

Abstract
Untreated HIV-1 infection progresses through acute and asymptomatic stages to AIDS. Although each of the three stages has well-known clinical, virologic, and immunologic characteristics, much less is known of the molecular mechanisms underlying each stage. In this study, we report lymphatic tissue microarray analyses, revealing for the first time stage-specific patterns of gene expression during HIV-1 infection. We show that although there is a common set of key genes with altered expression throughout all stages, each stage has a unique gene expression signature. The acute stage is most notably characterized by increased expression of hundreds of genes involved in immune activation, innate immune defenses (e.g., RIG-1, MDA-5, TLR7 and TLR8, PKR, APOBEC3B, 3F, 3G), adaptive immunity, and in the proapoptotic Fas-Fas ligand pathway. Yet, quite strikingly, the expression of nearly all acute stage genes return to baseline levels in the asymptomatic stage, accompanying partial control of infection. This transition from acute to asymptomatic stage is tied to increased expression of a diverse array of immunosuppressive genes (e.g., CLEC12B, ILT4, galectin-3, CD160, BCMA, FGL2, LAG3, GPNMB). In the AIDS stage, decreased expression of numerous genes involved in T cell signaling identifies genes contributing to T cell dysfunction. These common and stage-specific gene expression signatures identify potential molecular mechanisms underlying the host response and the slow, natural course of HIV-1 infection.
Untreated HIV-1 infection typically progresses from acute through a clinically asymptomatic stage to full-blown AIDS over the course of 5–10 years. In the acute stage (defined as individuals infected within 4 mo of documented sero-conversion to HIV), the rapid and profound damage to the host's immune system is manifest primarily as massive depletion of CD4+ T cells, especially in the gut (1, 2). Yet, despite this initial damage, most untreated patients enter a long period of clinical latency, the asymptomatic stage (defined as individuals infected for at least 4 mo with a CD4+ T cell count >200 cells/μl), in which the pace of immune damage slows. Eventually, however, with continued CD4+ T cell depletion and destruction of lymphatic tissue (LT)4 architecture, most infected patients progress to AIDS (defined as infected individuals with a CD4+ T cell count <200 cells/μl) (3).
Each stage has well-described clinical, virologic, and immunological characteristics, but the molecular mechanisms underlying these stage-specific features remain poorly understood. Thus, elucidating the molecular basis responsible for these key virologic and immunologic characteristics of the three stages of infection should improve our understanding of HIV-1 pathogenesis, particularly those genes and pathways that are important in determining the level of virus replication, the degree of immune activation, and the extent/pace of CD4+ T cell loss, as well as potentially identify new therapeutic targets.
Microarrays have been used for some time in the study of HIV-1 infection, including monitoring gene expression in cell lines, PBMCs, and PBMC subsets (4, 5). Although these previous studies are informative, they likely do not best represent the gene expression occurring in lymphoid tissue in vivo. To that end, we used Affymetrix Human Genome U133 Plus 2.0 Arrays to examine gene expression in LTs of HIV-1-positive subjects in each clinical stage compared with uninfected subjects (Table I), focusing on LTs because transcriptional profiles at this major site of virus production and pathology (6) should depict host-pathogen interactions directly relevant to pathogenesis. We show that there are stage-specific signatures of genes and pathways in LTs that highlight both a dramatically extensive early host response in acute infection and rapid resolution of that response during the asymptomatic stage, and we identify genes involved in T cell dysfunction in AIDS.
Table I.
Clinical characteristics of study subjects
| Patient | Disease Stage | Gender | Age (years) | Race | Blood CD4+ T Cell Count (cells/μl) | Plasma HIV-1 RNA Level (copies/ml) |
|---|---|---|---|---|---|---|
| 1476 | Uninfected | Female | 28 | Hispanic | 704 | Undetectable |
| 1472 | Uninfected | Female | 52 | Caucasian | 837 | Undetectable |
| 1442 | Uninfected | Female | 45 | Caucasian | 485 | Undetectable |
| 1452 | Uninfected | Male | 40 | Hispanic | 742 | Undetectable |
| 1467 | Uninfected | Male | 26 | Caucasian | 326 | Undetectable |
| 1324 | Acute | Male | 41 | Caucasian | 494 | 14,696 |
| 1430 | Acute | Male | 26 | Caucasian | 683 | 3,610 |
| 1458 | Acute | Male | 51 | Caucasian | 400 | 439,000 |
| 1329 | Acute | Male | 59 | Caucasian | 370 | 484,694 |
| 1389 | Acute | Male | 32 | Caucasian | 824 | 32,173 |
| 1449 | Acute | Male | 30 | Caucasian | 333 | >100,000 |
| 1435 | Acute | Male | 42 | Caucasian | 410 | >100,000 |
| 1391 | Acute | Male | 37 | African American | 414 | 24,718 |
| 1455 | Acute | Male | 23 | African American | 209 | 19,400 |
| 1463 | Asympta | Male | 23 | African American | 259 | 27,200 |
| 1335 | Asympt | Male | 32 | Caucasian | 400 | 15,284 |
| 1431 | Asympt | Male | 34 | Caucasian | 333 | 71,200 |
| 1464 | Asympt | Male | 34 | Caucasian | 202 | 122,000 |
| 1456 | Asympt | Male | 49 | Caucasian | 478 | 174,000 |
| 1408 | Asympt | Male | 39 | Caucasian | 685 | 20,014 |
| 1436 | Asympt | Male | 63 | Caucasian | 248 | 46,400 |
| 1407 | Asympt | Male | 35 | Caucasian | 372 | 31,922 |
| 1459 | Asympt | Male | 36 | Caucasian | 286 | >100,000 |
| 1438 | AIDS | Male | 49 | Caucasian | 147 | 4,960 |
| 1406 | AIDS | Male | 45 | African American | 188 | 10,684 |
| 1462 | AIDS | Male | 43 | Caucasian | 81 | 35,000 |
| 1327 | AIDS | Female | 40 | African American | 112 | 12,046 |
Note. Undetectable, assay limit of detection (50 copies/ml).
Asympt, Asymptomatic stage.
Materials and Methods
Lymph node biopsies, RNA extractions, synthesis of biotin-labeled cRNA probes, and microarray hybridization followed previously published procedures (7).
Lymph node biopsy specimens
Inguinal lymph node biopsies from 22 untreated HIV-1-infected subjects at different clinical stages and 5 uninfected subjects were obtained for this University of Minnesota Institutional Review Board-approved microarray study. Each lymph node biopsy was placed into a Falcon tube and snap frozen by dropping it into liquid nitrogen.
RNA extraction, synthesis of biotin-labeled cRNA probes, and microarray hybridization
Frozen lymph nodes were homogenized with a power homogenizer (Heat Systems Ultrasonic) in TRIzol (catalog no. 15596-018; Invitrogen) without thawing. Total RNA was isolated according to the manufacturer's protocol and further purified with a RNeasy Mini Kit (catalog no. 74104; Qiagen). Double-stranded cDNA and biotin-labeled cRNA probes were synthesized from 5 μg of total RNA with a MessageAmp II aRNA kit (catalog no. AM1757; Ambion). The cRNA probes were column purified and fragmented with a fragmentation kit (catalog no. 8740; Ambion).
Fifteen micrograms of fragmented cRNA was hybridized to an Affymetrix Human Genome U133 Plus 2.0. After hybridization, chips were washed, stained with streptavidin-PE, and scanned with GeneChip Operating Software at the Biomedical Genomics Center at the University of Minnesota. The experiments from each RNA sample were duplicated in the preparation of each cRNA probe and microarray hybridization.
Microarray data analysis
Cel. files were uploaded into the Expressionist program (Genedata; Pro version 4.5) and the expression level for each of the 47,000 transcripts in the arrays were analyzed by using the robust multi-array analysis algorithm. The expression levels from duplicated chips of the same individual's RNA were correlated and averaged. The expression data from all individuals were exported as Excel files for statistical analysis.
Tests for differences between the stages were conducted using the two-sample t test, assuming the variance of the measurements are the same in the two groups. Fold differences in the level of gene expression between any two stages were estimated with the ratio of the means in the two stages. All computations were conducted using the software S-plus version 3.4 from MathSoft.
After statistical analysis, data were sorted based on these transcript cutoffs: p ≤ 0.05 and fold change ≥1.7. Significantly changed genes and transcripts were uploaded into the NetAffix Analysis Center (http://www.affymetrix.com/analysis/index.affx) to query gene ontology information and into Ingenuity Pathways Analysis (Ingenuity Systems, www.ingenuity.com) for gene annotation and pathway analysis.
The data discussed in this publication have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus (40) and are accessible through GEO Series accession no. GSE16363 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE16363).
Real-time RT-PCR and immunohistochemical staining
Significantly altered genes (STAT1, RANTES, IFN-γ) in the microarray analysis were confirmed both by LightCycler real-time PCR and by immunohistochemical staining as published previously (7).
Results
We found that HIV-1-infected subjects (22 individuals) were clearly distinguishable from uninfected subjects (5 individuals) and that each disease stage had a distinct gene expression signature. As is evident in Fig. 1, the stages differed compared with uninfected subjects in both the number of genes with altered expression as well as the direction of change in gene expression.
FIGURE 1.
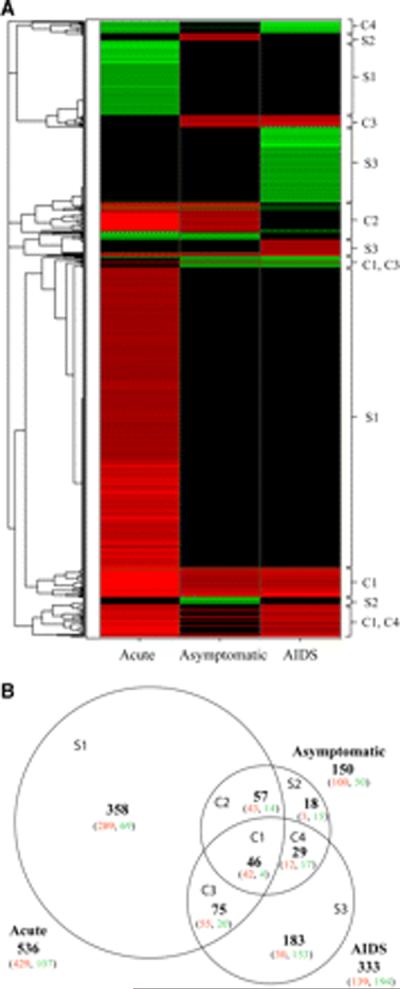
Classification of altered genes in LT from HIV-1-infected patients into disease stages and representative hierarchical clustering reveals stage-specific gene signatures. Genes with altered expression (≥1.7 times the level at uninfected baseline, p < 0.05) at the three stages of HIV-1 infection were characterized as the following: unique to the acute stage (S1), asymptomatic stage (S2), or AIDS stage (S3); C1, common to all stages; C2, common to acute and asymptomatic stages; C3, common to asymptomatic and AIDS stages; and C4, common to acute and AIDS stages. A, Hierarchical cluster analysis of altered genes from HIV-1-infected subjects (stage indicated at the bottom). Green, red, and black areas indicate, respectively, decreased, increased, and no significant change in expression compared with uninfected controls. B, Venn diagram depicting the number of genes with altered expression. The size of each region is proportional to the number of altered genes. Green and red numbers indicate, respectively, decreased and increased expression.
Altogether, from the array's nearly 47,000 transcripts, we found 766 altered genes. Only ~6% (46 genes) of the altered genes were common to all stages (region C1 in Fig. 1), with another ~21% (161 genes) common to two stages (C2, C3, and C4) and ~73% (559 genes) unique to one stage (S1, S2, and S3). Strikingly, ~70% of the 766 genes {536} were altered in the acute stage (S1, C1, C2, and C3), with ~67% (358 genes) of those unique to this stage (S1) and ~81% (289 genes) of those unique genes had increased expression. In sharp contrast, the asymptomatic stage (S2) had few (18 genes) uniquely altered genes. The AIDS stage (S3) had 183 uniquely altered genes, with ~84% (153 genes) decreased in expression.
Based on gene ontology/annotations from the NetAffx Analysis Center, Ingenuity Pathways Analysis, and extensive examination of published literature, we classified ~80% of all altered genes (621 genes) into functional categories shown in Fig. 2 and supplemental Table I.5
FIGURE 2.

In human LT of HIV-1-infected subjects, the general functional categories of genes with altered expression (≥1.7 times the level at baseline, p < 0.05). A, Common to all stages (C1); B, unique to the acute stage (S1); C, unique to the asymptomatic stage (S2); and D, unique to the AIDS stage (S3). Green and red letters indicate, respectively, decreased and increased expression. The size of each sector in a pie diagram is proportional to the number of genes in its category (in parentheses). All genes and their names derived from abbreviations can be found in supplemental Table I.
Common
Common to all stages (region C1) were 46 genes (Fig. 2A), one-half (23 genes) of which have known functions in immune defenses. STAT1 expression was highly increased, as were defense genes regulated by STAT1 such as IFN-γ and a number of IFN-related antiviral genes like 2′, 5′-oligoadenylate synthetase 1 (OAS1), ISG15, IFI27, and the guanylate-binding proteins (GBP1, 5). Genes involved in immune activation and cell proliferation accounted for ~25% (12 genes) of common genes, including cell cycle-related genes such as cyclin B1. Expression of most of these genes increased. Also, expression increased for genes encoding the important anti-HIV-1 chemokines RANTES and MIP-1β and their cognate receptor CCR5, as well as eight genes encoding proteins associated with NK cell and CD8+ T cell-mediated cytotoxicity, such as CD8, granzymes, and cytotoxic and regulatory T cell molecule.
Acute stage
The acute stage had by far the most (358 genes) uniquely altered genes (region S1), with ~81% (289 genes) increased in expression. Fig. 2B shows the functional categories of 291 unique acute stage genes; the rest appear in supplemental Table I.
Immune activation accounted for ~40% (144 genes) of unique acute stage genes. Notable are: 1) proinflammatory mediators (e.g., IL-32 and IL-1R antagonist); 2) signal transduction molecules (e.g., the T cell-related gene calmodulin 3); 3) transcriptional regulators (e.g., T-box 21 and the thymus hormone thymopoietin); 4) activators of cell proliferation (e.g., Ki-67 and proliferating cell nuclear Ag); 5) mediators of cell cycle progression (e.g., nucleophosmin/B23 and cell division-cycle-associated proteins); and 6) moderators of pathologic processes associated with immune activation (e.g., metallothioneins and thioredoxins to counter free radical damage).
Countering the predominance of increased expression of immune activation genes was an apparent paradoxical increase in genes whose function is clearly immunosuppressive. This may reflect a mechanism to balance the necessary activation of host defenses while avoiding the immunopathologic effects of immune activation but also may be detrimental in dampening the immune response in the face of continuing viral replication. Genes with increased expression in the acute stage linked to immunosuppression included the C-type lectin domain family 12, member B (CLEC12B), glycoprotein (transmembrane) nmb (GPNMB), and galectin-3, all inhibitors of macrophage activation and inflammatory processes, CD160, and LAG3), both negative regulators of T cell activation, ILT4, a receptor involved in the tolerization of dendritic cells (DCs), and IDO, an enzyme with far-reaching immunosuppressive effects regulating the immune system. The single immunoregulatory gene decreased in expression was RANKL, implicated in the maintenance and survival of DCs and peripheral CD4+CD25+ regulatory T cells.
Immune defenses accounted for ~7% (25 genes) of unique acute stage genes, representative of a broad, innate immune response in which the IFN system is the most prominent component of the host's response to HIV-1, e.g., increased expression of genes encoding OAS2 and 3, myxovirus resistance 2, polynucleotide phosphorylase, IFN-induced transmembrane protein 1 (9–27), and numerous others. Additional genes of innate immunity included increased expression of three complement and FcR genes as well as a number of antiviral molecules and pattern recognition receptors—genes coding for TLRs 7 and 8 and the C-type lectin receptors Dectin-1 and mannose receptor, the cytoplasmic RNA helicase proteins RIG-I and MDA-5, dsRNA-dependent protein kinase (PKR), and the APOBECs, including not only the known suppressors of HIV-1 replication, APOBEC3G and -3F, but also -3B. In addition, expression of four genes coding for cytotoxic effectors such as granzymes and killer-specific secretory protein of 37 kDa increased, consistent with innate and adaptive defenses by NK cells and CTL.
Apoptosis accounted for ~4% (14 genes) of unique acute stage genes. Quite selectively, among numerous canonical apoptotic pathways, only the Fas-Fas ligand-mediated cell death pathway was activated (Fig. 3). Proapoptotic genes increased in this pathway included Fas ligand, caspases 3 and 4, BH3-interacting domain death agonist (BID), and truncated BID, highlighting the importance of the Fas-Fas ligand pathway in LT during acute stage infection.
FIGURE 3.

Illustration of altered genes in the acute stage related to canonical apoptotic signaling pathways reveals activation of the Fas-Fas ligand pathway. Ingenuity Pathways Analysis software was used to identify altered genes related to canonical apoptotic pathways; red indicates genes significantly increased in expression and black indicates no significant change in gene expression.
Finally, tissue repair/remodeling and metabolism accounted for, respectively, ~9% (33 genes) and ~24% (87 genes) of unique acute stage genes. Notable are chitinase 3 like-2, decorin, and dermatopontin, genes that could be plausibly involved in collagen formation/deposition, an important pathologic process observed in HIV-1 infection (8). Also, ~90% (78 genes) of metabolism genes were increased in expression, suggesting accelerated cell growth and turnover.
Asymptomatic stage
In contrast to the acute stage, the asymptomatic stage (region S2) had relatively few altered genes (18 genes; Fig. 2C), reflecting a return to baseline levels of nearly all acute stage genes and a major shift in host response to the virus. We return in the Discussion to the mechanisms and significance of this striking change.
AIDS stage
Unlike the predominance of increased expression in the acute stage, genes unique to the AIDS stage (region S3) were largely decreased, ~84% of 183 (Fig. 2D). These included genes involved in immune activation, apoptosis, and tissue repair/remodeling. Interestingly, a substantial number of decreased genes were involved in the CD28 T cell costimulatory pathway, key molecules such as CD28, Txk (Tec family of nonreceptor tyrosine kinases), the lymphocyte-specific serine/threonine kinase death-associated protein-related apoptotic kinase 2 (DRAK2/STK17b), and the Ca2+-binding protein calmodulin, suggesting, in addition to significant T cell loss, T cell stimulation may be compromised during AIDS.
The small number of AIDS stage genes increased in expression included the antiviral chemokine MIP-1α, the innate antimicrobial molecule S100A9, the DCl and monocyte-derived macrophage differentiation marker decysin, and the cell surface proteoglycan syndecan-1.
Discussion
The major finding of this study is that there are stage-specific transcriptional signatures in LTs during HIV-1 infection. These signatures identify constellations of gene expression relevant to understanding the molecular mechanisms involved in the pathogenesis of each stage.
In the acute stage of infection, the large number of altered genes, most of them increased in expression (~81%), provides a signature consistent with the dramatic pace and magnitude of events in this stage seen clinically, virologically, and immunologically. Although the explosive replication of virus to peak levels in the first month of infection is known to elicit immune activation and host defenses that partially control infection, the transcriptional signatures now provide a comprehensive picture of the genes in LTs involved in immune activation, partial control of infection, and moderation of the pathologic consequences of immune activation.
Another general feature of this acute stage transcriptional signature is that it is a mirror image (opposite) of the transcriptional signature of infected individuals' treated with highly active anti-retroviral therapy. In response to treatment and the control of viral replication, genes related to immune activation and innate/adaptive defenses decreased, in contrast to the increased expression in untreated acute infection shown here (i.e., immune defense genes such as IFN-γ, OAS, MX2, IDO, RIG-1, granzymes and immune activation genes such as STAT1, FGL2). This result supports a model proposed by Li et al. (7), namely, that suppression of viral replication via treatment would relieve the stimulus driving gene expression regulating immune activation and defenses that were partially controlling infection, thus leading to decreased expression of these genes. The acute stage signature now confirms that prediction.
Not surprisingly, the host rapidly responds to HIV-1 infection by increasing expression of a large number of genes related to innate and adaptive defenses during the acute stage, such as IFN-related genes and those associated with NK cell- and CD8+ T cell-mediated cytotoxicity (Fig. 2B). However, what was surprising was the breadth and distinctive nature of this response and its restriction to acute stage infection. For example, APOBEC3B, -3F, and -3G were only increased during this stage. The potent anti-HIV-1 properties via cytidine deamination-dependent (9) and -independent mechanisms (10) of APOBEC3F and -3G have been well documented, but APOBEC3B, previously considered an intrinsic cellular defense against endogenous retrotransposons (11), has only recently been shown to have anti-HIV-1 activity in vitro (12, 13). The transcriptional signatures now document in vivo deployment of three of the APOBEC proteins as a first line of defense against HIV-1.
A number of key pathogen recognition receptors also were increased in expression only during acute stage infection, e.g., RIG-I, MDA-5, PKR, and TLRs 7 and 8. These proteins work in concert to detect and inhibit invading pathogens by recognizing various forms of viral RNA, transducing a downstream signal that activates accessory proteins such as NF-κB and IFN regulatory factor 3, leading to the production of proinflammatory cytokines and type I IFN (14, 15). In addition to the production of type I IFNs, engagement of TLR7, which is highly expressed in plasmacytoid DCs (16), can enhance HIV-1-specific T cell responses (17) while TLR8 signaling can inhibit HIV-1 replication in susceptible target cells (18). The restricted up-regulation of TLRs 7 and 8 in acute stage infection is consistent with their importance in early detection of viral infection and the concept of robust expression of sentinel-like molecules to create an antiviral state in the initial stages of HIV-1 infection.
Not surprisingly as well, the largest number of altered genes during acute infection were related to immune activation, encompassing categories from cell cycle to proinflammatory mediators to genes involved in the proliferation of cells. However, what the acute stage signature surprisingly reveals is the distinctive nature of genes in LTs involved in immune activation and their restriction to this stage. For example, among all of the potential proinflammatory cytokines, we only observed increased expression of the gene encoding cytokine IL-32.
IL-32 is an inflammatory cytokine induced by TH1-type cytokines in various cell types (19, 20) and has only recently received increased attention as an important component in autoimmune and inflammatory diseases through induction of other proinflammatory cytokines such as TNF-α (19–21), IL-1β (20, 21), IL-8 (19), and MIP-2 (19, 21). Additionally, one report (22) has implicated IL-32 in activation-induced cell death in T cells. This is the first study showing IL-32 expression in LTs in HIV-1 infection. IL-32 likely contributes to the exacerbation of inflammation during HIV-1 infection and may have an important, albeit heretofore unappreciated role in T cell death.
Interestingly, a number of pattern recognition receptors recognizing microbial products were also up-regulated during acute infection. Recent evidence has implicated microbial translocation from the gastrointestinal tract during HIV-1 infection as a factor driving systemic immune activation (23). The up-regulation of genes such as dectin-1 and galectin-3 may be key switches utilized by the host to recognize and alert the immune system to deal with an increase in foreign microbial products. Dectin-1 plays a key role in recognizing β-glucan-containing microorganisms and, once engaged, leads to the production of IL-23, a key cytokine involved in the generation of anti-bacterial Th17 cells (24). Galectin-3 has recently been shown to be a negative regulator of LPS-induced inflammation (25). The up-regulation of these genes during acute infection suggests that the host may be responding to an increased presence of bacteria, a compensatory mechanism to curtail aberrant immune activation.
Among several apoptotic pathways, the acute stage signature strongly points to a principal role for Fas-Fas ligand-mediated apoptosis in LTs of HIV-1-infected subjects during this stage. Key apoptosis-related genes map strictly to the Fas-Fas ligand pathway, including downstream effectors such as BID, truncated BID, and caspase 3, all increased in expression (Fig. 3).
In the asymptomatic stage, the rapid return to levels of gene expression indistinguishable from uninfected HIV-1 uninfected subjects and the paucity of altered genes unique to this stage, 18 genes vs 358 unique acute stage genes, is remarkable, given evidence of ongoing viral replication and lymphoid tissue damage during this stage despite its relative clinical quiescence (6, 26). This transition begins in the acute stage with increased expression of numerous immunosuppressive genes such as CLEC12B, ILT4, galectin-3, CD160, BCMA, FGL2, LAG3, GPNMB, and IDO at a time of immune activation, consistent with a role in moderating the immunopathological consequences of sustained immune activation and thereby contributing to slow disease progression characteristic of HIV-1, SIV, and other lentiviral infections. However, these moderating effects may come at a price, in dampening the immune response to infection and, in the long term, contributing to a dysfunctional and increasingly “exhausted” immune system (27).
The defining feature of the AIDS stage signature was decreased expression of most (~84%) of the 183 unique genes. Although decreased expression could be due to loss of cells expressing these genes, decreased expression in a particular cell type, or both, the net effect is the same dysfunctional host defenses due to decreased expression of various genes, such as: 1) CD244/2B4, a cell surface molecule expressed on NK and memory CD8+ T cells that determines the level of activation or inhibition based on the degree of receptor expression and extent of its ligation (28); 2) CCR7, the principal chemokine receptor responsible for migration of naive and memory T cells and DCs into lymph nodes (29); and 3) a number of genes involved in T cell activation/costimulation, most prominently the costimulatory signal-transmitting receptor CD28 (30) and other molecules associated with this pathway such as the T cell-specific tyrosine kinase Txk (31), DRAK2/STK17b, a serine/threonine kinase expressed specifically in B and T cells that modulates the threshold required for activation/costimulation (32), and Ca2+/calmodulin-dependent protein kinase IV, a key kinase likely involved in signal transduction events downstream of Txk (33).
Some of the changes in gene expression during AIDS may also contribute to CD4+ T cell depletion and limit reconstitution through the microenvironment-impairing deposition of collagen (8), such as increased expression of the gene encoding a cell surface proteoglycan, syndecan-1, which colocalizes with fibrosis-promoting-TGF-β in fibrotic and inflamed areas of cirrhotic human liver (34).
We interpret the small number of genes with altered expression common to all stages as evidence that there is a key set of genes required by the host to partially contain the virus. Many of the altered genes that are common to all stages of infection are related to immune activation and immune defenses, most prominently, CD38, a known marker for immune activation and poor prognosis when highly expressed in HIV-1 infection (35, 36), and genes encoding products associated with NK cells, IFNs, CD8+ T cell-mediated cytotoxicity, and antiviral chemokines. Altered expression of these genes highlights the importance of innate immunity and the IFN system throughout HIV-1 infection.
Our microarray study revealed very little overlap (<5%) with recent small interfering RNA-knockdown screens (37–39) designed to identify important host factors required for HIV-1 infection. This is likely due to differences between in vitro systems utilizing laboratory-adapted strains of virus and cell lines vs biopsied lymph nodes from HIV-1-infected individuals. Moreover, microarray assays measure change at the mRNA level and would thus not detect posttranscriptional changes in gene expression.
This microarray study is a descriptive approach to glean key insights into systems biology. The genes we identified in stage-specific, transcriptional signatures are a good starting point for further investigations of their roles in HIV-1 pathogenesis using in situ technologies to identify the types of cells in which gene expression has been altered and immunohistochemistry to identify potential changes in protein expression. In addition, it will be fruitful to examine the relationships between gene expression and infected cells, CD4+ T cell depletion, and collagen deposition, one pathological consequence of infection. Ultimately, these studies may be used not only to better understand viral pathogenesis during the various stages of HIV-1 disease but also to identify adjunctive approaches to improving treatment and immune reconstitution.
Acknowledgments
We thank Colleen O'Neill and Tim Leonard for help with the manuscript and figures.
Footnotes
This work was supported by Public Health Services Grant R01 AI056997 from the National Institute of Allergy and Infectious Diseases.
Abbreviations used in this paper: LT, lymphatic tissue; DC, dendritic cell; BID, BH3-interacting domain death agonist.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, Reilly C, Carlis J, Miller CJ, Haase AT. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434:1148–1152. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- 2.Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 3.Fauci AS. Host factors and the pathogenesis of HIV-induced disease. Nature. 1996;384:529–534. doi: 10.1038/384529a0. [DOI] [PubMed] [Google Scholar]
- 4.Giri MS, Nebozhyn M, Showe S, Montaner LJ. Microarray data on gene modulation by HIV-1 in immune cells: 2000–2006. J. Leukocyte Biol. 2006;80:1031–1043. doi: 10.1189/jlb.0306157. [DOI] [PubMed] [Google Scholar]
- 5.Hyrcza MD, Kovacs C, Loutfy M, Halpenny R, Heisler L, Yang S, Wilkins O, Ostrowski M, Der SD. Distinct transcriptional profiles in ex vivo CD4+ and CD8+ T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J. Virol. 2007;81:3477–3486. doi: 10.1128/JVI.01552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haase AT. Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 1999;17:625–656. doi: 10.1146/annurev.immunol.17.1.625. [DOI] [PubMed] [Google Scholar]
- 7.Li Q, Schacker T, Carlis J, Beilman G, Nguyen P, Haase AT. Functional genomic analysis of the response of HIV-1-infected lymphatic tissue to antiretroviral therapy. J. Infect. Dis. 2004;189:572–582. doi: 10.1086/381396. [DOI] [PubMed] [Google Scholar]
- 8.Schacker TW, Nguyen PL, Beilman GJ, Wolinsky S, Larson M, Reilly C, Haase AT. Collagen deposition in HIV-1 infected lymphatic tissues and T cell homeostasis. J. Clin. Invest. 2002;110:1133–1139. doi: 10.1172/JCI16413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu Y-L, Greene WC. Multifaceted antiviral actions of APOBEC3 cytidine deaminases. Trends Immunol. 2006;27:291–297. doi: 10.1016/j.it.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation: comparisons with APOBEC3G. J. Biol. Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 11.Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. USA. 2006;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng G, Greenwell-Wild T, Nares S, Jin W, Lei KJ, Rangel ZG, Munson PJ, Wahl SM. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood. 2007;110:393–400. doi: 10.1182/blood-2006-10-051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doehle BP, Schafer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 15.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 16.Ito T, Wang YH, Liu YJ. Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by Toll-like receptor (TLR) 7 and TLR9. Springer Semin. Immunopathol. 2005;26:221–229. doi: 10.1007/s00281-004-0180-4. [DOI] [PubMed] [Google Scholar]
- 17.Lore K, Betts MR, Brenchley JM, Kuruppu J, Khojasteh S, Perfetto S, Roederer M, Seder RA, Koup RA. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J. Immunol. 2003;171:4320–4328. doi: 10.4049/jimmunol.171.8.4320. [DOI] [PubMed] [Google Scholar]
- 18.Schlaepfer E, Audige A, Joller H, Speck RF. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J. Immunol. 2006;176:2888–2895. doi: 10.4049/jimmunol.176.5.2888. [DOI] [PubMed] [Google Scholar]
- 19.Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFα. Immunity. 2005;22:131–142. doi: 10.1016/j.immuni.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Shoda H, Fujio K, Yamaguchi Y, Okamoto A, Sawada T, Kochi Y, Yamamoto K. Interactions between IL-32 and tumor necrosis factor α contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res. Ther. 2006;8:R166. doi: 10.1186/ar2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joosten LAB, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TRD, Barrera P, van de Loo FAJ, Dinarello CA, van den Berg WB. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 2006;103:3298–3303. doi: 10.1073/pnas.0511233103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goda C, Kanaji T, Kanaji S, Tanaka G, Arima K, Ohno S, Izuhara K. Involvement of IL-32 in activation-induced cell death in T cells. Int. Immunol. 2006;18:233–240. doi: 10.1093/intimm/dxh339. [DOI] [PubMed] [Google Scholar]
- 23.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 24.Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol. Rev. 2008;226:112–131. doi: 10.1111/j.1600-065X.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Komai-Koma M, Gilchrist DS, Hsu DK, Liu FT, Springall T, Xu D. Galectin-3 is a negative regulator of lipopolysaccharide-mediated inflammation. J. Immunol. 2008;181:2781–2789. doi: 10.4049/jimmunol.181.4.2781. [DOI] [PubMed] [Google Scholar]
- 26.Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenisis of HIV infection: what the virus spares is as important as what it destroys. Nat. Med. 2006;12:289–295. doi: 10.1038/nm1380. [DOI] [PubMed] [Google Scholar]
- 27.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DAA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chlewicki LK, Velikovsky CA, Balakrishnan V, Mariuzza RA, Kumar V. Molecular basis of the dual functions of 2B4 (CD244) J. Immunol. 2008;180:8159–8167. doi: 10.4049/jimmunol.180.12.8159. [DOI] [PubMed] [Google Scholar]
- 29.Cyster JG. Leukocyte migration: scent of the T zone. Curr. Biol. 2000;10:R30–33. doi: 10.1016/s0960-9822(99)00253-5. [DOI] [PubMed] [Google Scholar]
- 30.Berridge MJ. Lymphocyte activation in health and disease. Crit. Rev. Immunol. 1997;17:155–178. doi: 10.1615/critrevimmunol.v17.i2.30. [DOI] [PubMed] [Google Scholar]
- 31.August A, Fischer A, Hao S, Mueller C, Ragin M. The Tec family of tyrosine kinases in T cells, amplifiers of T cell receptor signals. Int. J. Biochem. Cell Biol. 2002;34:1184–1189. doi: 10.1016/s1357-2725(02)00068-7. [DOI] [PubMed] [Google Scholar]
- 32.McGargill MA, Wen BG, Walsh CM, Hedrick SM. A deficiency in Drak2 results in a T cell hypersensitivity and an unexpected resistance to autoimmunity. Immunity. 2004;21:781–791. doi: 10.1016/j.immuni.2004.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson KA, Kane CD. Ca2+/calmodulin-dependent protein kinase IV and calcium signaling. Biometals. 1998;11:331–343. doi: 10.1023/a:1009276932076. [DOI] [PubMed] [Google Scholar]
- 34.Kovalszky I, Nagy P, Szende B, Lapis K, Szalay F, Jeney A, Schaff Z. Experimental and human liver fibrogenesis. Scand. J. Gastroenterol. 1998;33(Suppl. 228):51–55. doi: 10.1080/003655298750026561. [DOI] [PubMed] [Google Scholar]
- 35.Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, Debernardo R, Garate K, Rodriguez B, Margolis LB, Lederman MM. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood. 2007;109:4272–4279. doi: 10.1182/blood-2006-11-055764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onlamoon N, Tabprasit S, Suwanagool S, Louisirirotchanakul S, Ansari AA, Pattanapanyasat K. Studies on the potential use of CD38 expression as a marker for the efficacy of anti-retroviral therapy in HIV-1-infected patients in Thailand. Virology. 2005;341:238–247. doi: 10.1016/j.virol.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 37.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engleman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 38.Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GMC, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Micro. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]