

Abstract
An unexpected dichotomy was observed in the Ru-catalyzed asymmetric transfer hydrogenation of acyl phosphonates: reduction proceeded from the opposite face relative to that observed in the analogous reduction of α-keto esters. The first highly selective catalytic hydrogenation of acyl phosphonates was utilized in the dynamic kinetic resolution of α-aryl acyl phosphonates providing β-stereogenic α-hydroxy phosphonic acid derivatives.
The asymmetric synthesis of small molecules has profited from the development of well-defined homogenous catalysts.1 Asymmetric catalysis relies on the fundamental paradigm that privileged catalysts generate well-defined chiral spaces that provide an environment capable of effectively directing similarly structured small molecules for enantiofacial discrimination.2 This characteristic is practically useful insofar as a seminal advance can pave the way for useful extensions based on structurally-related congeners. Deviations from this principle are rare and important in understanding substrate/catalyst interactions.3 Herein, we disclose an unusual diametric reversal in diastereofacial selection in the asymmetric transfer hydrogenation of acyl phosphonates compared to the related α-keto esters. The reactions described provide access to new β-stereogenic-α-hydroxy phosphonic acid derivatives that have previously been inaccessible in stereoisomerically pure form.
Background/Rationale
α-Keto esters and acyl phosphonates A behave analogously in the bis(oxazoline)Cu(II)-catalyzed asymmetric hetero Diels-Alder reaction with vinyl ethers to provide dihydropyrans C and D, respectively (Figure 1, paths a and b).4 Activation of the dicarbonyl moiety via chelation is crucial in providing high levels of facial selectivity. We were interested in testing the notion that the α-keto ester/acyl phosphonate relationship could be exploited in the context of our laboratory’s ongoing work involving dynamic kinetic resolution5 by asymmetric transfer hydrogenation (DKR-ATH). We recently documented a new Ru(II)-catalyzed DKR-ATH of β-aryl-α-keto esters B providing hydride delivery from the Si-face to afford α-hydroxy esters E with high levels of diastereo- and enantioselectivity (path c).6 Extrapolating from precedent, the dynamic reduction of racemic α-aryl acyl phosphonate B was proposed to occur with analogous facial preference; however, in the event, the reduction occurred from the opposite diastereotopic face providing the quasidiastereomeric product F with excellent levels of selectivity (path d).7
Figure 1.

Reversal in enantiofacial selectivity.
Context
The leading methodology in the literature for the enantioselective preparation of α-hydroxy phosphonates is the addition of dialkyl phosphites to aldehydes (Pudovik reaction).8 Despite its synthetic utility as a C–P bondforming reaction, the absence of a diastereoselective variant hinders its incorporation in complex molecule synthesis. In principle, a complementary approach to the enantioselective Pudovik reaction is the asymmetric reduction of acyl phosphonates. Recently, Goulioukina and Beletskaya reported the first catalytic, asymmetric hydrogenation of acyl phosphonates, albeit with modest selectivity (up to 77.5:22.5 er).9 Despite the wealth of methodologies developed to access this important structural motif, methodologies designed to efficiently access β-stereogenic-α-hydroxy phosphonates are scarce.10 The development of the title reaction would provide a flexible entry point into new α-hydroxy phosphonic acid derivatives; this subunit appears in compounds exhibiting antibacterial, antiviral, antibiotic, pesticidal, and anticancer properties.11
Results
Employing α-aryl acyl phosphonate 1b as a test substrate, Noyori’s RuCl[(S,S)-TsDPEN](p-cymene) complex12 was found to provide hydroxy phosphonate 2b with modest anti/syn selectivity, but excellent levels of enantiocontrol for both diastereomers (Table 1, entry 1). Based on our group’s recent success in tuning the diastereoselectivity of the DKR-ATH of β-chloro-α-keto esters through the application of a bulky m-terphenylsulfonamide ligand,6b aminosulfonamide L2 was employed in the reduction of 1b in DMF and delivered a marked increase in diastereoselectivity (entry 2). Changing the solvent to DMSO resulted in a boost in diastereoselection up to 20:1 (entry 3). α-Naphthyl ethylenediamine-derived L3 was tested and found to engender even higher levels of diastereocontrol (entries 4 and 5). Both dimethyl and diethyl phosphonates were found to provide comparable levels of reactivity and selectivity (entries 5 and 6); however, the bulkier diisopropyl phosphonate 1c suffered from reduced reactivity presumably due to its increased steric requirements (entry 7).
Table 1.
Ligand/Substrate Optimization.a
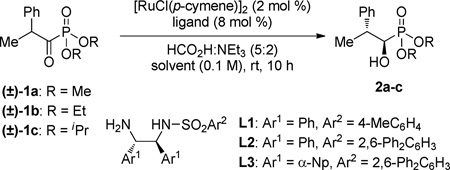 | ||||||
|---|---|---|---|---|---|---|
| entry | 1 | L | solvent | conv. (%)b |
anti:synb | erc |
| 1 | 1b | L1 | DMSO | >95 | 3:1 | >99.5:0.5 (98.5:1.5)d |
| 2 | 1b | L2 | DMF | >95 | 14:1 | 99:1 |
| 3 | 1b | L2 | DMSO | >95 | 20:1 | >99.5:0.5 |
| 4 | 1b | L3 | DMF | >95 | 22:1 | >99.5:0.5 |
| 5 | 1b | L3 | DMSO | >95 (93)e | 29:1 | >99.5:0.5 |
| 6 | 1a | L3 | DMSO | >95 (91)e | >30:1 | >99.5:0.5 |
| 7 | 1c | L3 | DMSO | 19 | – | – |
Reactions performed on 0.155 mmol scale;
Determined by 31P NMR analysis of the crude reaction mixture;
Determined by chiral HPLC analysis;
Syn isomer;
Isolated yield.
With optimized reaction conditions in hand, the reaction scope was examined (Table 2). A variety of electron-releasing and electron-withdrawing aryl groups were tolerated providing products in uniformly high yield and selectivity. Heteroaromatic substituents were also amenable to the reaction providing the N-Ts indoyl product 2k in 94% yield with excellent levels of diastereo- and enantiocontrol. Ortho-substitutents resulted in reduced reactivity necessitating elevated temperatures (45 °C) and longer reaction times to provide 2j in 6:1 dr and 98.5:1.5 er.13
Table 2.
Aromatic Substrate Scope.a
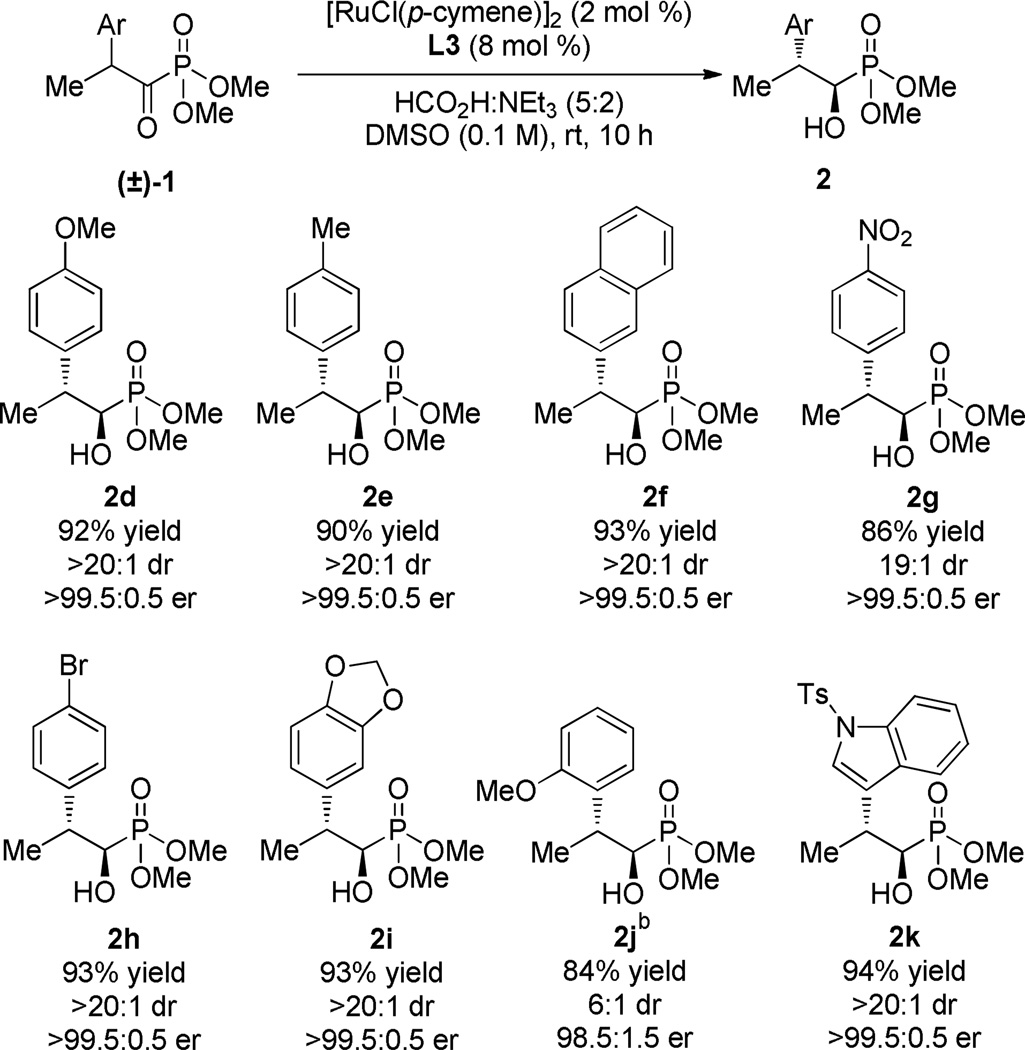 |
Reactions were performed on 0.155 mmol scale employing 5 equiv. HCO2H:NEt3 (5:2). Isolated yields of analytically pure material are reported. Diastereomer ratios were determined by 31P NMR analysis of the crude reaction mixture; enantiomer ratios were determined by chiral HPLC analysis.
Reaction performed at 45 °C for 20 h.
The identity of the α-aliphatic substituent was also investigated to probe the steric sensitivity of the system (Table 3). Linear aliphatic substituents were tolerated, providing products in equally high yield and selectivity and allowing for the incorporation of alkene and alkyne functional handles. The sterically demanding cyclopropyl acyl phosphonate reacted slower under the reactions conditions, requiring 36 h to provide 2p in 5:1 dr and excellent enantiocontrol.
Table 3.
Alkyl Substrate Scope.a
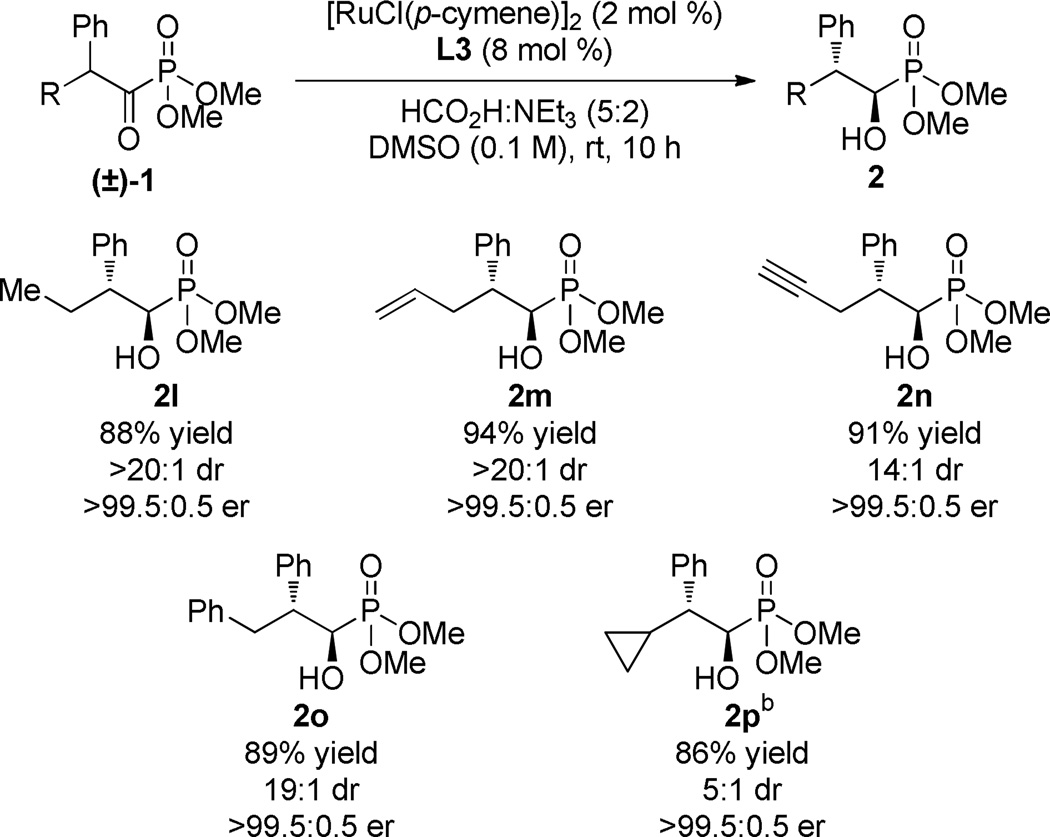 |
As in Table 2.
36 h.
To further probe the utility of this reaction, the bicyclic substrate 1q was subjected to the reduction conditions affording 2q in high yield and comparable levels of selectivity as the acyclic examples (Scheme 1). In contrast to ortho-substituted 2j, hydroxy phosphonate 2q was obtained with excellent levels of diastereoselectivity suggesting that the ortho-substituent occupies a sterically encumbering conformation when unconstrained causing nonideal substrate-catalyst interactions. The absolute stereochemistry of the products was established as (1R,2R) via x-ray crystallographic analysis of 2e and 2q confirming the anti orientation of the alcohol and aryl groups.14
Scheme 1.

DKR-ATH of cyclic substrate 1q.
The presence of a β-substituent was found to be unnecessary for high levels of enantioselectivity (Scheme 2). Despite the development of excellent catalysts for highly asymmetric Pudovik reactions into aromatic aldehydes, simple aliphatic aldehydes typically provide lower levels of selectivity. Although the reduction of aryl acyl phosphonate 3a under optimized reaction conditions provided (R)-4a15 with an er of only 92:8 er, the reduction of aliphatic acyl phosphonates proceeded to provide enantiopure products 4b–d in high yield.16 The excellent levels of enantiocontrol observed for 4b–d are a marked improvement over Pudovik-based methodologies, highlighting the potential utility and complementarity of this transfer hydrogenation in the preparation of enantiopure α-hydroxy phosphonic acids bearing one stereocenter.
Scheme 2.

ATH of acyl phosphonates.
The turnover in stereoselectivity will require further investigation to fully understand, but some initial observations can be offered that are relevant to the unusual effects we have uncovered (Figure 2). Despite being electronic congeners of α-keto esters, acyl phosphonates are tetrahedral rather than trigonal at the α-carbon, a circumstance that alters the steric environment at the ketone undergoing reduction. The impact of this geometric change is probably compounded by the fact that the carbonyl activation mode in the (amido)Ru(II) complex is dramatically different (outer sphere/bifunctional) than the bis(oxazoline)Cu(II) systems (inner sphere chelation control) where acyl phosphonates and α-keto esters experience identical influence from the chiral catalyst.
Figure 2.

Variables that potentially account for stereoselectivity inversion.
In summary, an unexpected reversal in facial selectivity was observed in the Ru-mediated asymmetric transfer hydrogenation of acyl phosphonates from their structural mimics, α-keto esters. This dichotomy in reactivity was exploited in the development of an extremely selective dynamic kinetic resolution of α-aryl acyl phosphonates providing β-stereogenic α-hydroxy phosphonic acid derivatives. The first highly selective catalytic reduction of acyl phosphonates also provides complementary access to challenging Pudovik adducts. The precise identification of key catalyst/substrate interactions, reactant orientations and activation modes will be important for understanding the divergence between α-keto esters and acyl phosphonates and for exploiting this finding in future applications.
Supplementary Material
ACKNOWLEDGMENT
The project described was supported by Award No. R01 GM084927 from the National Institute of General Medical Sciences. M.T.C. acknowledges the University of North Carolina at Chapel Hill Department of Chemistry for an Ernest L. Eliel Graduate Fellowship. X-ray crystallography performed by Dr. Peter S. White.
Funding Sources
No competing financial interests have been declared.
Footnotes
ASSOCIATED CONTENT
Experimental procedures, spectral, and HPLC data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Mitchinson A, Finelstein J. Nature. 2003;455:303–349. [Google Scholar]; (b) Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. Sausalito: University Science Books; 2009. [Google Scholar]
- 2.(a) Yoon TP, Jacobsen EN. Science. 2003;299:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]; (b) Pfaltz A, Drury WJ., III Proc. Natl. Acad. Sci. U.S.A. 2004;101:5723–5726. doi: 10.1073/pnas.0307152101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhou Q-L, editor. Privileged Chiral Ligands and Catalysts. Weinheim: Wiley-VCH; 2008. [Google Scholar]
- 3.For an example of substrate-induced diastereodivergence see: Huber JD, Leighton JL. J. Am. Chem. Soc. 2007;129:14552–14553. doi: 10.1021/ja076035h.
- 4.Selected examples of acyl phosphonates in asymmetric synthesis: Evans DA, Johnson JS. J. Am. Chem. Soc. 1998;120:4895–4896. Thorhauge J, Johnannsen M, Jorgensen KA. Angew. Chem. Int. Ed. 1998;37:2405–2407. Evans DA, Johnson JS, Burgey CS, Campos KR. Tetrahedron Lett. 1999;40:2879–2882. Evans DA, Johnson JS, Olhava EJ. J. Am. Chem. Soc. 2000;122:1635–1649. Evans DA, Scheidt KA, Fandrick KR, Lam HW, Wu J. J. Am. Chem. Soc. 2003;125:10780–10781. doi: 10.1021/ja036985c. Takenaka N, Abell JP, Yamamoto H. J. Am. Chem. Soc. 2007;129:742–743. doi: 10.1021/ja0668320. Evans DA, Fandrick KR, Song HJ, Scheidt KA, Xu R. J. Am. Chem. Soc. 2007;129:10029–10041. doi: 10.1021/ja072976i. Jiang H, Paixao MW, Monge D, Jorgensen KA. J. Am. Chem. Soc. 2010;132:2775–2783. doi: 10.1021/ja9097803. Liu T, Wang Y, Wu G, Song H, Zhou Z, Tang C. J. Org. Chem. 2011;76:4119–4124. doi: 10.1021/jo2002825. Kang YK, Suh KH, Kim DY. Synlett. 2011:1125–1128.
- 5. Noyori R, Ikeda T, Ohkuma M, Widhalm M, Kitamura H, Takaya S, Akutagawa S, Sayo N, Saito T. J. Am. Chem. Soc. 1989;111:9134–9135. For recent reviews: Steinreiber J, Faber K, Griengl H. Chem. Eur. J. 2008;14:8060–8072. doi: 10.1002/chem.200701643. Pellissier H. Tetrahedron. 2011;67:3769–3802.
- 6.(a) Steward KM, Gentry EC, Johnson JS. J. Am. Chem. Soc. 2012;134:7329–7332. doi: 10.1021/ja3027136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Steward KM, Corbett MT, Goodman CG, Johnson JS. J. Am. Chem. Soc. 2012;134:20197–20206. doi: 10.1021/ja3102709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For a full account of α-keto ester and acyl phosphonate substrates investigated, the reader is directed to the Supporting Information.
- 8.Selected examples: Wynberg H, Smaardijk AA. Tetrahedron Lett. 1983;24:5899–5900. Rath NP, Spilling CD. Tetrahedron Lett. 1994;35:227–230. Yokomatsu T, Yamagishi T, Shibuya S. Tetrahedron: Asymmetry. 1993;4:1783–1784. Arai T, Bougauchi M, Sasai H, Shibasaki M. J. Org. Chem. 1996;61:2926–2927. doi: 10.1021/jo960180o. Sasai H, Bougauchi M, Arai T, Shibasaki M. Tetrahedron Lett. 1997;38:2717–2720. Saito B, Katsuki T. Angew. Chem. Int. Ed. 2005;44:4600–4602. doi: 10.1002/anie.200501008. Saito B, Egami H, Katsuki T. J. Am. Chem. Soc. 2007;129:1978–1986. doi: 10.1021/ja0651005. Zhou X, Liu X, Yang X, Shang D, Xin J, Feng X. Angew. Chem. Int. Ed. 2008;47:392–394. doi: 10.1002/anie.200704116. Abell JP, Yamamoto H. J. Am. Chem. Soc. 2008;130:10521–10523. doi: 10.1021/ja803859p. Uraguchi D, Ito T, Ooi T. J. Am. Chem. Soc. 2009;131:3836–3837. doi: 10.1021/ja810043d. Suyama K, Sakai Y, Matsumoto K, Saito B, Katsuki T. Angew. Chem. Int. Ed. 2010;49:797–799. doi: 10.1002/anie.200905158.
- 9. Goulioukina NS, Bondarenko GN, Bogdanov AV, Beletskaya IP, Gavrilov KN. Eur. J. Org. Chem. 2009:510–515. Selected examples of stoichiometric asymmetric reductions: Gajda T. Tetrahedron: Asymmetry. 1994;5:1965–1972. Meier C, Laux WHG. Tetrahedron: Asymmetry. 1995;6:1089–1092. Selected examples of catalytic asymmetric hydrogenation of α,β-unsaturated phosphonates: Burk MJ, Stammers TA, Straub JA. Org. Lett. 1999;1:387–390. Rubio M, Suarez A, Alvarez E, Pizzano A. Chem. Commun. 2005:628–630. doi: 10.1039/b414288h. Fernandez-Perez H, Donald SMA, Munslow IJ, Benet-Buchholz J, Maseras F, Vidal-Ferran A. Chem. Eur. J. 2010;16:6495–6508. doi: 10.1002/chem.200902915. Tamura K, Sugiya M, Imamoto T, Yoshida K, Yanagisawa A. Org. Lett. 2010;12:4400–4403. doi: 10.1021/ol101936w. Zhang J, Dong K, Wang K, Ding K. Org. Biomol. Chem. 2012;10:1598–1601. doi: 10.1039/c1ob06835k.
- 10.Selected examples: Barco A, Benetti S, Bargamini P, De Risi C, Marchetti P, Pollini GP, Zanirato V. Tetrahedron Lett. 1999;40:7705–7708. Dodda R, Zhao CG. Org. Lett. 2006;8:4911–4914. doi: 10.1021/ol062005s. Gondi VB, Kagihara K, Rawal VH. Angew. Chem. Int. Ed. 2009;48:776–779. doi: 10.1002/anie.200804244. Perera S, Naganaboina VK, Wang L, Zhang B, Guo Q, Rout L, Zhao C-G. Adv. Synth. Catal. 2011;353:1729–1734. doi: 10.1002/adsc.201000835. Zhang H, Wen X, Gan L, Peng Y. Org. Lett. 2012;14:2126–2129. doi: 10.1021/ol300664d.
- 11.(a) Redmore D. In: Topics in Phosphorus Chemistry. Grayson M, Griffith EJ, editors. Vol. 8. New York: John Wiley; 1976. pp. 515–585. [Google Scholar]; (b) Yamamoto H, Inokawa S. Adv. Carbohydr. Chem. Biochem. 1984;42:135–191. [Google Scholar]; (c) Giannousis PP, Bartlett P. J. Med. Chem. 1987;30:1603–1609. doi: 10.1021/jm00392a014. [DOI] [PubMed] [Google Scholar]; (d) Stowasser B, Budt K-H, Li JQ, Peyman A, Ruppert D. Tetrahedron Lett. 1992;33:6625–6628. [Google Scholar]; (e) Patel DV, Rielly-Gauvin K, Ryono DE, Free CA, Rogers WL, Smith SA, DeForrest JM, Oehl RS, Petrillo EW., Jr J. Med. Chem. 1995;38:4557–4569. doi: 10.1021/jm00022a022. [DOI] [PubMed] [Google Scholar]; (f) Froestl W, Mickel SJ, Hall RG. J. Med. Chem. 1995;38:3297–3312. doi: 10.1021/jm00017a015. [DOI] [PubMed] [Google Scholar]; (g) Froestl W, Mickel SJ, von Sprecher G. J. Med. Chem. 1995;38:3313–3331. doi: 10.1021/jm00017a016. [DOI] [PubMed] [Google Scholar]; (h) Spangler LA, Mikołajczyk M, Burdge EL, Kiełbasiński P, Smith HC, Łyzwa P, Fisher JD, Omelańczuk J. J. Agric. Food Chem. 1999;47:318–321. doi: 10.1021/jf980527j. [DOI] [PubMed] [Google Scholar]; (i) Kafarski P, Lejczak B. Curr. Med. Chem.: Anti-Cancer Agents. 2001;1:301–312. doi: 10.2174/1568011013354543. [DOI] [PubMed] [Google Scholar]; (j) Shibuya S. Yakugaku Zasshi. 2004;124:725–749. doi: 10.1248/yakushi.124.725. [DOI] [PubMed] [Google Scholar]; (k) Berlicki L, Kafarski P. Curr. Org. Chem. 2005;9:1829–1850. [Google Scholar]
- 12.(a) Hashiguchi S, Fujii A, Ikariya T, Noyori R. J. Am. Chem. Soc. 1995;117:7562–7563. [Google Scholar]; (b) Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J. Am. Chem. Soc. 1996;118:2521–2522. [Google Scholar]; (c) Noyori R, Hashiguchi S. Acc. Chem. Res. 1997;30:97–102. [Google Scholar]
- 13.Under the standard reaction conditions (rt, 10 h), the reaction proceeded to 28% conversion with a 11:1 dr.
- 14.CCDC 908430 (2q), 908511 (2e), and 908431 (S8) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Centre via www.ccdc.cam.ac.uk/data_request/cif. The structure in Scheme 1 was generated with CYLview, 1.0b; Legault CY. Universite de Sherbrooke. 2009 ( http://www.cylview.org).
- 15.Zhu SF, Chen WQ, Zhang QQ, Mao HX, Zhou QL. Synlett. 2011:919–922. [Google Scholar]
- 16.L2 and L3 provided identical enantioselectivity.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.