oxygen affinity High/increased oxygen affinity states: Fetal red cells Decreased RBC 2,3-BPG Carboxyhemoglobinemia, HbCO Structural variants Low/decreased oxygen affinity states: Increased RBC 2,3-BPG Structural variants D. Methemoglobinemia Congenital methemoglobinemia: Structural variants Cytochrome b5 reductase deficiency Acquired (toxic) methemoglobinemia E. Posttranslational modifications Nonenzymatic glycosylation Amino-terminal acetylation Amino-terminal carbamylation Deamidation |
The majority of human hemoglobin mutants were discovered as an incidental finding, unassociated with any hematologic or clinical phenotype. However, a number of α- and β-globin mutants are associated with distinct clinical phenotypes. These fall into five broad categories: the sickle syndromes (SS, SC, Sβ0-thalassemia, and Sβ+-thalassemia); unstable mutants causing congenital Heinz body hemolytic anemia; mutants with high oxygen affinity resulting in erythrocytosis; low oxygen affinity mutants and the M hemoglobins causing cyanosis; and mutants associated with a thalassemia phenotype. Most of these disorders are inherited genetic defects, but there are some defects that are acquired or occur de novo.
THE THALASSEMIA SYNDROMES
The thalassemia syndromes are inherited disorders characterized by absence or markedly decreased accumulation of one of the globin subunits of hemoglobin. In the alpha (α)-thalassemias, there is absent or decreased production of α-globin subunits, whereas in the beta (β)-thalassemias, there is absent or reduced production of β-globin subunits. Rare thalassemias affecting the production of delta (δ)- or gamma (γ)-globin subunits have also been described but are not clinically significant disorders. Combined deficiency of δ + β-globin subunits, or of all of the β-like globin subunits also occurs. These disorders are described in detail in Thein (2013) and Higgs (2013).
THE β-THALASSEMIAS
The β-thalassemias can be subclassified into those in which there is total absence of normal β-globin subunit synthesis or accumulation, the β0-thalassemias, and those in which some structurally normal β-globin subunits are synthesized, but in markedly decreased amounts, the β+-thalassemias. The molecular basis of the β-thalassemias is very heterogeneous (see Thein 2013), with over 200 different mutations having been described. In general, the mutations causing β-thalassemia are point mutations affecting a single nucleotide, or a small number of nucleotides, in the β-globin gene. Rare deletion forms of β-thalassemia have also been described. One of these deletions is caused by “unequal” crossing over between the linked and partially homologous δ- and β-globin genes, resulting in the formation of a fusion δβ-globin gene, the Lepore gene, that has a low level of expression. Large deletions involving part or all of the β-globin gene cluster are responsible for the δβ-thalassemias, the εγδβ-thalassemias, and the hereditary persistence of fetal hemoglobin (HPFH) syndromes.
Despite the marked heterogeneity in the molecular basis of the β-thalassemias, the clinical phenotype of these disorders is relatively homogeneous because of their common pathophysiology: deficiency of HbA tetramers and excess accumulation of free α-subunits incapable of forming hemoglobin tetramers because of deficiency of β-like globin subunits (see Nienhuis and Nathan 2012). In heterozygotes (β-thalassemia trait or β-thalassemia minor), there is mild to moderate hypochromic microcytic anemia, without evidence of hemolysis, whereas in homozygotes or compound heterozygotes (β-thalassemia major), there is usually a severe transfusion-dependent hemolytic anemia associated with marked ineffective erythropoiesis resulting in destruction of erythroid precursor cells in the bone marrow.
A less common clinical phenotype is referred to as β-thalassemia intermedia. In this clinical disorder, there is a moderate to severe, partially compensated, hemolytic anemia that does not require chronic transfusion therapy to maintain a satisfactory circulating hemoglobin level in the affected patient, although occasional transfusions may be required if the anemia worsens because of associated complications. Such patients have a milder disease because there is less severe α- to non-α-globin subunit imbalance than in typical β-thalassemia major patients, resulting in less accumulation of free α-subunits that cause the ineffective erythropoiesis. There are different possible causes for such a lessened α- to non-α-globin subunit imbalance, including: inheritance of milder β+-thalassemia mutations with less severe than usual deficiency of β-globin subunit production; coinheritance of a form of α-thalassemia; or coinheritance of another genetic trait associated with increased production of the γ-subunit of fetal hemoglobin. Most patients with β-thalassemia intermedia carry two mutant β-globin genes: they have a genotype typical of β-thalasemia major, but the phenotype is modified by one of the factors listed above. However, rare cases of β-thalassemia intermedia are caused by heterozygosity for a single mutant β-globin gene associated with the production of a highly unstable β-globin subunit that causes RBC damage in a fashion similar to excess free α-subunits; this is the so-called “dominant β-thalassemia” (Thom et al. 2013).
The δβ-thalassemias are associated with total deficiency of β-globin subunit production, but are clinically milder than typical cases of β0-thalassemia, because there is an associated persistent high level of expression of the γ-subunit of fetal hemoglobin that decreases the degree of α-subunit excess. The εγδβ-thalassemias are associated with neonatal hemolytic anemia, but this resolves during the first few months of life and the associated phenotype in adults is that of β-thalassemia trait or β-thalassemia minor. The syndrome of hereditary persistence of fetal hemoglobin (HPFH) is not, strictly speaking, a form of β-thalassemia because it is not associated with significant α- to non-α-globin subunit imbalance, but is characterized by high levels of persistent γ-globin production and is frequently considered within the spectrum of δβ-thalassemia.
THE α-THALASSEMIAS
In contrast to the β-thalassemias, which are usually caused by point mutations of the β-globin gene, the α-thalassemia syndromes are usually caused by the deletion of one or more α-globin genes and are subclassified according to the number of α-globin genes that are deleted (or mutated): one gene deleted (α+-thalassemia); two genes deleted on the same chromosome or in cis (α0-thalassemia); three genes deleted (HbH disease); or four genes deleted (hydrops fetalis with Hb Bart's). Nondeletion forms of α-thalassemia have also been characterized but are relatively uncommon. These disorders are discussed in detail in Higgs (2013).
Clinically, the deletion of only one of the four α-globin genes is not associated with significant hematologic abnormalities and is sometimes called the “silent carrier” state for α-thalassemia. The deletion of two α-globin genes can occur in two forms: (1) on the same chromosomes, or in cis; or (2) on opposite chromosomes, or in trans, which is the homozygous state for the single gene deletion, or homozygous α+-thalassemia. The cis genotype is particularly common in Asian populations, whereas the trans genotype is highly prevalent in persons of black/African ancestry. The clinical phenotype is similar with both genotypes and consists of mild hypochromic, microcytic anemia, without hemolysis, somewhat analogous to that of β-thalasssemia trait, but somewhat less severe. The deletion (or markedly decreased expression) of three α-globin genes is associated with the syndrome of HbH disease, a compensated hemolytic anemia that usually does not require treatment by RBC transfusion. The basis of the hemolysis is the excess accumulation of β-globin subunits that self-associate to form β-chain tetramers or HbH. In contrast to the situation in the β-thalssemias in which excess α-globin subunits rapidly form insoluble aggregates, excess β-globin chains can form soluble tetramers (HbH). However, HbH is relatively unstable and does precipitate as RBCs age, forming inclusion bodies that damage RBCs and shorten their lifespan (see Higgs 2013). The deletion of all four α-globin genes is usually fatal during late pregnancy or shortly after birth. This condition is called hydrops fetalis with Hb Bart’s. Hb Bart’s is a tetramer of four γ-globin subunits and is ineffective as an oxygen transporter: it has a very high oxygen affinity, similar to that of myoglobin, and does not release oxygen to the tissues under physiologic conditions. Therefore, the infant, whose RBCs lack HbF or HbA and contain mostly Hb Bart’s, suffers severe hypoxia resulting in hydrops fetalis. Rare cases have been rescued by intrauterine transfusions, but these children subsequently require lifelong transfusion support similar to that required by children with β-thalassemia major.
One form of nondeletion α-thalassemia, called Hb Constant Spring (HbCS), is particularly prevalent in southeast Asia. It is caused by a chain termination mutation, which results in the synthesis of an elongated α-globin subunit that accumulates at very low levels in RBCs of affected individuals. When coinherited with the α0-cis deletion on the other chromosome, there results a form of HbH disease that is more severe than the typical HbH disease associated with full deletion of three α-globin genes (see Higgs 2013).
In addition to these forms of α-thalassemia inherited in a pattern consistent with Mendelian genetics, there are two α-thalassemia syndromes that are caused by de novo or acquired mutations affecting expression of the α-globin genes: (1) the α-thalassemia with mental retardation syndrome (ATR); and (2) acquired α-thalassemia (HbH disease) associated with myelodysplastic syndromes (ATMDS). These syndromes are described in detail in Gibbons (2012).
THE α-THALASSEMIA WITH MENTAL RETARDATION SYNDROME
There are two subtypes of this syndrome: (1) one is associated with very large deletions involving the α-globin genes and adjacent genes on chromosome 16, that are not inherited from a parent and appear to have arisen de novo during embryogenesis (the ATR-16 syndrome); and (2) the other is associated with structurally normal α-globin genes and is caused by germline mutations in a transcription factor gene located on the X chromosome (the ATR-X syndrome). The encoded transcription factor has been called ATRX and is an important regulator of α-globin gene expression.
ACQUIRED α-THALASSEMIA (HbH DISEASE) ASSOCIATED WITH MYELODYSPLASTIC SYNDROMES (MDS)
A small number of patients with MDS, a clonal bone marrow disorder characterized by disturbed hematopoiesis with cytopenias and abnormal myeloid differentiation, develop RBC abnormalities consistent with acquired HbH disease. The α-globin genes in such patients are structurally normal, but their expression is severely impaired. The molecular basis for the decreased α-globin gene expression in this condition has been determined to be acquired somatic mutations of the ATRX gene, the same gene that is mutated in the syndrome of α-thalassemia with mental retardation. The impairment of α-globin gene expression is more severe in the ATMDS syndrome than in the ATRX syndrome.
QUALITATIVE DISORDERS OF GLOBIN STRUCTURE
Sickle Cell Disorders
Sickle hemoglobin (HbS) results from an amino acid substitution at the sixth residue of the β-globin subunit: β6-Glu → Val. Approximately 8% of African Americans are heterozygous for this hemoglobin variant, a condition called sickle cell trait or HbAS. In equatorial Africa, where malaria is endemic, the prevalence of HbAS is much higher and can reach over 30% in some populations because of survival advantage of HbAS heterozygotes from complications of falciparum malaria. RBCs of persons with HbAS typically have 40% HbS and 56%–58% HbA. Individuals with HbAS are typically asymptomatic; severe hypoxia is required for them to experience manifestations of sickle cell disease, called sickling.
The basis of sickling in patients homozygous for the disorder, called sickle cell anemia or HbSS, is polymerization of deoxy-HbS resulting in the formation of multistranded fibers that create a gel and change the shape of RBCs from biconcave discs to elongated crescents. The polymerization/sickling reaction is reversible following reoxygenetion of the hemoglobin. Thus, an RBC can undergo repeated cycles of sickling and unsickling. There are two major pathophysiological consequences of sickling:
Repeated cycles of sickling damage the red blood cell membrane leading to abnormalities of permeability and cellular dehydration, eventually causing premature destruction of RBCs and a chronic hemolytic anemia.
Sickled RBCs are rigid, increase blood viscosity and obstruct capillary flow, causing tissue hypoxia and, if prolonged, cell death, tissue necrosis/infarction, and progressive organ damage. Acute episodes are often called vaso-occlusive pain crises.
The pathophysiology and clinical manifestations of sickle cell disease are described in detail in Schechter and Elion (2013) and Serjeant (2013). There can be a great deal of variability in the severity of the clinical manifestations of the various sickle cell syndromes in patients with the same β-globin gene genotype and even between siblings in the same family. This variability can be caused by the coinheritance of a number of different genetic modifiers of the disease, including coinheritance of α-thalassemia, quantitative trait loci affecting the level of production of fetal hemoglobin and other traits, as discussed in Lettre (2012). In general, the clinical severity of the different sickle cell syndromes correlates directly with the amount and concentration of HbS in the red cell. The kinetics of sickling are markedly concentration dependent, the speed of the reaction being inversely proportional to about the 30th power of the starting HbS concentration. Thus, a small increase in intracellular HbS concentration, such as that resulting from loss of intracellular water and cellular dehydration, can have a major effect on the speed of the polymerization reaction. The amount and type of other non-S hemoglobins in the red cell can also influence the extent or rate of sickling, and thus clinical severity. HbSC disease is associated with significant clinical manifestations. Why do SC patients often have vaso-occlusive manifestations as dramatic as those with SS disease, whereas AS individuals have virtually no morbidity? HbC and HbA copolymerize to an identical degree with HbS (Bunn et al. 1982). Two independent factors conspire to make HbSC a disease. The presence of HbC in the red cell greatly enhances potassium efflux and red cell dehydration, which increases corpuscular hemoglobin concentration (Bunn et al. 1982) and promotes polymerization. Sickling in SC patients is further enhanced by the fact that they have ∼50% HbS, whereas HbAS individuals have ∼40% HbS. This difference is because α-globin subunits bind more readily to negatively charged βA-subunits than to positively charged βC-subunits, as explained further at the end of this work. Other clinically significant sickle cell syndromes include: sickle/β0-thalassemia, sickle/β+-thalassemia, HbSD disease, and HbSO-Arab disease. HbD and HbO-Arab participate more readily than HbA in copolymer formation with HbS.
A striking example of how another hemoglobin can dramatically influence sickling is the condition that results from coinheritance of HbS with hereditary persistence of fetal hemoglobin: HbSF or S/HFPH. This condition, in which there is ∼70% HbS and ∼30% HbF, is clinically asymptomatic and is a notable exception to the general rule that clinical severity is directly related to the amount (or %) of HbS in the RBC. In this condition, the HbF is distributed in a pancellular fashion so that every RBC contains ∼30% HbF and, because HbF does not participate at all in polymer formation, this is sufficient to prevent sickling of all RBCs under physiologic conditions. This situation is in contrast to what occurs when variable amounts of HbF are present in HbSS disease or the sickle/β-thalassemia syndromes. In the latter disorders, the HbF is distributed in a heterocellular fashion, in which the HbF is present in only a subset of cells called F cells, leaving the other cells that lack HbF unprotected from the antisickling effects of HbF. Nevertheless, a marked increase in the proportion of F cells in such cases can have a beneficial effect on the clinical manifestation of these sickling disorders. Therefore, there is a great deal of interest and ongoing research to develop strategies for enhancing HbF synthesis and F-cell production as a therapeutic modality for sickling disorders (see Sankaran and Orkin 2013).
UNSTABLE HEMOGLOBIN VARIANTS
A substantial minority of Hb mutants have substitutions that alter the solubility of the molecule in the red cell. The intraerythrocytic precipitated material derived from the unstable abnormal Hb is detectable by a supravital stain as dark globular aggregates called Heinz bodies (HBs). These intracellular inclusions reduce the life span of the red cell and generate a hemolytic process of varied severity called congenital Heinz body hemolytic anemia. When a red cell hemolysate of an affected individual is heated to 50°C or treated with 17% isopropanol, a precipitate usually develops. The hemoglobin electrophoresis often reveals an abnormal banding pattern. Definitive diagnosis requires analysis of either globin structure or DNA sequence. For more information on congenital Heinz body hemolytic anemia and other hemoglobin variants summarized in this section, see Thom et al. (2013).
HEMOGLOBIN VARIANTS WITH ALTERED OXYGEN AFFINITY
Over 25 hemoglobin variants have been encountered in individuals with erythrocytosis. Amino acid substitutions cause an increase in oxygen affinity usually because of impairment in the stability of the T or “deoxy” quaternary structure. Erythrocytosis is usually familial and is the only well-documented clinical finding. Increased oxygen affinity results in a decrease in hemoglobin’s ability to release oxygen during blood flow in the microcirculation. Unlike individuals with secondary erythrocytosis caused by high altitude exposure or to cardiac or pulmonary hypoxemia, those having hemoglobin with high oxygen affinity have normal arterial oxygen tension. Therefore, they are not hypoxemic. However, the reduction in unloading of oxygen to tissues results in cellular hypoxia. Thus, the cells in the kidney that produce erythropoietin sense hypoxia and up-regulate Epo gene expression leading to expansion of the red cell mass. About half of the high-affinity variants can be detected by hemoglobin electrophoresis. Definitive diagnosis is established by demonstration of a “shift to the left” in the oxyhemoglobin binding curve.
Less often, individuals and their relatives will present with low O2 saturation and cyanosis owing to a hemoglobin variant with a marked reduction in oxygen affinity. The variants that have been most thoroughly characterized entail amino acid replacements that destabilize the R or “oxy” quarternary structure. Affected individuals have normal hemoglobin levels and no clinical manifestations other than the dusky skin color.
THE M HEMOGLOBINS
Rare families have attracted medical attention because affected individuals have cyanosis owing to the inheritance of a hemoglobin variant in which an amino acid substitution within the heme pocket results in stabilization of the heme iron in the oxidized Fe3+ form. As discussed in Schechter (2013), among the most highly conserved amino acids within hemoglobin subunits are two histidines, one at helix position F8, which binds to the heme iron and one at E7 in the pocket where oxygen and other ligands bind to heme. Replacement of either of these histidines by tyrosine in either the α-, β-, or γ-globin subunit results in an alteration in the electronic environment of the heme iron so that it is stabilized as Fe3+. Thus, the M hemoglobins include α58 His → Tyr, α87 His → Tyr, β63 His → Tyr, β92 His → Tyr, γ63 His → Tyr, and γ92 His → Tyr. In addition, β67 Val → Glu results in very similar biochemical features and clinical presentation. Affected individuals have cyanosis similar in hue to those with methemoglobinemia in which the heme iron in normal hemoglobin has been oxidized to Fe3+. They have either normal hemoglobin levels or mild anemia and no clinical manifestations other than their skin color.
IMPACT OF SUBUNIT ASSEMBLY ON HEMOGLOBIN PHENOTYPE
The composition of the normal and mutant hemoglobins in red cells of heterozygotes provides insight into the assembly of human hemoglobins (Bunn and McDonald 1983). The great majority of β-subunit mutants are synthesized at the same rate as βA and have normal stability. Therefore, heterozygotes would be expected to have equal amounts of normal and mutant hemoglobin. However, measurements of the proportion of normal and abnormal hemoglobins in heterezygotes have revealed unexpected variability. Figure 1 shows a comparison of 105 stable β-subunit mutants. Positively charged mutants, such as hemoglobins S, C, D-Los Angeles, and E5, constitute significantly less than half of the total hemoglobin in heterozygotes and are reduced further in the presence of α-thalassemia, because of increased competition between normal and mutant β-subunits for limiting amounts of α-subunits. In contrast, many of the negatively charged mutants are present in amounts exceeding that of HbA. In two heterozygotes who had a negatively charged mutant (HbJ-Baltimore or HbJ-Iran) in conjunction with α-thalassemia, the proportion of the mutant hemoglobin was further increased because these βJ-subunits out-compete normal βA-subunits for limiting amounts of α-subunits. Analyses of the proportion of β-subunit mutant in heterozygotes suggest that alterations in surface charge contribute to different rates of assembly of the hemoglobin tetramer. This hypothesis is supported by in vitro mixing experiments with normal and mutant subunits showing that when α-subunits are present in limiting amounts (mimicking α-thalassemia), negatively charged mutants are formed much more readily than are positively charged mutants (Mrabet et al. 1986; Adachi et al. 1998).
Figure 1.

Impact of surface charge on hemoglobin assembly. (A) Effect of charge on the proportion of abnormal hemoglobin in individuals heterozygous for 105 stable β-globin variants. Each data point represents a mean value for a given variant. Substitutions involving a histidine residue were scored as a change of one half charge. The “−1” group differs significantly from the “+1” group (P < 0.001). (B) Effect of α-thalassemia on the proportion of seven positively charged β-subunit variants (blue) and three negatively charged variants (red). (Updated from Bunn and McDonald 1983 and Bunn 1987.)
This electrostatic model of hemoglobin assembly has clinical implications. Differences in rates of assembly explain the higher proportion in HbS in sickle C (SC) disease than in sickle trait (AS), a difference that contributes significantly to HbS polymerization and consequent vaso-occlusion in SC patients (Bunn et al. 1982). HbS-Oman is a rare stable mutant with both the sickle (β6Glu → Val) substitution and β121Glu → Lys, a substitution that stabilizes the sickle polymer. Thus, β-subunits of HbS-Oman have a gain of three positive charges. Accordingly, in red cells of heterozygotes, it is present in much less abundance than HbA. However, because of its markedly enhanced polymerization, heterozygotes with only 20% HbS-Oman and one-gene deletion α-thalassemia (α-/αα) have hemolytic anemia and signs and symptoms of sickle vaso-occlusion. In contrast, HbS-Oman heterozygotes with more severe α-thalassemia (α-/α-) have only 13% HbS-Oman and a normal clinical and hematologic phenotype (Nagel et al. 1998).
The impact of surface charge on hemoglobin assembly also explains the differences in the levels of HbA2 (Bunn 1987) and HbF (Adams et al. 1985; Chui et al. 1990) that accompany certain hematological disorders. For example, α-hemoglobin subunits bind more readily to β than to the positively charged δ. Accordingly, HbA2 levels are decreased in the presence of limiting levels of α-hemoglobin, e.g., α-thalassemia as well as iron deficiency in which there is an acquired impairment of α-globin synthesis.
POSTTRANSLATIONAL ALTERATIONS IN HEMOGLOBIN STRUCTURE
A wide range of posttranslational modifications of hemoglobin structure have been identified, some of which result in significant perturbations of function.
Nonenzymatic Glycation
The concentration of glucose in the RBC cytosol is nearly that of the plasma. Glucose is in equilibrium between the cyclic hemiacetal structure and the open aldehyde structure. The latter is able to react covalently with amino groups of a wide range of proteins. Because this reaction favors amino groups with relatively low pKas, the most abundant glycated hemoglobin is at the amino termini of α- and β-globin. In normal red cells, ∼5% of hemoglobin has a glucose adduct at the β-amino terminus designated HbAIc:
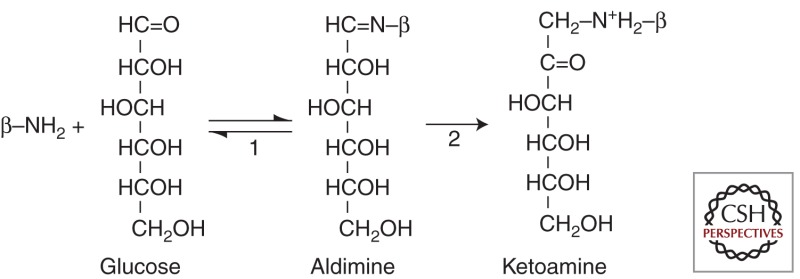
Reaction 1 shows the formation of the Schiff base or aldimine adduct. This product then rearranges to the more stable (virtually irreversible) ketoamine linkage (reaction 2). HbAIc and other less abundant glucose adducts are formed in a linear manner over the 120-day life span of the red cell. Therefore, HbAIc levels are low in individuals with hemolysis. Because the level of HbAIc is an accurate reflection of the average concentration of plasma glucose over the preceding 2 months, it has proven to be very useful in monitoring therapeutic control in patients with diabetes.
Amino-Terminal Acetylation
Shortly following translation of most proteins, the amino-terminal (initiator) methionine is cleaved and often the adjacent amino acid is acetylated at the amino terminus. These posttranslational modifications are highly dependent on the sequence of neighboring amino acids. All human globins undergo cleavage of the amino-terminal methionine but none undergo amino-terminal acetylation except for ∼20% of γ-globin subunits (HbFI). However, rare human hemoglobin variants have been identified that have amino acid replacements in the amino-terminal region that cause both retention of the initiator methionine and N-acetylation.
Amino-Terminal Carbamylation
Urea in the plasma is in equilibrium with ammonium and cyanate ions. Cyanate is able to bind covalently to the amino termini of α- and β-globin subunits to form adducts. Carbamylated hemoglobin is not detectable in normal red cells but can accumulate to measurable levels in patients with uremia.
Deamidation
In many proteins, the amino group on certain asparagine residues, particularly when adjacent to a histidine, dissociates leaving aspartic acid. Deamidation does not occur on asparagines in normal globin subunits. However, seven human variants have been identified in which partial or complete deamidation occurs. Four of these entail replacements by asparagine. The deamidation does not appear to impact function in any of these variants.
ACQUIRED ALTERATIONS IN HEMOGLOBIN FUNCTION
Methemoglobinemia
Certain drugs and toxins can cause enhanced oxidation of heme iron to form methemoglobin. Above levels of 10%, methemoglobin individuals appear cyanotic. The extent of hemoglobin oxidation is determined by spectrophotometric analysis of the blood. Methemoglobinemia impairs oxygenation of tissues because the Fe3+ heme is incapable of binding to oxygen, and, even more importantly, the presence of one or two oxidized hemes per tetramer stabilizes the R quaternary structure and thereby increases the affinity of the remaining hemes for oxygen. Central nervous and cardiovascular symptoms and signs begin to appear when methemoglobin exceeds 30%. Much lower levels are seen in individuals with congenital methemoglobinemia owing to deficiency in cytochrome b5 reductase. Both toxic and congenital methemoglobinemia can be effectively treated by administration of the redox dye methylene blue.
Carboxyhemoglobinemia
Carbon monoxide (CO) binds to heme iron of hemoglobin with an affinity 210 greater than that of oxygen. Normal red cells contain a very small amount of carboxyhemoglobin owing to the release of CO from macrophages following the uptake of senescent red cells and degradation of heme by heme oxygenase. Higher, albeit nontoxic, levels of HbCO are present in patients with hemolytic anemias. Inhalation of CO from incomplete combustion of hydrocarbons can result in toxic carboxyhemoglobinemia. As in methemoglobinemia, CO poisoning causes morbidity and mortality by ischemia owing to a left shift in the oxyhemoglobin binding curve, thereby impairing the unloading of oxygen to cells and tissues. Patients with acute CO poisoning generally develop CNS and cardiovascular symptoms when levels exceed 20%. Levels of 40%–60% HbCO result in stupor, coma, and death. All symptomatic patients should be promptly removed from further exposure to CO and treated with oxygen. If possible, those with severe toxicity should be treated with hyperbaric oxygen administration or exchange transfusion.
Alterations in Oxygen Affinity
As mentioned above, protons (H+) and 2,3-BPG are potent allosteric modulators of oxygen affinity in normal RBC. Because of their preferential binding to the T or “deoxy” quaternary structure of hemoglobin, they lower the affinity for oxygen and therefore shift the oxyhemoglobin binding curve “to the right.” Except under clinical circumstances resulting in either acidosis or alkalosis, plasma pH and red cell pH are tightly regulated at 7.4 and 7.2, respectively. In contrast, levels of red cell 2,3-BPG can vary considerably in a wide range of clinical settings. Patients with low levels, such as those who have received large amounts of stored RBCs will have increased oxygen affinity, which could compromise tissue oxygenation. Increased levels of RBC 2,3-BPG are much more commonly observed, primarily in patients with some form of hypoxia. As a result, the “right shift” in the oxyhemoglobin dissociation curve significantly enhances the unloading of oxygen to tissues and is therefore a major mode of compensation, particularly in patients with severe anemia.
For more detailed information, see Dailey and Meissner (2013).
For more detailed information on hemoglobin function, see Schechter (2013).
Levels of HbE in AE heterozygotes are lower than the others because the synthesis of βE-globin is impaired owing to a defect in mRNA splicing (see Fucharoen and Weatherall 2012 and Thein 2013).
Editors: David Weatherall, Alan N. Schechter, and David G. Nathan
Additional Perspectives on Hemoglobin and Its Diseases available at www.perspectivesinmedicine.org
REFERENCES
*Reference is also in this collection.
- Adachi K, Yamaguchi T, Pang J, Surrey S 1998. Effects of increased anionic charge in the β-globin chain on assembly of hemoglobin in vitro. Blood 91: 1438–1445 [PubMed] [Google Scholar]
- Adams JD, Coleman MB, Hayes J, Morrison WT, Steinberg MH 1985. Modulation of fetal hemoglobin synthesis by iron deficiency. N Engl J Med 313: 1402–1405 [DOI] [PubMed] [Google Scholar]
- Bunn HF 1987. Subunit assembly of hemoglobin: An important determinant of hematologic phenotype. Blood 69: 1–6 [PubMed] [Google Scholar]
- Bunn HF, McDonald MJ 1983. Electrostatic interactions in the assembly of haemoglobin. Nature 306: 498–500 [DOI] [PubMed] [Google Scholar]
- Bunn HF, Noguchi CT, Hofrichter J, Schechter GP, Schechter AN, Eaton WA 1982. Molecular and cellular pathogenesis of hemoglobin SC disease. Proc Natl Acad Sci 79: 7527–7531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui DH, Patterson M, Dowling CE, Kazazian HJ, Kendall AG 1990. Hemoglobin Bart’s disease in an Italian boy. Interaction between α-thalassemia and hereditary persistence of fetal hemoglobin. N Engl J Med 323: 179–182 [DOI] [PubMed] [Google Scholar]
- *.Dailey HA, Meissner PN 2013. Erythroid heme biosynthesis and its disorders. Cold Spring Harb Perspect Med 10.1101/cshperspect.a011676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Gell DA, Zhou S, Gu L, Kong Y, Li J, Hu M, Yan N, Lee C, Rich AM, et al. 2004. Molecular mechanism of AHSP-mediated stabilization of α-hemoglobin. Cell 119: 629–640 [DOI] [PubMed] [Google Scholar]
- *.Fucharoen S, Weatherall DJ 2012. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med 2: a011734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Higgs DR 2013. The molecular basis of α-thalassemia. Cold Spring Harb Perspect Med 3: a011718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihm AJ, Kong Y, Hong W, Russell JE, Rouda S, Adachi K, Simon MC, Blobel GA, Weiss MJ 2002. An abundant erythroid protein that stabilizes free α-haemoglobin. Nature 417: 758–763 [DOI] [PubMed] [Google Scholar]
- *.Lettre G 2012. The search for genetic modifiers of disease severity in the β-hemoglobinopathies. Cold Spring Harb Perspect Med 2: a015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollan TL, Yu X, Weiss MJ, Olson JS 2010. The role of α-hemoglobin stabilizing protein in redox chemistry, denaturation, and hemoglobin assembly. Antioxid Redox Signal 12: 219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollan TL, Khandros E, Weiss MJ, Olson JS 2012. The kinetics of α-globin binding to α hemoglobin stabilizing protein (AHSP) indicate preferential stabilization of a hemichrome folding intermediate. J Biol Chem 287: 11338–11350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrabet NT, McDonald MJ, Turci S, Sarkar R, Szabo A, Bunn HF 1986. Electrostatic attraction governs the dimer assembly of human hemoglobin. J Biol Chem 261: 5222–5228 [PubMed] [Google Scholar]
- *.Musallam KM, Tahar AT, Rachmilewitz EA 2012. β-Thalassemia intermedia: A clinical perspective. Cold Spring Harb Perspect Med 2: a013482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel RL, Daar S, Romero JR, Suzuka SM, Gravell D, Bouhassira E, Schwartz RS, Fabry M, Krishnamoorthy R 1998. HbS-Oman heterozygote: A new dominant sickle syndrome. Blood 92: 4375–4382 [PubMed] [Google Scholar]
- *.Nienhuis AW, Nathan DG 2012. Pathophysiology and clinical manifestations of the β-thalassemias. Cold Spring Harb Perspect Med 2: a011726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Sankaran VG, Orkin SH 2013. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med 3: a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Schechter AN 2013. Hemoglobin function. Cold Spring Harb Perspect Med 3: a011650 [Google Scholar]
- *.Schechter AN, Elion J 2013. Pathophysiology of sickle cell disease. Cold Spring Harb Perspect Med (to be published) [Google Scholar]
- *.Serjeant G 2013. Natural history of sickle cell disease. Cold Spring Harb Perspect Med 10.1101/cshperspect. a011783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Thein SL 2013. Molecular basis of β-thalassemia. Cold Spring Harb Perspect Med 10.1101/cshperspect.a011700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Thom CS, Dickson CF, Gell DA, Weiss MJ 2013. Hemoglobin variants: Biochemical properties and clinical correlates . Cold Spring Harb Perspect Med 10.1101/cshperspect.a011858 [DOI] [PMC free article] [PubMed] [Google Scholar]