

Abstract
Small angle X-ray scattering (SAXS) is a powerful method for investigating macromolecular structure in solution. SAXS data provide information about the size and shape of a molecule with a resolution of approximately 2–3 nm. SAXS is particularly useful for the investigation of nucleic acids, which scatter X-rays strongly due to the electron-rich phosphate backbone. Therefore, SAXS has become an increasingly popular method for modeling nucleic acid structures, an endeavor made tractable by the highly regular helical nature of nucleic acid secondary structures. Recently, we used SAXS in combination with NMR to filter and refine all-atom models of a U2/U6 small nuclear RNA complex. In this unit we present general protocols for sample preparation, data acquisition, and data analysis and processing. Additionally, examples of correctly and incorrectly processed SAXS data and expected results are provided.
UNIT INTRODUCTION
This unit contains a general procedure for characterizing nucleic acid structure using small angle X-ray scattering (SAXS). SAXS is a powerful method for analyzing the global shape of macromolecules in solution. Because SAXS measures the contrast in scattering between the macromolecule and buffer, it is particularly well suited for nucleic acids which have high contrast due to the electron-rich phosphate backbone. SAXS provides essential information about the size and shape of the molecule and can be used to generate low resolution models for comparison with homologous crystal or NMR structures. All-atom models can also be filtered and refined against SAXS data.
Determination of novel nucleic acid structures is of great importance for understanding their biological roles; however, it poses a considerable challenge for traditional structural methods. Nucleic acids can be difficult to crystallize and NMR structure determination is challenging due to low proton density, resulting in fewer observable signals, and spectral overlap due to limited chemical shift dispersion. Because of these challenges, characterization of nucleic acids by low resolution methods is becoming increasingly popular. Examples of nucleic acid studies using SAXS include the investigation of conformational changes in a DNA Holliday junction (Nollmann et al., 2004); measurement of single stranded DNA and RNA persistence lengths (Chen et al., 2012b), time-resolved studies of RNA folding pathways (Pollack and Doniach, 2009; Roh et al., 2010; Russell et al., 2000) and structural studies of ribozymes (Kazantsev et al., 2011; Lipfert et al., 2008), tRNA and tRNA-like structures (Grishaev et al., 2008; Hammond et al., 2009; Zuo et al., 2010), riboswitches (Ali et al., 2010; Baird and Ferré-D’Amaré, 2010; Chen et al., 2012a; Garst et al., 2008; Lipfert et al., 2007) and spliceosomal RNAs (Burke et al., 2012).
SAXS is highly complementary to both X-ray crystallography and NMR. Few nucleic acid molecules exceeding 75 nucleotides (nts) have been solved by NMR due to the challenges of spectral overlap and fast relaxation. In contrast, SAXS is appropriate for nucleic acids from 25 to hundreds of nucleotides in size. Very small nucleic acids (< 25 nts) may not be amenable to SAXS as their size is at or below the resolution limit (~20–30 Å). SAXS requires significantly less sample than NMR and data collection can take as little as a second or less at synchrotron radiation sources. Using synchrotron radiation, nucleic acid dynamics can be measured with SAXS in real time (Pollack, 2011). SAXS also allows for selection of ensembles of models that predict the range of motions sampled in solution (Bernado et al., 2007; Pelikan et al., 2009).
STRATEGIC PLANNING
The workflow for this method is outlined in Fig. 1. The end goal of SAXS characterization is often a low resolution envelope (referred to as an ab initio model). Additional information about the secondary structure and long range contacts from other methods such as chemical probing, NMR or bioinformatics is indispensable to obtaining a high quality ensemble of structural models. For a recent review on combining NMR and SAXS data to investigate RNA structures, see (Wang et al., 2010).
Figure 1.

General scheme for characterization of RNA using small angle X-ray scattering. Ovals indicate procedures discussed in other publications (see Strategic Planning and Commentary).
Sample Preparation
It is crucial to obtain a pure, monodisperse sample before data collection to ensure that all subsequent data processing and modeling steps are valid. Formation of unwanted higher order complexes or sample degradation can distort scattering data and result in low quality structural models (Rambo and Tainer, 2010). Unfortunately, poor sample quality is not always apparent until after the data analysis process. Initial data analysis will provide some information about sample quality, but it is essential to assess the sample using other methods as well (outlined in Basic Protocol 1). Dynamic light scattering (DLS) is a powerful method for assessing the size and homogeneity of macromolecules (Ferré D’Amaré, 1997; Jachimska et al., 2008), and can be used to ascertain sample quality prior to SAXS data collection. DLS determines the diffusion coefficient of a molecule by measuring time-dependent fluctuations in the intensity of light scattering. The diffusion coefficient is used to calculate the hydrodynamic radius and size distribution of particles in the sample (Jachimska et al., 2008).
The initial steps in sample preparation, construct design, RNA transcription and purification have been described in detail elsewhere (Hennig et al., 2001; Milligan and Uhlenbeck, 1989; Nelissen et al., 2012). The following steps in purification employ the same buffer that will be used for SAXS data collection, so it is useful to decide on the buffer composition early. SAXS samples often contain Tris as the buffering agent due to its ability to scavenge hydroxyl radicals generated by exposure of the solvent to X-rays. Phosphate buffer is generally not recommended because it reduces the contrast between the sample and buffer scattering. In order to minimize the effects of interparticle repulsion, nucleic acid samples require at least 100 mM monovalent ionic strength (sodium chloride or potassium chloride) and 1–40 mM magnesium chloride (optional), which is often important for folding. Other small molecules, such as ligands, may also be added.
One robust method for obtaining a homogeneous, monodisperse sample is to use high resolution size exclusion chromatography (see Unit 17.3) (Rambo and Tainer, 2010). RNA molecules can form higher order complexes after in vitro transcription, and these complexes (which are often bi- or tri-molecular) must be separated from the desired RNA species. It is important to choose a size exclusion column based on the size and properties of the molecule. This technique is also useful for isolating homogeneous nucleic acid complexes or nucleic acid-protein complexes.
Buffer matching and accurate concentration determination are also crucial to SAXS characterization. Effective buffer matching is achieved through extensive dialysis. An accurate concentration is required for data processing and can be determined by hydrolyzing a small amount of the sample in KOH and measuring the absorbance at 260 nm (A260) (Cavaluzzi and Borer, 2004).
Data acquisition
SAXS data acquisition can be performed using either a synchrotron or bench top X-ray source. Synchrotron sources have much greater X-ray flux and therefore allow for much faster data collection with higher signal to noise (Fig. 2A). This kind of source is ideal for kinetic experiments or for samples that are unavoidably dilute or degrade very quickly. Additionally, high flux synchrotron radiation allows for collection of wide angle X-ray scattering (WAXS) which can be used to evaluate buffer matching. Typically, experimental arrangements at synchrotron sources include a flow cell apparatus to move the sample across the X-ray beam, which minimizes radiation damage of the sample during data collection. However, due to this arrangement, larger sample volumes (~250 μl) may be required. Alternatively, bench top SAXS instruments are also appropriate for use with nucleic acids, despite having lower flux than synchrotron sources (Fig. 2A). Bench top X-ray sources may be point or line collimated. Line collimated sources can generate high flux across a larger volume of sample due to a slit-shaped beam geometry; however, data processing requires a deconvolution step, called “desmearing” (Soliman et al., 1998). Point collimated sources focus a narrow X-ray beam on only a small area of the sample, and do not require desmearing. In some cases, data acquisition for the sample may be very long (e.g. up to 12 hours for a dilute sample using a bench top X-ray source). In this case, data collection time for the buffer can be reduced but must be scaled based on X-ray intensity at the time of the experiment. The requisite sample volume for a capillary used in bench top SAXS instruments can be as low as 20–30 μl.
Figure 2.
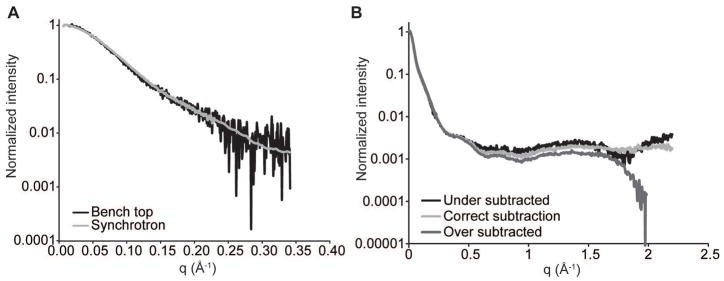
A. X-ray scattering of a 28.2 kDa RNA at 1 mg/ml concentration from either a synchrotron source (Advanced Photon Source or APS) or bench top source (Bruker Nanostar) for 4 hours (see Basic Protocol 2). Synchrotron data were averaged over a total of 3 min. from 4 individual 45 sec. exposures. The bench top data were collected from a single 4 hour exposure. B. SAXS and WAXS data from the same RNA demonstrating accurate and inaccurate buffer subtraction (see Basic Protocol 3).
Biological SAXS typically requires data in the range of 0.1° < 2θ < 5° or 0.01 < q < 0.35 Å−1 for structure characterization (Fig. 2A), where q is a transform of the scattering angle:
and λ is the X-ray wavelength (usually 1.54 Å). This data range can be obtained with a sample to detector distance of 67–100 cm. Collection of WAXS data (0.3 < q < 2 Å−1) will reveal a scattering peak resulting from the buffer. This peak is useful for adjusting buffer matching, but is not absolutely necessary if concentration determination and buffer matching are performed properly. WAXS is typically detected at a sample to detector distance of 30 cm. If both SAXS and WAXS are being collected, a longer SAXS sample to detector distance of 200–400 cm can be used to obtain scattering at smaller angles (q < 0.01 Å−1) as scattering at wider angles is redundant with WAXS. Simultaneous SAXS and WAXS data collection will ensure consistency in the overlapped scattering region used to merge the two data sets. Often, an arrangement with two detectors is available for this purpose at synchrotron facilities.
Data analysis
Initial analysis of SAXS data should be performed directly after data acquisition to rapidly assess sample quality and experimental setup. This includes buffer subtraction, Guinier analysis and Kratky analysis (Putnam et al., 2007; Rambo and Tainer, 2010). Buffer subtraction may need to be fine-tuned at a later time, especially if WAXS data are available. Guinier analysis will provide an approximate radius of gyration and is a good method for sample quality assessment (see Basic Protocol 3). Kratky analysis provides information about the general conformation of the molecule, such as the extent of folding and whether the molecule is globular, extended or unfolded.
After data acquisition, more complex data analysis such as determination of the pair distance distribution function (PDDF) and ab initio modeling can be performed. The PDDF is the sum of all inter-atomic pair distances in the molecule and is determined by an indirect Fourier transform of the scattering data using the program Gnom (Svergun, 1992). Determination of the PDDF provides the maximum dimension of the molecule (Dmax – note: also sometimes called Rmax) as well as an idealized scattering curve that is extrapolated to q = 0 Å−1. This scattering curve is used to calculate a low resolution ab initio envelope of the nucleic acid composed of dummy atoms or beads. The ab initio model provides the global fold of the molecule and can be fitted to existing structures or models. Additionally, ≥ 11–12 base-pair extensions can be added to different helices in the nucleic acid to identify structural features (Baird and Ferré-D’Amaré, 2010; Burke et al., 2012; Zuo et al., 2010), provided such extensions do not disrupt the overall fold. These extensions will manifest as ≥ 30 Å elongations in the envelope.
All-atom modeling
In parallel with data collection and analysis, secondary structure, if not known a priori, must be determined. This can be achieved with computational tools such as MFOLD (Zuker, 2003), MC-FOLD (Parisien and Major, 2008), or RNAstructure (Harmanci et al., 2011). However, nucleic acids frequently form alternative conformations that are similar in free energy. Therefore, the predicted secondary structure must often be determined experimentally using biochemical methods (Low and Weeks, 2010; Mathews et al., 2004), phylogenetic covariation analysis (Juan and Wilson, 1999), or NMR for nucleic acids that are on the order of 100 nts or less (Hennig et al., 2001). The secondary structure informs the overall conformation of the RNA (Lescoute and Westhof, 2006) and can be used to build a large library of models to filter against SAXS data (see Commentary). While obtaining a low resolution envelope for a highly flexible or heterogeneous molecule is very challenging, flexibility can be modeled by a combination of molecular modeling, molecular dynamics simulations and SAXS prediction tools that have ensemble modeling capabilities, such as ensemble optimization method or EOM (Bernado et al., 2007; Fang et al., 2000; Pelikan et al., 2009).
BASIC PROTOCOL 1: SAMPLE PREPARATION
Large quantities of RNA can be prepared by either in vitro transcription (Hennig et al., 2001; Milligan and Uhlenbeck, 1989; Scott and Hennig, 2008) or in E. coli using recombinant technology (Nelissen et al., 2012). Typically, RNAs > 80 nts are transcribed in vitro from a linearized plasmid containing the template for the RNA sequence, created using standard cloning techniques (Sambrook and Russell, 2001). The level of purification achieved using these methods may be adequate for some RNAs; however, further steps are required for most RNAs to obtain the quality of sample necessary for SAXS characterization. Depending on the number of experiments, X-ray source and sample handling arrangement, 0.5–3 mg of nucleic acid should be sufficient. If purified using denaturing methods, we recommend re-folding RNAs after purification to eliminate formation of higher order multimeric species. Alternatively, RNAs may be purified using non-denaturing methods, circumventing the need for a refolding step (Pereira et al., 2010).
Materials
Tris base, 99% pure
Ethylenediamine tetraacetic acid, disodium salt dihydrate (EDTA), > 99% pure
Sodium chloride (NaCl) or potassium chloride (KCl), >99% pure
Magnesium chloride (MgCl2), >99% pure (optional)
Potassium hydroxide (KOH), 89% pure
0.22 μm filter flask, 1 L, Millipore or GE Healthcare
0.20 μm syringe filters, Sartorius Stedim
0.22 μm SpinX cellulose acetate filters, CoStar
Centrifugal filters, regenerated cellulose, 15 ml and 0.5 ml, Amicon® Ultra
Dialysis cassettes, Slide-A-Lyzer® Thermo Scientific, 0.5–3 ml capacity
100 μl syringe with 23 mm, 22 gauge, blunt end needle, Hamilton (to minimize sample retention)
GE Healthcare HiLoad™ 16/60 Superdex 75 or 200 prep grade column
FPLC system (such as ÄKTAFPLC™) equipped with on-line UV monitor at 254 nm, 100 μl loading loop and fraction collector
Centrifuge capable of holding 50 ml conical tubes
Microcentrifuge
Nanodrop ND-1000 spectrophotometer or other UV spectrophotometer
Analyze sample homogeneity and re-fold (optional)
-
1
Resuspend nucleic acid sample in the appropriate volume of SAXS buffer.
Add enough buffer to obtain a concentration of 5–10 mg/ml. -
2
Assay a small aliquot (0.1–0.50 nmol or 10–15 μg) for folding homogeneity by non-denaturing polyacrylamide gel electrophoresis (PAGE), which can be performed in the same manner as denaturing PAGE (Unit 10.4) without the denaturing agent (formamide or urea), using 29:1 acrylamide: bisacrylamide, 88 mM tris borate.
A 6% (w/v) polyacrylamide solution works well for most RNAs 50–200 nt. Toluidine blue (tolonium chloride) staining is usually adequate for detection of most species. -
3
Examine the gel. If only one band is visible, proceed to step 8.
The presence of additional, slowly migrating species is likely indicative of bi- or tri-molecular complex formation. If these bands are minor (~ <10%), size exclusion chromatography (step 8) may be adequate to remove these species. The following refolding steps will be necessary for those samples with significant folding heterogeneity. -
4
Precipitate the RNA in 0. 3 M sodium acetate pH 5.2 and 3 volumes of ethanol, and resuspend the sample in a large volume of buffer such that the RNA concentration is < 10 μM.
Concentration and ionic strength may need to be adjusted depending on the properties of the nucleic acid. In our experience, refolding in low ionic strength buffers (e.g. 10 mM Tris pH 7.0, 50 μM EDTA) can successfully reduce the formation of multimeric species. Use an elongated tube, such as a 15 ml Falcon tube, to maximize surface area for efficient heating and cooling of samples. -
5
Heat the sample to 90°C for 2 minutes.
-
6
Immediately place the sample on ice and allow to cool completely.
-
7
Repeat steps 2–3 to assay the folding homogeneity of the sample.
Repeat this procedure as necessary, varying buffer composition or sample concentration to achieve homogeneous folding.
Purify the sample by size exclusion chromatography
-
8
Prepare 4 L of the buffer that will be used for SAXS data collection (see Strategic Planning) and filter using a 0.22 μm filter flask.
2 L will be used for size exclusion chromatography and 2 L will be used later for dialysis. Store at 4°C for up to one month. -
9
Concentrate the folded RNA using a centrifugal filtration device to a volume of 110–120 μl according to the manufacturer’s instructions.
-
10
At the same time, equilibrate the size exclusion column with at least 3 column volumes of SAXS buffer.
-
11
Inject the sample using a 100 μl loop with the recommended low retention syringe and needle.
10–20 μl of sample will be retained in the needle and can be flushed out with buffer and either loaded onto the column or saved. -
12
Elute the sample using the same buffer and collect 1 ml fractions.
Calibration with protein standards can be very misleading for prediction of elution volume. Nucleic acid molecules of the same molecular weight as proteins standards often elute at a significantly different volume than expected. -
13
Remove 10–15 μl of each fraction with absorbance at 254 nm and analyze by non-denaturing PAGE as in step 2.
-
14
Pool the fractions that contain only the desired species.
-
15
Concentrate the pooled fractions using a centrifugal filter until the concentration of the nucleic acid is ~ 3 mg/ml as determined by UV absorbance at 260 nm (A260).
A260 extinction coefficients are: A: 15.02 mM−1cm−1; C: 7.07 mM−1cm−1; G: 12.08 mM−1cm−1 and U: 9.66 mM−1cm−1 (Cavaluzzi and Borer, 2004). The molar masses of nucleotide monophosphates are AMP: 347.22 g/mol; CMP: 323.20 g/mol; GMP: 363.22 g/mol; GTP: 523.18 g/mol and UMP: 324.18 g/mol. Total volume should be 0.5–1 ml. Proceed immediately to step 16.
Dialyze the sample to achieve buffer matching
-
16
Prepare a dialysis cassette in 1 L of SAXS buffer according to the manufacturer’s instructions.
-
17
Using a 1 ml syringe, load 0.5–1 ml of RNA sample into the dialysis cassette according to the manufacturer’s instructions.
-
18
Dialyze for 8–12 hours with stirring at 4°C.
-
19
Exchange the buffer for 1 L of fresh SAXS buffer and continue dialysis for another 8–12 hours at 4°C.
-
20
Remove the sample from the dialysis cassette.
Ideally, the RNA should be used as soon as possible after purification (Rambo and Tainer, 2010); however, this is not always practical. RNA can be stored at this point at 4°C for a few hours, but should be flash frozen (see step 25) as soon as possible. -
21
Filter 50 ml of the dialysate from step 19 through a 0.2 μm syringe filter.
Store up to 1 week at 4°C or for longer periods at −20°C.
Prepare a dilution series and determine sample concentration
-
22
Remove 1 μl of the RNA sample and dilute accordingly to obtain an approximate concentration based on the A260.
-
23
Using the filtered dialysate from step 21, dilute the RNA sample serially such that the lowest concentration is near 0.5 mg/ml.
The sample volume will depend on the sample handling arrangement for the instrument. Contact the facility before sample preparation to obtain this information. Serial dilutions may also be prepared at the facility and concentrations determined after returning from data collection given that the sample can be recovered. -
24
Remove 1 μl from each sample in the dilution series and add to an eppendorf tube containing 1 μl of 1 N KOH with 8 μl of ddH2O. Incubate the reserved KOH solutions at room temperature overnight to hydrolyze.
-
25
At this point, flash freeze the samples from step 23 by dropping into liquid nitrogen. Store at < −80°C until data collection.
-
26
Obtain an exact concentration of the hydrolyzed samples by measuring the A260 and using the extinction coefficients listed above.
-
27
Immediately before data collection, thaw samples and load into 0.2 μm spin filters.
-
28
Centrifuge at >15,000 × g for 1 minute. The final sample will be the flow through.
BASIC PROTOCOL 2: SAXS DATA ACQUISITION
Introduction
Data acquisition procedures vary widely depending on the X-ray source (synchrotron or bench top) and the exact instrumental setup. It is best to consult with the facility you will be using ahead of time to obtain information about experimental setup, sample volume and other important parameters. The protocol outlined below is the general workflow during data acquisition; however, this procedure may change depending on the instrument and facility.
Materials
Silver(I) behenate (AgBe)
Distilled deionized water
1–1.5 mm quartz capillary (supplied with instrument)
Glassy carbon (optional, supplied with instrument)
Purified nucleic acid from Basic Protocol 1
Reserved filtered dialysate from Basic Protocol 1
SAXS instrument – approximately 67–100 cm sample to detector distance
Additional detector for WAXS (optional) – 30 cm sample to detector distance
Standard sample: lysozyme (Fluka), glucose isomerase (Hampton research) or standard RNA sample (Table 1)
Table 1.
SAXS standard samples
Steps
- Before data collection, obtain scattering from a calibration standard such as AgBe.AgBe has a well characterized D-spacing of 58.38 Å that results in a scattering pattern composed of distinct rings (Huang et al., 1993). This is used as a standard to precisely measure the sample to detector distance.
- If using a bench top instrument, measure the scattering intensity of the X-ray source through glassy carbon.The incident X-ray beam cannot be detected due to the beam stop. Glassy carbon is a strongly scattering material that allows for quantitation of beam intensity (Zhang et al., 2010). This value is important for standardization of the intensity of the X-ray source from one data set to the next.
- Before starting data collection, ensure that the capillary is properly aligned in the beam. Test the capillary position by exposing the empty capillary to X-rays for a short interval.Improper alignment will result in asymmetric parasitic scattering around the beam stop. Ideally, the same capillary should be used for buffer and sample.
- Fill the capillary with ddH2O and perform a short scan to determine the water scattering (optional).This scan be anywhere from 0.1–20 seconds for synchrotron radiation and up to 5 minutes for a bench top source. H2O scattering is often subtracted from all subsequent scans (including buffer and sample scans) to improve background subtraction.
- Load the appropriate amount of buffer into the capillary.Because the sample chamber is under vacuum in bench top instruments, the sample can sometimes be evacuated from the capillary if it is not properly sealed. Take a 10 second scan with glassy carbon between the sample and the detector to determine the absorbance of the buffer – typically a 10 fold decrease in transmission is observed if the sample is still in the capillary.
- Acquire buffer scattering data.This buffer should be the dialysate from step 21 of Basic Protocol 1. Data acquisition time should be the same for buffer and sample and is typically 1–20 seconds at a synchrotron source or 1–6 hours on a bench top source.
Acquire scattering for the most dilute sample (see step 6 above for acquisition time suggestions).
Repeat steps 6–7 for standard samples and all experimental samples.
- Transform the 2-dimensional data from the detector to a 1-dimensional scattering curve (Fig. 2A) based on the calibration performed at the beginning of data acquisition (step 1).This will also convert the angle (θ) to a q value. This step can be performed using a number of different software tools, which are usually provided by the facility. Otherwise, Data Squeeze (www.datasqueezesoftware.com) is available for free and has the same capabilities. Desmearing may be necessary at this step if using a line collimated X-ray source.
BASIC PROTOCOL 3: SAXS DATA PROCESSING AND ANALYSIS
Introduction
SAXS data processing is an iterative process in which the quality of the data and the characteristics of the sample are assessed at every step. It is advantageous to perform all data processing steps on a well characterized, standard molecule as a positive control to ascertain that calibration has been performed properly. Example data sets for the tetraloop-receptor RNA complex (Table 1, PDB ID: 2JYJ) are available on-line for download from the Current Protocols website. Most data processing steps will use Primus (Konarev et al., 2003), a program in the ATSAS suite of software; however, programs like Igor Pro or Gnuplot may also be used for early steps in processing, such as buffer subtraction and Guinier analysis.
DAMMIF (Franke, 2009) is currently the best program for calculating low resolution ab initio structures (or “envelopes”) of nucleic acids. Despite its prevalent use, DAMMIF is limited in use with nucleic acids because it is optimized for use with proteins. Specifically, DAMMIF is dependent on prediction of the scattering curve of the low resolution envelope using form factors derived from protein composition. When used with nucleic acids, the result is inflation of the low resolution envelope. Despite this issue, DAMMIF can be used to approximate nucleic acid envelopes; however, each molecule will require different parameters. Therefore, it is important to become familiar with the documentation (http://www.embl-hamburg.de/biosaxs/manual_dammif.html). To obtain a final, averaged envelope, the DAMMIF models are superimposed, averaged and filtered by a suite of programs (DAMAVER, DAMSEL, DAMSTART, DAMFILT, DAMSUP and SUPCOMB) called using DAMAVER. Both DAMMIF and DAMAVER are part of the ATSAS software package, and most efficiently implemented on a Linux system with multiple processors; however, they can also be used in the Windows and Mac OS X operating systems.
Materials
ATSAS software package (www.embl-hamburg.de/biosaxs/software.html) for the appropriate operating system(s)
Sample data files: 2JYJ_APS_SAXS_WAXS_1mgml.dat and 2JYJ_Nanostar_SAXS_1mgml_4h.dat (downloaded from currentprotocols.com)
Subtract the buffer scattering from the sample scattering
-
1
Adjust the buffer scattering to exclude volume occupied by the nucleic acid by multiplying by the adjusted buffer density (α).
Calculate α based on the partial specific volume of RNA in solution according to the following equation:where the nucleic acid (NA) concentration is in mg/ml (Shpungin et al., 1980). -
2
If WAXS is available, adjust α such that the intensity of the buffer subtracted scattering at q > 1.8 Å−1 is approximately constant (Fig. 2B).
-
3
Repeat step 1–2 for all dilutions. Be sure to note the name of the file containing the buffer subtracted data.
Perform a Guinier fit to obtain Rg and check for concentration effects
-
4
Plot only data less than approximately q = 0.05 Å−1.
-
5
Plot the data as q2 versus ln (I).
In Primus this is done by clicking the “Guinier” button in the tools dialogue. This plot should be linear (Fig. 3) according to the equationWhere I(0) is the scattering intensity at q = 0 Å−1 and Rg is the radius of gyration. A nonlinear Guinier plot is indicative of interparticle interactions (aggregation or repulsion), sample heterogeneity or both (Fig. 3). If the Guinier plot is not linear, it is essential to reassess sample quality at this point. -
6
Adjust the plot range until qmax*Rg ~ 1.2 for roughly globular molecules or 0.8 for rod shaped molecules.
-
7
Record the resulting Rg and I(0) values and compare between dilutions.
I(0) is proportional to the molecular weight of the molecule. I(0) and Rg should ideally remain constant within error between concentrations unless structure factor contributions (i.e. repulsion) are affecting the scattering curve (Fig. 3).
Figure 3.
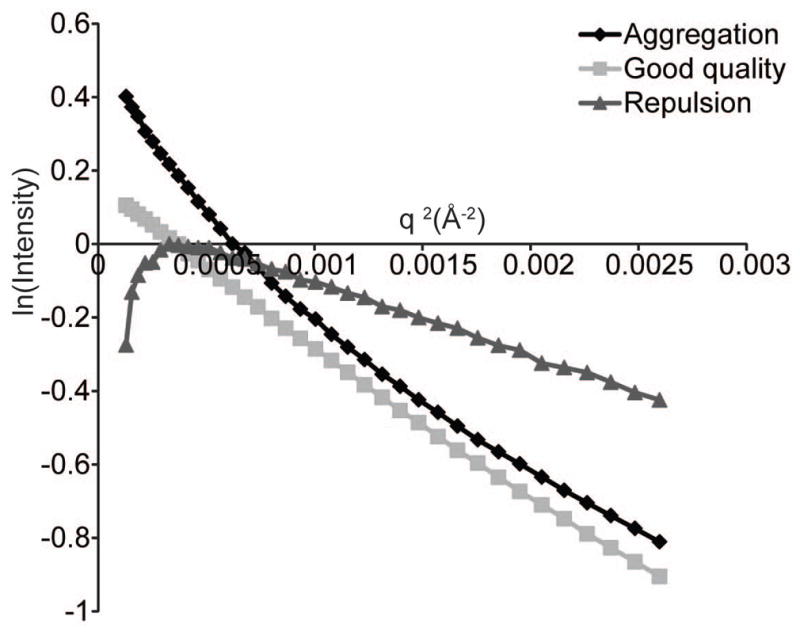
Guinier plot of an aggregated protein (black diamonds), an RNA with interparticle repulsion (gray triangles) or an RNA sample of good quality (gray squares) (see Basic Protocol 3).
Determine the general conformation of the molecule using a Kratky plot
-
8
Plot q versus q2I in the range q < 0.4 Å−1.
In version 2.3 or older, click Sasplot and then select View → Y*X2 : X. In version 2.4, select plots, then Kratky plot. -
9
Examine the plot: Compact molecules (rod-shaped or globular) will display one peak (Fig. 4), extended molecules will display a peak with a shoulder (Fig. 4), and unfolded molecules will increase continuously with no peak.
Figure 4.

Kratky plot of three different RNA molecules with unique global folds (see Basic Protocol 3).
Extrapolate an idealized curve at zero concentration
-
10
In Primus, load all background subtracted curves in the dilution series.
-
11
Enter the concentration of each dilution (see steps 24 and 26 of Basic Protocol 1).
-
12
Plot only the portion of the curve that has exact overlap between the different concentrations.
This will likely exclude approximately q < 0.05–0.10 Å−1 and q > 0.40 Å−1. -
13
Calculate a theoretical curve with no concentration dependence by clicking “Zeroconc” in version 2.3 or older.
Frequently, RNA experiences interparticle repulsion that manifests as inconsistent values of I(0) and Rg for different dilutions. This step will remove contributions from repulsion. Note: this feature is not available in version 2.4 -
14
Alternatively, merge the small angles (q < 0.4 Å−1) of a low concentration data set with the wider angles (0.35 < q < 2 Å−1) of a high concentration data set.
This method is preferable if aggregation is a problem at higher concentrations.
Determine the pair distance distribution function (PDDF)
-
15
Load the highest quality buffer subtracted scattering curve or the zero concentration extrapolated curve (steps 10–14) in Primus. Remove all data points at q > 0.30–0.33 Å−1.
The range of data used at this step will depend on the signal to noise ratio at larger values of q. Exclude the noisiest data by decreasing the maximum q value. -
16
Click Gnom (version ≤ 2.3) or Tools → Distance distribution (version >2.4)
-
17
Estimate the maximum dimension of the particle (Dmax or Rmax). 3*Rg is a good place to start. Do not restrain P(Rmax) = 0 to start. Click “run”.
-
18
Examine the resulting fit to the scattering curve.
Ensure that there are no excessive oscillations in the predicted curve due to over-fitting noise at larger q values. Also ensure that the predicted curve fits the experimental data well at small values of q. Poor fitting in this region can indicate an incorrect Dmax or contributions from interparticle interactions. -
19
Examine the resulting PDDF.
The PDDF should fall off smoothly at large values of “r” toward zero. Notice where p(r) curve becomes constant and choose this as a new Dmax value. If the curve does not level off at larger values of r, try increasing Dmax. -
20
Repeat steps 17–19; however, this time use the newly determined Dmax value and restrain P(Rmax) = 0.
-
21
Adjust Dmax in increments of 1–2 Å until the curve falls smoothly to zero and does not sample any negative values (Fig. 5).
Figure 5.
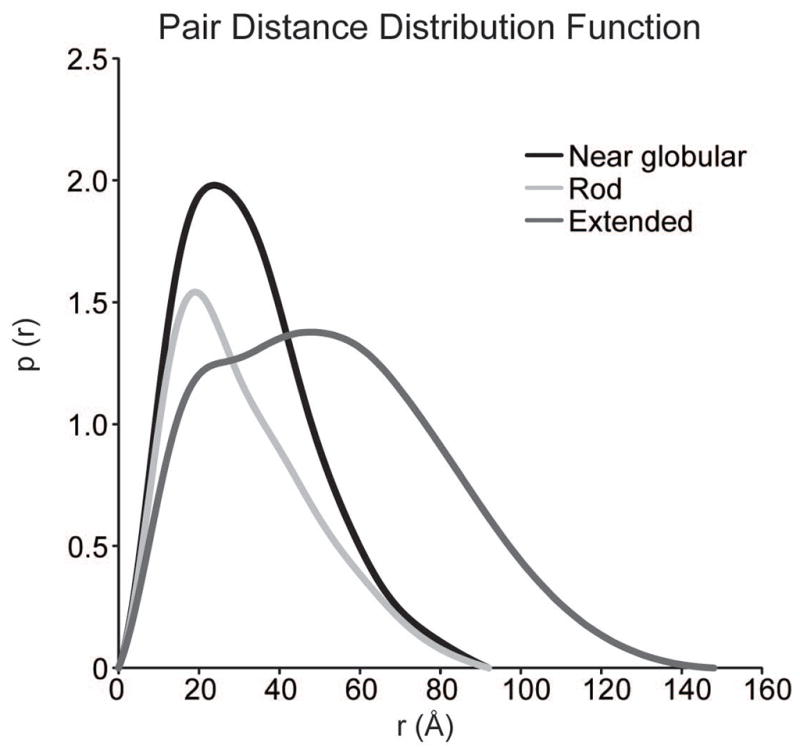
Pair distance distribution functions of the same three RNA molecules as in Fig. 4.
Generate ab initio models
-
22
Using the output file generated by steps 15–21 (will be a .out file), run DAMMIF 10–20 times using parameters appropriate for your molecule (see introduction above).
-
23
Move the resulting ab initio models (*-1.pdb files) to a new directory. Navigate to this directory.
-
24
Type the command damaver/a.
This will run the DAMAVER suite of programs to average and filter the ab initio models. In Windows, the folder containing damaver must be specified before the command. -
25
Examine the DAMAVER output file “damsel.log”.
This log file contains the normalized spatial discrepancy (NSD) values for each pair of models. The NSD is a measure of similarity between a set of points (in this case the ab initio models). In general, the mean NSD (NMSD) tends toward 0 for highly similar objects and exceeds 1 for systematically different objects (Kozin and Svergun, 2001). For SAXS models, the NMSD should fall between 0.7–0.9 for a unique solution to the scattering curve. An NMSD larger than 0.9 could be indicative of many things, including sample heterogeneity or a high degree of flexibility. -
26
Examine the resulting filtered model (damfilt.pdb) using a molecular model visualization program such as Pymol and compare it to any existing structures of relevant domains or homology models.
-
27
(Optional) Overlay all-atom models with the average ab initio model using the Supcomb20 algorithm (ATSAS software) (Kozin and Svergun, 2001).
COMMENTARY
Background
Development of SAXS instrumentation and technology
Characterization of macromolecules by small angle scattering gained traction in the 1970s with studies of the ribosome in solution using neutron scattering (SANS) (Engelman et al., 1975). The first RNA molecules analyzed by SAXS were tRNAs from yeast (Lake, 1967) and E. coli (Ninio et al., 1972); however, characterization was limited to inference of size and shape features from the scattering curve. The development of SAXS instrumentation leapt forward dramatically as synchrotron X-ray sources became more widely available starting in the late 80s. In the late 90s, computational abilities caught up with SAXS instrumentation and some of the first low resolution models determined by small angle scattering were obtained of the ribosome (Svergun et al., 1997). The biological small angle scattering group led by Dmitri Svergun at EMBL-Hamburg has drastically improved the availability of software for processing and analyzing SAXS, improving the accessibility of this method for new users.
Application of SAXS characterization to nucleic acids
In recent years, characterization of biomolecules by SAXS has become increasingly popular. Due to heightened interest, SAXS has become more accessible both in terms of instrumentation and the availability of software for data processing and analysis. SAXS is a versatile technique and has been used to study RNA folding of complex molecules such as RNase P (Fang et al., 2000) and the Tetrahymena ribozyme (Das et al., 2003) and to characterize the free and bound conformations of several riboswitches (Ali et al., 2010; Baird and Ferré-D’Amaré, 2010; Chen et al., 2012a; Garst et al., 2008; Lipfert et al., 2007; Lipfert et al., 2010). The combination of NMR and SAXS has been used to characterize the structure of the turnip crinkle virus ribosome binding element (Zuo et al., 2010) and the U2/U6 snRNA complex (Burke et al., 2012), which is a part of the active spliceosome. A few nucleic acid-protein complexes, such as the ribosome (see above) and the picoRNA viral replication complex (Claridge et al., 2009), have also been characterized by SAXS and/or SANS.
Beyond low resolution characterization
Although SAXS provides low resolution structural information, in many cases this is enough to infer important information about the structure of nucleic acids. Cryo-electron microscopy (cryo-EM) and tomography (cryo-ET) also provide a similar level of resolution to SAXS; however, SAXS has some advantages over these methods. First, SAXS is applicable to much smaller molecules and complexes than cryo-EM/T, which makes it more compatible with NMR. Second, SAXS can provide more information about highly flexible or unfolded molecules. Single molecule cryo-EM studies rely on averaging many molecules with the same properties to achieve sufficient signal. In highly flexible molecules, this averaging leads to loss of information in flexible regions (Stark and Luhrmann, 2006). Cryo-ET studies combine images of different orientations of a molecule, resulting in a three-dimensional “snapshot” of the molecule, and may be more appropriate for heterogeneous samples (Miyazaki et al., 2010). SAXS retains information about all possible orientations and conformations of a molecule in solution. If an adequate diversity of models can be generated, SAXS can be used to extract an ensemble of models that represents the range of conformational diversity in solution.
Molecular modeling and molecular dynamics simulations are important tools for generating all-atom structural models in conjunction with SAXS. Nucleic acids are well-suited for all-atom modeling due to the consistency of the basic helical structural unit between sequences. Knowledge of secondary structure is required, but can be determined by a number of methods (see Strategic Planning). Long-range contacts, such as pseudoknots and tetraloop-receptor interactions, can also be predicted through bioinformatics and biochemical methods (Low and Weeks, 2010). Modeling software employs either a fragment-based approach (Parisien and Major, 2008) or coarse-grained approach (Jonikas et al., 2009). Homology modeling is also a good choice when the structure of a similar molecule is already known (Grishaev et al., 2008). A potentially challenging step in modeling nucleic acid structure is adequate sampling of conformational space. This can be achieved using molecular dynamics simulations (Fang et al., 2000) or normal mode analysis, on a server like elNémo (Suhre and Sanejouand, 2004).
Once a large array of models has been generated, they can be filtered against the experimental scattering data. The SAXS curve of a model is predicted by summing all existing atom pair distances using the Debye equation:
Where dij is the distance between atoms i and j, N is the number of atoms in the molecule and f(q) is the form factor for a type of atom. While this is a computationally intensive process, several web-based software tools approximate the form factor for a group of atoms and are appropriate for nucleic acids, such as FAST-SAXS RNA (Yang et al., 2010) and the FoXS server (Schneidman-Duhovny et al., 2010). Ensembles of models can also be fit to the experimental SAXS curve using multiple ensemble simulation (Pelikan et al., 2009) or ensemble optimization methods (Bernado et al., 2007). Ensemble modeling is especially powerful for characterizing dynamics that may be functionally important.
Critical parameters and troubleshooting
Of utmost importance is sample preparation and quality. Nucleic acid samples must be as homogeneous as possible and free of degradation. Additionally, buffer matching and concentration determination must be as accurate as possible (see Basic Protocol 1). Because nucleic acids are often soluble even when misfolded or aggregated and because SAXS is a low resolution method, issues in sample preparation may not be evident until far into the data analysis process. This problem is compounded by the fact that SAXS data processing is a complex and non-linear process that often requires optimization of several different parameters. Therefore, it is essential to assay sample quality as much as possible by denaturing and non-denaturing PAGE, size exclusion chromatography, dynamic light scattering and, if possible, sample specific activity assays. Standard samples, such as lysozyme or a well characterized RNA (Zuo et al., 2008), are also very important (see Basic Protocol 2, Table 1). Standards should be characterized at a minimum every time the SAXS instrument is rearranged, which is often every data collection at a synchrotron X-ray source and intermittently at a bench top source. Frequent characterization of standard samples will increase the chances of detecting issues in detector image processing, calibration and conversion of the detector image to a 1-D scattering curve.
Sometimes, even when sample preparation and data processing are performed correctly, it is not possible to obtain a good quality, unique ab initio model (Kazantsev et al., 2011). In this situation, the nucleic acid may be very flexible or poorly structured. As described above, ensemble modeling may be one possible route for a flexible nucleic acid. Alternatively, construct design may also be reconsidered. Nucleic acid structure can be drastically altered by small changes in sequence or truncations of important sequence. For instance, often two guanine residues are incorporated at the 5′ end of an RNA sequence for transcription with T7 RNA polymerase. If these residues are non-native, use of a hammerhead ribozyme at the 5′ end (Price et al., 1995) may be considered to generate fully native sequence. A hammerhead ribozyme downstream of the transcript will resolve 3′ end heterogeneity, which is useful when single nucleotide resolution purification is challenging (e.g. transcripts > 80 nts) (Walker et al., 2003).
Time considerations
Up to 1 month should be allowed for sample preparation. Ideally, size exclusion chromatography and dialysis should be performed immediately before data collection. Data acquisition times vary widely depending on the X-ray source. Typical synchrotron data collection for several samples may take 1 or 2 days including instrument setup. Bench top data collection times are up to 24 hours per sample depending on concentration and the stability of the sample. Initial data processing should be performed concomitantly with data acquisition to ascertain sample quality and whether additional dilution points are necessary. The time required for data processing and ab initio modeling is highly variable and can take from a week to a month depending on the properties of the nucleic acid.
Figure 6.
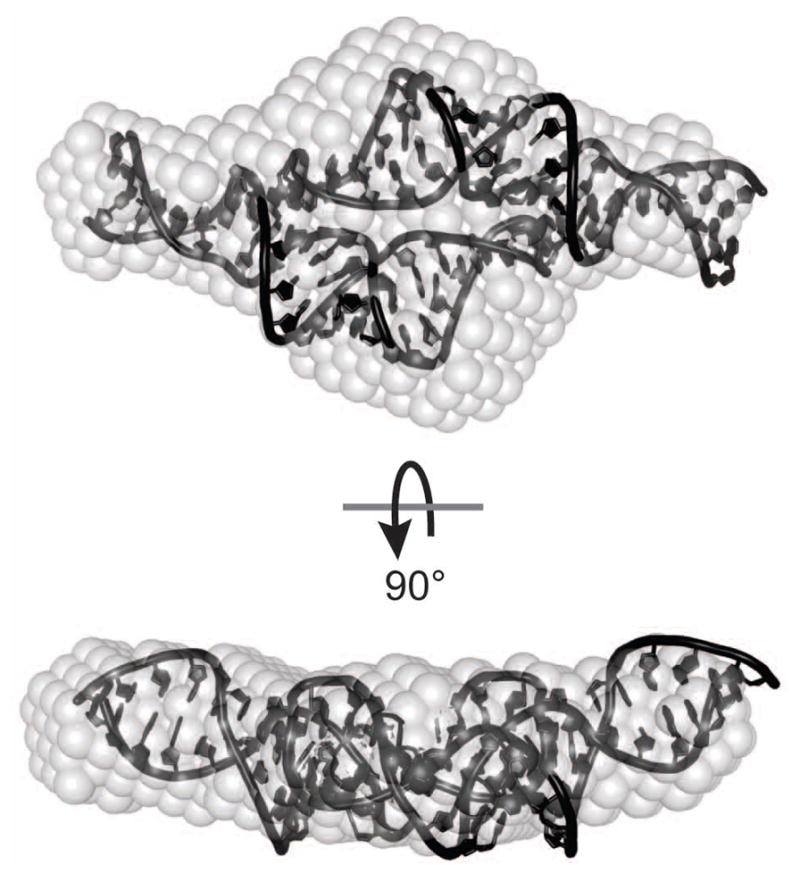
Ab initio structure (gray spheres) of a 28.2 kDa RNA with the jointly refined NMR/SAXS structure (Zuo et al., 2008) superimposed using the Supcomb20 algorithm (Kozin and Svergun, 2001).
Acknowledgments
We thank Xiaobing Zuo and Steven Weigand at APS and Brian Jones at Bruker AXS for assistance with data collection, and Yun Xing-Wang, Alex Grishaev and Jill Trewhella for helpful discussions. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. J.E.B. was supported by NIH Predoctoral training grant T32 GM07215-34. This work was supported by NIH grant R01 GM065166 to S.E.B.
References
- Ali M, Lipfert J, Seifert S, Herschlag D, Doniach S. The ligand-free state of the TPP riboswitch: a partially folded RNA structure. Journal of Molecular Biology. 2010;396:153–165. doi: 10.1016/j.jmb.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NJ, Ferré-D’Amaré AR. Idiosyncratically tuned switching behavior of riboswitch aptamer domains revealed by comparative small-angle X-ray scattering analysis. Rna. 2010;16:598–609. doi: 10.1261/rna.1852310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. Journal of the American Chemical Society. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- Burke JE, Sashital DG, Zuo X, Wang YX, Butcher SE. Structure of the yeast U2/U6 snRNA complex. Rna. 2012;18:673–683. doi: 10.1261/rna.031138.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaluzzi MJ, Borer PN. Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 2004;32:e13. doi: 10.1093/nar/gnh015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Zuo X, Wang YX, Dayie TK. Multiple conformations of SAM-II riboswitch detected with SAXS and NMR spectroscopy. Nucleic Acids Res. 2012a;40:3117–3130. doi: 10.1093/nar/gkr1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Meisburger SP, Pabit SA, Sutton JL, Webb WW, Pollack L. Ionic strength-dependent persistence lengths of single-stranded RNA and DNA. Proc Natl Acad Sci U S A. 2012b;109:799–804. doi: 10.1073/pnas.1119057109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claridge JK, Headey SJ, Chow JY, Schwalbe M, Edwards PJ, Jeffries CM, Venugopal H, Trewhella J, Pascal SM. A picornaviral loop-to-loop replication complex. J Struct Biol. 2009;166:251–262. doi: 10.1016/j.jsb.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Kwok LW, Millett IS, Bai Y, Mills TT, Jacob J, Maskel GS, Seifert S, Mochrie SG, Thiyagarajan P, Doniach S, Pollack L, Herschlag D. The fastest global events in RNA folding: electrostatic relaxation and tertiary collapse of the Tetrahymena ribozyme. Journal of Molecular Biology. 2003;332:311–319. doi: 10.1016/s0022-2836(03)00854-4. [DOI] [PubMed] [Google Scholar]
- Engelman DM, Moore PB, Schoenborn BP. Neutron scattering measurements of separation and shape of proteins in 30S ribosomal subunit of Escherichia coli: S2-S5, S5-S8, S3-S7. Proc Natl Acad Sci U S A. 1975;72:3888–3892. doi: 10.1073/pnas.72.10.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Littrell K, Yang XJ, Henderson SJ, Siefert S, Thiyagarajan P, Pan T, Sosnick TR. Mg2+-dependent compaction and folding of yeast tRNAPhe and the catalytic domain of the B. subtilis RNase P RNA determined by small-angle X-ray scattering. Biochemistry. 2000;39:11107–11113. doi: 10.1021/bi000724n. [DOI] [PubMed] [Google Scholar]
- Ferré D’Amaré AR, Burley, Stephen K. Dynamic light-scattering as a tool for evaluating crystallizability of macromolecules. Methods Enzymol. 1997;276:157–166. doi: 10.1016/S0076-6879(97)76056-7. [DOI] [PubMed] [Google Scholar]
- Franke DaS, DI DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garst AD, Heroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem. 2008;283:22347–22351. doi: 10.1074/jbc.C800120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishaev A, Ying J, Canny MD, Pardi A, Bax A. Solution structure of tRNAVal from refinement of homology model against residual dipolar coupling and SAXS data. J Biomol NMR. 2008;42:99–109. doi: 10.1007/s10858-008-9267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JA, Rambo RP, Filbin ME, Kieft JS. Comparison and functional implications of the 3D architectures of viral tRNA-like structures. Rna. 2009;15:294–307. doi: 10.1261/rna.1360709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci AO, Sharma G, Mathews DH. TurboFold: iterative probabilistic estimation of secondary structures for multiple RNA sequences. BMC Bioinformatics. 2011;12:108. doi: 10.1186/1471-2105-12-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig M, Williamson JR, Brodsky AS, Battiste JL. Recent advances in RNA structure determination by NMR. In: Beaucage Serge L, et al., editors. Current protocols in nucleic acid chemistry. Unit 7. Chapter 7. 2001. p. 7. [DOI] [PubMed] [Google Scholar]
- Huang TC, Toraya H, Blanton TN, Wu Y. X-Ray-Powder Diffraction Analysis of Silver Behenate, a Possible Low-Angle Diffraction Standard. J Appl Crystallogr. 1993;26:180–184. [Google Scholar]
- Jachimska B, Wasilewska M, Adamczyk Z. Characterization of globular protein solutions by dynamic light scattering, electrophoretic mobility, and viscosity measurements. Langmuir. 2008;24:6866–6872. doi: 10.1021/la800548p. [DOI] [PubMed] [Google Scholar]
- Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. Rna. 2009;15:189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan V, Wilson C. RNA secondary structure prediction based on free energy and phylogenetic analysis. Journal of Molecular Biology. 1999;289:935–947. doi: 10.1006/jmbi.1999.2801. [DOI] [PubMed] [Google Scholar]
- Kazantsev AV, Rambo RP, Karimpour S, Santalucia J, Jr, Tainer JA, Pace NR. Solution structure of RNase P RNA. Rna. 2011;17:1159–1171. doi: 10.1261/rna.2563511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Crystallogr. 2001;34:33–41. [Google Scholar]
- Lake JA. Yeast transfer RNA: a small-angle x-ray study. Science. 1967;156:1371–1373. doi: 10.1126/science.156.3780.1371. [DOI] [PubMed] [Google Scholar]
- Lescoute A, Westhof E. Topology of three-way junctions in folded RNAs. Rna. 2006;12:83–93. doi: 10.1261/rna.2208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Das R, Chu VB, Kudaravalli M, Boyd N, Herschlag D, Doniach S. Structural transitions and thermodynamics of a glycine-dependent riboswitch from Vibrio cholerae. Journal of Molecular Biology. 2007;365:1393–1406. doi: 10.1016/j.jmb.2006.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Ouellet J, Norman DG, Doniach S, Lilley DM. The complete VS ribozyme in solution studied by small-angle X-ray scattering. Structure. 2008;16:1357–1367. doi: 10.1016/j.str.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Sim AY, Herschlag D, Doniach S. Dissecting electrostatic screening, specific ion binding, and ligand binding in an energetic model for glycine riboswitch folding. Rna. 2010;16:708–719. doi: 10.1261/rna.1985110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JT, Weeks KM. SHAPE-directed RNA secondary structure prediction. Methods. 2010;52:150–158. doi: 10.1016/j.ymeth.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Irobalieva RN, Tolbert BS, Smalls-Mantey A, Iyalla K, Loeliger K, D’Souza V, Khant H, Schmid MF, Garcia EL, Telesnitsky A, Chiu W, Summers MF. Structure of a conserved retroviral RNA packaging element by NMR spectroscopy and cryo-electron tomography. Journal of Molecular Biology. 2010;404:751–772. doi: 10.1016/j.jmb.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen FH, Leunissen EH, van de Laar L, Tessari M, Heus HA, Wijmenga SS. Fast production of homogeneous recombinant RNA--towards large-scale production of RNA. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninio J, Luzzati V, Yaniv M. Comparative small-angle x-ray scattering studies on unacylated, acylated and cross-linked Escherichia coli transfer RNA I Val. Journal of Molecular Biology. 1972;71:217–229. doi: 10.1016/0022-2836(72)90347-6. [DOI] [PubMed] [Google Scholar]
- Nollmann M, Stark WM, Byron O. Low-resolution reconstruction of a synthetic DNA holliday junction. Biophys J. 2004;86:3060–3069. doi: 10.1016/s0006-3495(04)74355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- Pelikan M, Hura GL, Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen Physiol Biophys. 2009;28:174–189. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira MJ, Behera V, Walter NG. Nondenaturing purification of co-transcriptionally folded RNA avoids common folding heterogeneity. PLoS One. 2010;5:e12953. doi: 10.1371/journal.pone.0012953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack L. Time resolved SAXS and RNA folding. Biopolymers. 2011;95:543–549. doi: 10.1002/bip.21604. [DOI] [PubMed] [Google Scholar]
- Pollack L, Doniach S. Time-resolved X-ray scattering and RNA folding. Methods Enzymol. 2009;469:253–268. doi: 10.1016/S0076-6879(09)69012-1. [DOI] [PubMed] [Google Scholar]
- Price SR, Ito N, Oubridge C, Avis JM, Nagai K. Crystallization of RNA-protein complexes. I. Methods for the large-scale preparation of RNA suitable for crystallographic studies. Journal of Molecular Biology. 1995;249:398–408. doi: 10.1006/jmbi.1995.0305. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Improving small-angle X-ray scattering data for structural analyses of the RNA world. Rna. 2010;16:638–646. doi: 10.1261/rna.1946310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, Guo L, Kilburn JD, Briber RM, Irving T, Woodson SA. Multistage collapse of a bacterial ribozyme observed by time-resolved small-angle X-ray scattering. Journal of the American Chemical Society. 2010;132:10148–10154. doi: 10.1021/ja103867p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R, Millett IS, Doniach S, Herschlag D. Small angle X-ray scattering reveals a compact intermediate in RNA folding. Nat Struct Biol. 2000;7:367–370. doi: 10.1038/75132. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning : a laboratory manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 2001. [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LG, Hennig M. RNA structure determination by NMR. Methods in molecular biology (Clifton, NJ) 2008;452:29–61. doi: 10.1007/978-1-60327-159-2_2. [DOI] [PubMed] [Google Scholar]
- Shpungin IL, Perevozchikov VA, Serdiuk IN, Zaccai G. Isolation and physical study of the 13S fragment of 16S RNA and its complex with ribosomal protein S4. Mol Biol (Mosk) 1980;14:939–950. [PubMed] [Google Scholar]
- Soliman M, Jungnickel BJ, Meister E. Stable desmearing of slit-collimated SAXS patterns by adequate numerical conditioning. Acta Crystallogr A. 1998;54:675–681. [Google Scholar]
- Stark H, Luhrmann R. Cryo-electron microscopy of spliceosomal components. Annu Rev Biophys Biomol Struct. 2006;35:435–457. doi: 10.1146/annurev.biophys.35.040405.101953. [DOI] [PubMed] [Google Scholar]
- Suhre K, Sanejouand YH. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. [Google Scholar]
- Svergun DI, Burkhardt N, Pedersen JS, Koch MH, Volkov VV, Kozin MB, Meerwink W, Stuhrmann HB, Diedrich G, Nierhaus KH. Solution scattering structural analysis of the 70 S Escherichia coli ribosome by contrast variation. II. A model of the ribosome and its RNA at 3.5 nm resolution. Journal of Molecular Biology. 1997;271:602–618. doi: 10.1006/jmbi.1997.1191. [DOI] [PubMed] [Google Scholar]
- Walker SC, Avis JM, Conn GL. General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res. 2003;31:e82. doi: 10.1093/nar/gng082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Zuo X, Wang J, Yu P, Butcher SE. Rapid global structure determination of large RNA and RNA complexes using NMR and small-angle X-ray scattering. Methods. 2010;52:180–191. doi: 10.1016/j.ymeth.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Parisien M, Major F, Roux B. RNA structure determination using SAXS data. J Phys Chem B. 2010;114:10039–10048. doi: 10.1021/jp1057308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ilavsky J, Long GG, Quintana JPG, Allen AJ, Jemian PR. Glassy Carbon as an Absolute Intensity Calibration Standard for Small-Angle Scattering. Metall Mater Trans A. 2010;41A:1151–1158. [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X, Wang J, Foster TR, Schwieters CD, Tiede DM, Butcher SE, Wang YX. Global molecular structure and interfaces: refining an RNA:RNA complex structure using solution X-ray scattering data. Journal of the American Chemical Society. 2008;130:3292–3293. doi: 10.1021/ja7114508. [DOI] [PubMed] [Google Scholar]
- Zuo X, Wang J, Yu P, Eyler D, Xu H, Starich MR, Tiede DM, Simon AE, Kasprzak W, Schwieters CD, Shapiro BA, Wang YX. Solution structure of the cap-independent translational enhancer and ribosome-binding element in the 3′ UTR of turnip crinkle virus. Proc Natl Acad Sci U S A. 2010;107:1385–1390. doi: 10.1073/pnas.0908140107. [DOI] [PMC free article] [PubMed] [Google Scholar]