

Abstract
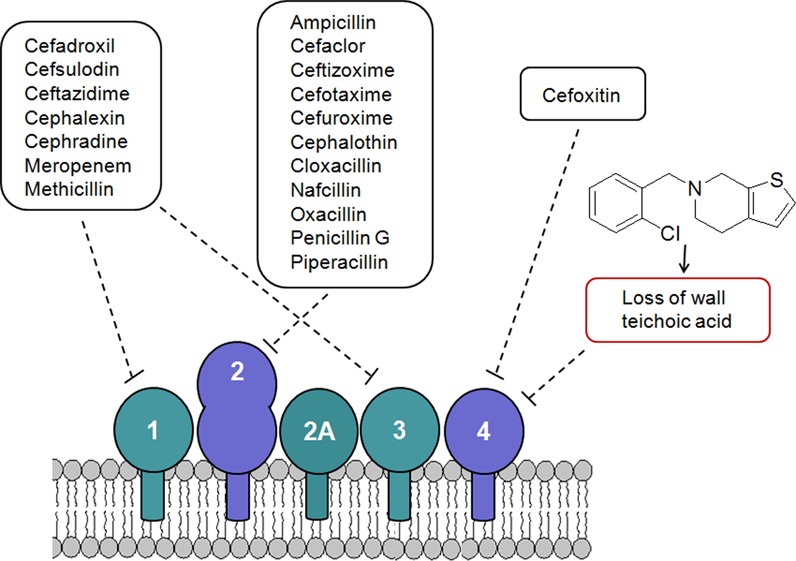
Rising drug resistance is limiting treatment options for infections by methicillin-resistant Staphylococcus aureus (MRSA). Herein we provide new evidence that wall teichoic acid (WTA) biogenesis is a remarkable antibacterial target with the capacity to destabilize the cooperative action of penicillin-binding proteins (PBPs) that underlie β-lactam resistance in MRSA. Deletion of gene tarO, encoding the first step of WTA synthesis, resulted in the restoration of sensitivity of MRSA to a unique profile of β-lactam antibiotics with a known selectivity for penicillin binding protein 2 (PBP2). Of these, cefuroxime was used as a probe to screen for previously approved drugs with a cryptic capacity to potentiate its activity against MRSA. Ticlopidine, the antiplatelet drug Ticlid, strongly potentiated cefuroxime, and this synergy was abolished in strains lacking tarO. The combination was also effective in a Galleria mellonella model of infection. Using both genetic and biochemical strategies, we determined the molecular target of ticlopidine as the N-acetylglucosamine-1-phosphate transferase encoded in gene tarO and provide evidence that WTA biogenesis represents an Achilles heel supporting the cooperative function of PBP2 and PBP4 in creating highly cross-linked muropeptides in the peptidoglycan of S. aureus. This approach represents a new paradigm to tackle MRSA infection.
Since their first appearance in the early 1960s, methicillin-resistant Staphylococcus aureus (MRSA) strains have spread worldwide and have become one of the most menacing of human pathogens.1,2 For much of this period, outbreaks of MRSA were confined to hospitals; however, over the past decade, the prevalence of MRSA in the community has increased alarmingly. USA300 and USA400 isolates now represent the most predominant cause of community-acquired infections in the United States, Canada, and Europe.3,4 While penicillin and other β-lactams such as methicillin were once very effective antibiotics in treating staphylococcal infections, the widespread resistance of MRSA to this class of antibiotics has made treatment increasingly difficult. Besides common resistance to methicillin and β-lactams in general, S. aureus has also become resistant to so-called “drugs of last resort” including vancomycin, daptomycin, and linezolid.5−7
β-Lactams target the synthesis of peptidoglycan (PG), a stress-bearing cell wall polymer of a disaccharide of N-acetylmuramic acid and N-acetylglucosamine. The former is bonded to a short amino acid stem (l-alanine-d-iso-glutamine-l-lysine-d-alanine-d-alanine) that is bridged to neighboring stems via a pentaglycyl segment in S. aureus (Supplementary Figure 1). Transglycosylases and transpeptidases mediate the final assembly of peptidoglycan, forming glycosyl bonds between the disaccharides and cross-links between the neighboring stem peptides using pentaglycine bridges, respectively. These enzymes are collectively known as penicillin-binding proteins (PBPs), with their transpeptidase region making up the cellular target of β-lactam antibiotics. β-Lactam resistance in MRSA involves the acquisition of PBP2A, encoded in mecA, to the complement of the four native staphylococcal PBPs. The function of the transpeptidase domain of PBP2A is similar to that of the bifunctional transglycosylase/transpeptidase PBP2 but has a remarkably low affinity for β-lactam antibiotics.8 PBP2A is thought to take over the biosynthetic role of β-lactam-inhibited PBP2 in cross-linking peptidoglycan when assisted by the transglycosylase domain of native PBP2.9
The bacterial cell wall remains a prime target for anti-MRSA drug discovery and the β-lactam class of antibiotics continues to be explored, albeit with limited success, for example, with the development of analogues capable of inhibiting PBP2A.10,11 Others strategies to treat MRSA have centered on the development of new drug classes with orthogonal mechanisms to the β-lactams such as linezolid, daptomycin, and new glycopeptides.12 Nevertheless resistance to these novel classes has already been reported.5−7 With limited treatment options for MRSA, there is a pressing need for new agents that will avoid existing resistance mechanisms.
To this end, there is a growing interest in identifying auxiliary genes that are required for β-lactam resistance as potential novel targets. Previous work has suggested that PBP2A is not the sole determinant for β-lactam resistance.13−16 Specifically, although not essential for viability, PBP4 has been shown to play a key role in β-lactam resistance in strains of community-acquired MRSA (CA-MRSA), which was linked to its unique function in producing highly cross-linked peptidoglycan species during cell wall synthesis.17 Thus cooperative functions among PBPs, in particular PBP2 and PBP2A9 as well as PBP2 and PBP4,17,18 during cell wall synthesis are also critical to the expression of β-lactam resistance in MRSA. Moreover, growing evidence suggests that a complex network of gene products involved in PG metabolism,14 synthesis,19,20 and regulation21,22 play key roles in the expression of β-lactam resistance in strains of MRSA. Most recently, antisense technology was used to systematically probe 245 essential genes in MRSA for their role in β-lactam resistance.23 Several non-obvious genes were noted including ftsZ, an important cell division protein. A follow up study revealed that the known FtsZ inhibitor, PC190723, was in fact synergistic with imipenem and effective against MRSA.24
There is likewise emerging evidence that wall teichoic acids (WTAs), anionic polyol phosphate polymers covalently attached to PG, have a role in the expression of β-lactam resistance in MRSA. In 1994, a transposon mutant (llm) that suppressed methicillin resistance in S. aureus was isolated and mapped to the 3′-terminal region of tarO, which encodes the first step of WTA synthesis.25 In S. aureus, the assembly of WTA polymers begins with two non-essential steps for cell viability, encoded in tarO and tarA, and continues with late-acting gene products that are indispensible upon initiation of polymer synthesis.26 Specifically, the TarO protein initiates the assembly of WTAs with the transfer of N-acetylglucosamine-1-phosphate to a membrane-anchored undecaprenyl-phosphate carrier lipid while TarA successively adds an N-acetylmannosamine moiety. This product is ultimately elaborated into a long polymer containing ribitol phosphate repeats that is attached to PG (26) (Supplementary Figure 1). The function of WTAs is incompletely understood but shown to be important in processes such as cell division27−29 and virulence30 and, more recently, in the expression of β-lactam resistance. In the aforementioned antisense study, gene tarL, encoding a late acting WTA biosynthetic enzyme, was identified to lead to β-lactam sensitivity upon depletion.23 In addition, recent evidence has shown that TarS, a glycosyltransferase that attaches β-O-N-acetyl-d-glucosamine residues to WTA polymers, is also required in maintaining β-lactam resistance in MRSA.31 Finally, tunicamycin, a common probe of WTA synthesis, has been shown to sensitize MRSA to β-lactam antibiotics.28 Here, sensitization was attributed to defects in the assembly of PG machinery, due to possible mislocalization of either PBP2 or PBP2A.28 While tunicamycin is an imperfect inhibitor of wall teichoic acid synthesis–it is a non-selective glycosyltransferase inhibitor that also inhibits PG synthesis and has significant eukaryotic toxicity32–these observations are intriguing and consistent with a role for WTA in β-lactam resistance.
Herein we provide new evidence that wall teichoic acid (WTA) biogenesis is indeed a remarkable antibacterial target in MRSA. We found that deletion of gene tarO restored the sensitivity of MRSA to cefuroxime and other β-lactam antibiotics with signature selectivity for PBP2. In addition, we have discovered a novel inhibitor of wall teichoic acid synthesis in S. aureus that strongly potentiates β-lactam antibiotics against MRSA in vitro and in vivo. Where previous studies have suggested that WTA has an important role in localizing PBP4, we provide evidence here that WTA biogenesis plays a key role among the cooperative function of PBPs 2 and 4. The present work offers new insights into the complex biology underlying cell wall synthesis in S. aureus and provides a promising example of how antibiotic drug resistance may be targeted with existing drugs.
Results and Discussion
Deletion of tarO Sensitizes MRSA to β-Lactams
Given their intimate link to β-lactam resistance, we sought to gain a better understanding of the precise mechanism by which WTA polymers mediate β-lactam resistance. We generated a deletion of the tarO gene in MRSA strains, both community- (CA-) and hospital-acquired (HA-), to investigate their sensitivity to β-lactams. Phosphate analysis of isolated cell wall of the epidemic strains CA-MRSA USA300 ΔtarO and HA-MRSA EMRSA 15 ΔtarO, which measures the levels of phosphate-rich WTA polymers, confirmed that the strains were devoid of WTA (Supplementary Table 1). Comparison of the parental strains to their respective ΔtarO deletion strains following treatment with an extensive panel of antibiotics revealed a high sensitivity to β-lactams, while the activity of other classes of antibiotics remained unaffected (Figure 1). Interestingly, only certain β-lactams were highly sensitized in the deletion background, while others retained their resistant phenotype. For example, we observed a greater than 64-fold change in the CA-MRSA ΔtarO strain with cefuroxime and oxacillin and as high as a 512-fold change in the HA-MRSA ΔtarO strain with ceftizoxime. Very minor changes in MIC values were obtained with β-lactams such as cefsulodin and meropenem.
Figure 1.

CA- and HA-MRSA ΔtarO deletion strains impaired for WTA synthesis are sensitized to β-lactam antibiotics. Sensitivity profiles of diverse antibiotics in CA-MRSA USA300 (black bars) and HA-MRSA EMRSA15 (white bars) relative to their ΔtarO deletion strains. Fold change refers to the MIC of the antibiotic in the parent strain divided by MIC in the deletion strain. The highest sensitivity was exclusively observed with certain β-lactam antibiotics.
While a potential connection between WTA expression and PG assembly has been inferred,28,33,34 only recently has a possible mechanism been uncovered. In the absence of WTA synthesis, PBP4 of S. aureus RN4220 was shown to be mislocalized away from the division septum and thus unable to perform its role of cross-linking PG.35 Concordantly, in CA-MRSA strains, PBP4 was shown to be responsible for the production of highly cross-linked peptidoglycan and essential for β-lactam resistance.17,36 These two observations suggested a possible mechanism for the β-lactam sensitivity seen in ΔtarO strains, namely, the impairment of PBP4 function in peptidoglycan cross-linking. Thus, we examined the level of PG cross-linking in CA-, HA-MRSA, and respective ΔtarO deletion strains. To ensure the observations were due specifically to the deletion of tarO and therefore the loss of WTA, a plasmid expressing tagO, the B. subtilis orthologue, was used to complement the HA-MRSA EMRSA15 ΔtarO deletion strain (Supplementary Figure 2). Indeed, the ΔtarO strain was found to have decreased levels (approximately 30%) of highly cross-linked muropeptide species as compared to the parental strain and a higher amount of monomeric, dimeric, and trimeric muropeptides (Supplementary Figure 2), establishing a link between WTA synthesis and β-lactam sensitivity. We posit that a strain devoid of WTA leads to the mislocalization of PBP4, compromising its role as a transpeptidase in PG cross-linking, and specifically in the case of CA-MRSA USA300 results in sensitivity to certain β-lactams.
Combination Screening Identifies Ticlopidine
Given the therapeutic potential of blocking WTA biogenesis in restoring the efficacy of β-lactams against MRSA, we set out to identify a novel inhibitor of WTA synthesis. As a source of chemical matter we employed a library of approximately 2,080 previously approved drugs (PADs). There has been considerable interest in recent years in the concept of screening for new uses for previously approved drug molecules. The interest stems from a growing understanding that small molecules with proven therapeutic activity for a particular use often have uncharacterized potential for alternate therapeutic uses.37,38 Implicit is the advantageous potential of any such molecule to rapidly advance into clinical development by leveraging a proven track record in humans and a deep history of study. Importantly, potentiators of β-lactam activity would be highly desirable components of therapeutic combinations against MRSA.
Thus, we mounted a screen of 2,080 previously approved drugs (Supplementary Table 2) for compounds capable of potentiating the activity of cefuroxime, a broad spectrum β-lactam that was highly sensitized in the ΔtarO strain, against the clinically relevant CA-MRSA strain USA300 (Supplementary Figure 3a). Our screening efforts yielded several active compounds (Supplementary Figure 3b). To identify potential inhibitors of WTA, active combinations were tested against the CA-MRSA USA300 ΔtarO strain. Here, we were looking for suppression of synergy since this strain lacks WTA and is not susceptible to WTA inhibitors. One compound, ticlopidine (Figure 2a), an antiplatelet drug (Ticlid) that inhibits the binding of adenosine 5′-phosphate to its platelet receptor in humans, was effective in restoring the efficacy of cefuroxime against MRSA (Figure 2b) and this synergistic interaction was reversed in the ΔtarO strain (Figure 2c). Ticlopidine, while not active on its own as an antibiotic, was potently synergistic with cefuroxime. Indeed, the fractional inhibitory concentration (FIC) index, a common measure of synergy,39 for this pair against CA-MRSA USA300 was ≤0.063 (Figure 2b). Further, ticlopidine dramatically potentiated the activity of cefuroxime against 9 of 10 MRSA strains,40 including CA-MRSA strain USA300 (64-fold) and HA-MRSA strain USA200 (32-fold) (Table 1). Notably, we did not observe synergy in the common lab strain of S. aureus, RN4220 (Table 1), consistent with a lack of sensitivity to β-lactams in a ΔtarO deletion of this strain (Supplementary Table 3). Strong synergies with cefuroxime were also observed with commercially available on-patent analogues of ticlopidine, namely, clopidogrel (Plavix) and prasugrel (Effient), against CA-MRSA USA300 (FIC index ≤0.125 and ≤0.5, respectively) (Supplementary Table 4). To assess in vivo efficacy, we administered ticlopidine and cefuroxime in a Galleria mellonella model of MRSA infection. The larval stage of the Greater Wax Moth is a widely used model to assess S. aureus virulence and allows for a testing throughput that is otherwise impossible with small mammals but nevertheless representative of in vivo activity.41,42 In this model, a significantly (P < 0.001) higher fraction of larvae survived MRSA infection following combined treatment, compared to that with cefuroxime and ticlopidine alone (Figure 2d). After 14 days, the survival rate increased from 3.3% in untreated larvae to 53.3% when treated with the combination; treatment with cefuroxime and ticlopidine alone led to 16.7% and 10% survival, respectively (Figure 2d). Thus, cefuroxime and ticlopidine at sub-efficacious doses acted synergistically to provide G. mellonella protection from bacterial infection.
Figure 2.

Ticlopidine potentiates the activity of the β-lactam antibiotic cefuroxime against CA-MRSA USA300, but not CA-MRSA USA300 ΔtarO. (a) Chemical structure of ticlopidine. (b) Microdilution checkerboard analysis showing the combined effect of cefuroxime and ticlopidine against CA-MRSA USA300 where the extent of inhibition is shown as a heat plot. Synergistic effects are evident as both molecules alone have MICs that exceed 256 μg/mL and result in an FIC index of ≤0.063. (c) Suppression of the synergy in CAMRSA USA300 ΔtarO, leading to an additive interaction with FIC index of ≤2. (d) Galleria mellonella virulence assay. Survival curve of G. mellonella infected with CA-MRSA USA300 receiving no drug treatment (control, CTRL) or a treatment with 0.3 mg/kg cefuroxime (CEF) or 0.3 mg/kg ticlopidine (TIC) or a combination of both at 0.3 mg/kg each (CEF+TIC). After 14 days, treatment with the combination lead to significantly increased survival, compared to no drug or antibiotic treatment alone (P < 0.001).
Table 1. In Vitro Interactions between Ticlopidine and Cefuroxime in Various S. aureus Species.
| straina | MIC cefuroxime (μg/mL) | FICb cefuroxime | MIC ticlopidine (μg/mL) | FICb ticlopidine | FIC indexc |
|---|---|---|---|---|---|
| Newmand | 2 | 0.125 | >256 | 0.125 | ≤0.250 |
| HA-MRSA USA600e | ≥1024 | 0.008 | >256 | 0.032 | ≤0.040 |
| HA-MRSA USA100/800/NYe | 1024 | 0.125 | >256 | 0.125 | ≤0.250 |
| HA-MRSAe | >2048 | 0.250 | >256 | 0.032 | ≤0.282 |
| HA-MRSA USA200/EMRSA16e | 1024 | 0.032 | >256 | 0.063 | ≤0.095 |
| HA-MRSA USA500e | 32 | 1 | >256 | 1 | ≤2 |
| HA-MRSAe | >2048 | 0.250 | >256 | 0.016 | ≤0.266 |
| CA-MRSA USA400/MW2e | 256 | 0.063 | >256 | 0.063 | ≤0.125 |
| HA-MRSA EMRSA15e | 512 | 0.063 | >256 | 0.063 | ≤0.125 |
| HA-MRSAe | 2048 | 0.125 | >256 | 0.063 | ≤0.188 |
| CA-MRSA USA300e | 512 | 0.032 | >256 | 0.032 | ≤0.063 |
| RN4220f | 0.5 | 1 | >128 | 1 | ≤2 |
| SA178R1g | 0.5 | 1 | >128 | 1 | ≤2 |
HA, hospital-associated isolate; CA, community-associated isolate.
Fractional inhibitory concentration (FIC) = [X]/MICX, where [X] is the lowest inhibitory concentration of drug in the presence of the co-drug.
FIC index = FICcefuroxime + FICticlopidine.
Reference (50).
Reference (40).
Reference (51).
Reference (26).
Characterization of the Mode of Action of Ticlopidine
To further probe the mechanism of action of ticlopidine, we investigated its interactions with an extensive panel of diverse antibiotics using CA-MRSA USA300. Ticlopidine restored the efficacy of several β-lactam antibiotics, while the activity of other classes of antibiotics remained unaffected (Supplementary Table 5). Indeed, the sensitization profile was strikingly similar to that seen in the ΔtarO strains (Figure 1), although the sensitivity was generally higher in the deletion strain, likely because it was completely devoid of WTA (as shown by phosphate analysis in Supplementary Table 1). Most remarkable was the lack of antibacterial activity of ticlopidine on its own and its capacity to render MRSA highly susceptible to a number of β-lactams. We reasoned that if ticlopidine inhibited WTA synthesis, its lack of antibacterial activity would be consistent with inhibition of the early steps in the pathway catalyzed by TarO or TarA. WTA biosynthetic genes exhibit complex dispensability patterns.26,43 WTA genes encoding the initiating enzymes, TarO and TarA, are dispensable for growth in vitro while the downstream late-acting genes have an essential phenotype. Idiosyncratically, the late-acting genes become dispensable in strains with a deletion in either tarO or tarA, presumably because accumulation of WTA intermediates is toxic to the cell.26,29,33,43
To test the hypothesis that ticlopidine inhibited either of these initiating enzymes we initially employed genetic and physiological approaches. First, we took advantage of the conditional dispensability patterns of late steps in WTA assembly. As an early step inhibitor, ticlopidine should have an antagonistic interaction with the late step (TarG) inhibitor, targosil. (44) TarG, an essential gene product, is the transmembrane component of the ABC transporter that exports WTAs to the cell surface. Indeed, ticlopidine rendered targosil completely inactive against both CA-MRSA USA300 and the S. aureus lab strain RN4220 (Figure 3a and Supplementary Figure 4). In additional genetic experiments, we tested the capacity of ticlopidine to suppress the lethal phenotype associated with late gene tarH. Using a strain where tarH expression was under the control of a xylose-induced promoter, we found that ticlopidine could partially rescue growth in the absence of xylose (40% relative to growth in 2% xylose) at the highest concentration tested (Figure 3b). We next investigated ticlopidine's ability to directly decrease WTA synthesis by measuring phosphate content in the cell wall of S. aureus with increasing concentrations of ticlopidine. In both CA-MRSA USA300 (Figure 3c) and SA1781 (a RN4220 derivative) (Supplementary Figure 5), WTA incorporation began to decrease at ticlopidine concentrations of 64 μg/mL, similar to the concentration at which it begins to potentiate the activity of cefuroxime. Greater inhibition was noted at 200 μg/mL of ticlopidine, but residual WTA remained (∼50%), while ΔtarO strains were completely devoid of phosphate. We note here that the partial suppression seen was consistent with the partial inhibition of WTA synthesis evident in our cell wall phosphate content assays. Furthermore, bacteriophage Ø11, which uses WTA as receptor-binding sites, infected S. aureus RN450 but not ticlopidine- or tunicamycin-treated RN450 (Supplementary Figure 6). WTA is known to contribute to the resistance of PG against lysozyme.45 We found that wild-type PG was completely resistant to degradation by lysozyme, while PG from ticlopidine-treated cells and ΔtarO cells were indeed sensitive to degradation (Supplementary Figure 7). In sum, our genetic and physiological experiments were consistent with ticlopidine targeting either TarO or TarA.
Figure 3.

Ticlopidine inhibits the initiation of wall teichoic acid biosynthesis in S. aureus. (a) At concentrations of 8 μg/mL, ticlopidine begins to antagonize the activity of targosil, a late-stage inhibitor, against CA-MRSA USA300. (b) Ticlopidine can suppress the lethality associated with late WTA stage deletion. Shown is the percent growth of a tarH conditional deletion strain normalized to the growth upon induction with 2% xylose in the presence of increasing concentrations of ticlopidine. Ticlopidine can recover approximately 40% of the growth at the highest concentration of 256 μg/mL. (c) Ticlopidine shows a dose-dependent decrease in the phosphate content of cell wall isolated from CA-MRSA USA300, with approximately 50% less phosphate when ticlopidine is present at 200 μg/mL. CA-MRSA USA300 tarO is completely devoid of WTA compared to the parental strain. (d) Membrane-based in vitro assay following the generation of a radiolabeled reaction product of TarO, undecapreny-P-P-[14C]GlcNAc, as a result of the incorporation of [14C]GlcNAc onto undecaprenyl-P-P. Assay was performed on membranes derived from E. coli cells (wecA) expressing recombinant TarO from S. aureus. The reaction product was monitored by thin layer chromatography (TLC) and shown to be dependent on the presence of recombinant TarO (Supplementary Figure 8). Ticlopidine inhibited the activity of TarO, yielding an IC50 value of 238 ± 14 μM.
To test directly the hypothesis that ticlopidine inhibited either TarO or TarA, we used in vitro biochemical assays. While ticlopidine showed no impact on the transferase activity of TarA (Supplementary Figure 9) we found substantial inhibition of TarO activity (Figure 3d). Further, the relatively weak inhibition constant (IC50 of 238 μM or 71 μg/mL) was consistent with the partial phenotypes noted in our phenotypic and genetic assays.
A Basis for the Synergy among Ticlopidine and β-Lactams
To further investigate the basis of synergy of ticlopidine and cefuroxime, we sought to understand the characteristic β-lactam sensitivity profiles that were shared on treatment of MRSA in combination with ticlopidine or alone using the ΔtarO MRSA strains. A survey of the PBP selectivity of β-lactams tested in our studies revealed that sensitization was seen particularly with those having high binding affinities for PBP2 of S. aureus (Figure 1 and Supplementary Table 5). Given the impact of the loss of WTA on PBP4 localization,35 PBP2 selectivity can be rationalized on the basis of previous reports of cooperativity between PBP2 and PBP4 in creating highly cross-linked PG in MRSA strains contributing to β-lactam resistance18 (Figure 4, panel i). A strain lacking WTA, such as with treatment with ticlopidine, would be compromised in its PG cross-linking due to mislocalized PBP4 and be highly sensitive to β-lactams such as cefuroxime that target its partner protein PBP2 (Figure 4, panel iii). Resistance would persist if the β-lactam in combination with ticlopidine targets PBP1 or PBP3, as sufficient cross-linking would maintain the resistant phenotype (Figure 4, panel ii). Further, this rationale is consistent with the idea that PBP2A is not the sole determinant for β-lactam resistance in MRSA.9,15,17 It suggests that the synergy between ticlopidine and cefuroxime might also be observed in a wild-type, methicillin-sensitive strain such as S. aureus Newman, which lacks PBP2A. Indeed, synergy is evident in this strain (Table 1). Remarkably, this work also suggests that β-lactam resistance might be reversed by targeting PBP2 and PBP4 with combinations of existing β-lactams. We tested this using the PBP4-selective β-lactam cefoxitin.46 When cefoxitin was combined with the PBP2-selective β-lactams cefuroxime, ceftizoxime, oxacillin, and penicillin, the MICs of these antibiotics decreased 32- to 128-fold. Conversely, combining cefoxitin with β-lactams having a low affinity for PBP2 led to only a 2- to 8-fold change in the MIC (Supplementary Table 6).
Figure 4.
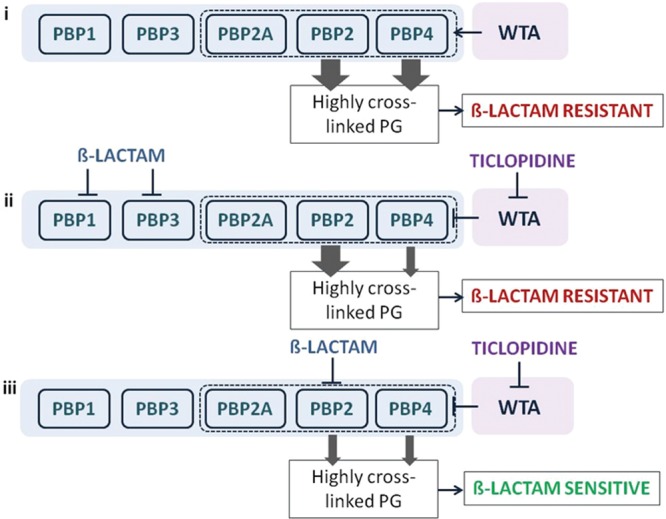
A synthetic lethal interaction when targeting TarO and native PBP2. Indirect Inhibition of PBP4, via inhibition of TarO, and inhibition of PBP2 function account for the synergistic interaction among ticlopidine and cefuroxime. (i) When not challenged with β-lactams, WTA synthesis will guide the proper localization of PBP4 and, together with PBP2, will provide highly cross-linked muropeptide species (thick arrow) that contribute to high-level β-lactam resistance. (ii) Treatment with a β-lactam with low affinity for PBP2 and ticlopidine leads to an additive interaction as sufficient highly cross-linked peptidoglycan are present to maintain β-lactam resistance (one thick arrow), even when PBP4 function is affected by the lack of WTA (one thin arrow). (iii) Due to their cooperative function, PBP4 (when challenged with ticlopidine) and PBP2 (when challenged with β-lactam with high affinity for PBP2), will be impaired in their capacities to produce a highly cross-linked cell wall (two thin arrows), contributing to enhanced β-lactam susceptibility and thus a synergistic interaction.
Conclusions
The WTA biosynthetic pathway has long been speculated as a viable target for antibacterial intervention. While dispensable for viability, WTA is known to be a critical determinant of cell shape in Bacillus subtilis,27,29 virulence in S. aureus,30 and recently, for proper cell division.28 Furthermore, the essential phenotypes of late WTA biosynthetic genes, although paradoxical, suggest that these enzymes may well be reasonable targets for new antibiotics. Emerging evidence of a role for wall teichoic acids in β-lactam resistance in MRSA has fueled further interest in targeting this pathway, including a renewed interest in tunicamycin, a natural product nucleoside antibiotic that has been shown to inhibit TarO.28 Unfortunately, tunicamycin is a promiscuous inhibitor of bacterial and eukaryotic phosphosugar transferases, hindering its use as a selective probe and therapeutic. In the work reported here, we have carried out a meticulous chemical genetic study of the importance of wall teichoic acids to community- and hospital-acquired MRSA strains to reveal signature interactions with β-lactam antibiotics having a known selectivity for PBP2. These findings led to the discovery that ticlopidine, a well-known antiplatelet drug, had the cryptic capacity to block WTA synthesis through the inhibition of TarO. Importantly, synergistic interactions of ticlopidine and PBP2-binding β-lactams provide for efficacious combinations to limit the growth of MRSA strains in vitro and in vivo. Thus ticlopidine represents a promising new lead with a strong record of examination in humans but also provides an exciting new probe of WTA synthesis. In our hands, ticlopidine was an extremely useful probe along with existing β-lactam antibiotics to further elaborate the role of WTA in supporting the cooperative role of PBP2 and 4 in the expression of β-lactam resistance in MRSA. As such, this work offers new insights into the complex biology underlying cell wall synthesis in S. aureus and provides a promising example of how antibiotic drug resistance might be targeted with existing drugs.
Methods
Peptidoglycan Purification and Analysis
Peptidoglycan from CA-MRSA USA300, HA-MRSA EMRSA15, their ΔtarO deletion, and HA-MRSA EMRSA15ΔtarO pLI50-tagO was prepared from exponentially growing cells as previously described.47 The purified peptidoglycan was digested with mutanolysin (Sigma), an N-acetylmuramidase that cuts glycan strands between the N-acetylmuramic and N-acetylglucosamine residues. The resulting muropeptides were reduced with sodium borohydride (Sigma) and analyzed by reverse-phase HPLC using a Hypersil ODS column (Thermo Electron Corporation). The eluted muropeptides were detected and quantified by determination of their ultraviolet absorption at 206 nm, using the Shimadzu LC solution software. The area of eluted UV-absorbing peaks, corresponding to the different muropeptides, was quantified and shown as a percentage of the total area of the chromatogram.
Combination Screening
CA-MRSA USA300 was screened against the PAD library in the presence of cefuroxime. The screening protocol was based on CLSI guidelines. Screening was carried out in 96-well plates, in duplicate, using Mueller Hinton Broth (MHB) with 2% DMSO and a library compound concentration of 10 μM. The concentration of cefuroxime was 16 μg/mL, a quarter of its MIC value obtained under the same conditions. Background controls (8 wells per plate) contained only media, and DMSO and growth controls, also 8 wells per plate, contained media, DMSO, and inoculum. Plates were incubated at 37 °C for 20 h, and optical density was read at 600 nm using an EnVision plate reader (Perkin-Elmer). The percentage growth for each test well was calculated as (OD – mean background)/(mean growth – mean background) × 100 and normalized to the percent growth attributed by the PAD alone to obtain a growth ratio such that a ratio of 1.0 is indicative of no difference.
Checkerboard Analyses and FIC Index Determination
FICs were determined by setting up standard checkerboard broth microdilution assays with 8 (or 9) serially diluted concentrations of each drug, using the same conditions as screening. At least 3 replicates were done for each combination, and the means were used for calculation. The MIC for each drug was the lowest [drug] showing <10% growth. The FIC for each drug was calculated as the [drug in the presence of co-drug] for a well showing <10% growth, divided by the MIC for that drug. The FIC index is the sum of the two FICs. Interactions with FIC index of <0.5 were deemed synergistic.
Phosphate Analysis
Strains were inoculated from an overnight culture and grown in 100 mL of MHB to OD600 of 0.8–0.9 at 37 °C, 250 rpm with various compound treatment. Isolation of cell wall and phosphate analysis were performed as previously described.48
In Vitro TarO Inhibition Assay
For this assay, UDP-[14C]GlcNAc (0.1 mCi/mL) was purchased from American Radiolabeled Chemicals, Ultima Gold liquid scintillation cocktail from Perkin-Elmer, and silica gel 60 TLC plates from EMD Chemicals. TarO activity was assayed in 100 μL reactions containing Reaction Buffer (50 mM Tris pH = 8, 10 mM MgCl2, 1 mM EDTA), 300 μM UDP-GlcNAc, 0.1 μCi UDP-[14C]GlcNAc, 0.1% (w/v) TritonX-100, 0.8% (v/v) DMSO, and varied amounts of membranes (75 μg to 1 mg of protein). Reactions were quenched with the addition of 1250 μL of CHCl3/MeOH (3:2). Lipid-linked products were extracted according to the following method: Quenched reactions were incubated for 2.5 h followed by vortexing for 3 min. Insoluble material was removed by centrifugation (13000g, 2 min), and 150 μL of 40 mM MgCl2 was added to the supernatant. Samples were vortexted for 5 min, and the upper aqueous layer was removed. The lower, organic layer was washed twice with 400 μL of pure solvent upper phase (CHCl3/MeOH/H2O/1 M MgCl2 in H2O [18:294:282:1]). The final product was analyzed by either TLC or scintillation counting. TLC analysis was preformed as in ref (49).
Acknowledgments
We would like to thank N. Magarvey for providing bacteriophage Ø11. The work was supported by a Canada Research Chair Award to E.D.B., the Canadian Cystic Fibrosis Foundation, and an operating grant from CIHR to E.D.B. (MOP-81330). Work in the laboratory of G.D.W. was supported by a Canada Research Chair Award to G.D.W. and CIHR operating grant to G.D.W. (MT-13536). Work in the laboratory of M.G.P. was funded by FCT grants PTDC/BIA-BCM/099152/2008 and PEst-OE/EQB/LA0004/2011. P.M.P. was supported by FCT fellowship SFRH/BD/41119/2007.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12. [DOI] [PubMed] [Google Scholar]
- Klevens R. M.; Morrison M. A.; Nadle J.; Petit S.; Gershman K.; Ray S.; Harrison L. H.; Lynfield R.; Dumyati G.; Townes J. M.; Craig A. S.; Zell E. R.; Fosheim G. E.; McDougal L. K.; Carey R. B.; Fridkin S. K. (2007) Invasive methicillin-resistant Staphylococcus aureus infections in the United States. J. Am. Med. Assoc. 298, 1763–1771. [DOI] [PubMed] [Google Scholar]
- Baba T.; Takeuchi F.; Kuroda M.; Yuzawa H.; Aoki K.; Oguchi A.; Nagai Y.; Iwama N.; Asano K.; Naimi T.; Kuroda H.; Cui L.; Yamamoto K.; Hiramatsu K. (2002) Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359, 1819–1827. [DOI] [PubMed] [Google Scholar]
- Diep B. A.; Gill S. R.; Chang R. F.; Phan T. H.; Chen J. H.; Davidson M. G.; Lin F.; Lin J.; Carleton H. A.; Mongodin E. F.; Sensabaugh G. F.; Perdreau-Remington F. (2006) Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet 367, 731–739. [DOI] [PubMed] [Google Scholar]
- Howden B. P.; Johnson P. D.; Charles P. G.; Grayson M. L. (2004) Failure of vancomycin for treatment of methicillin-resistant Staphylococcus aureus infections. Clin. Infect. Dis. 39, 1544.; author reply 1544–1545. [DOI] [PubMed] [Google Scholar]
- Mangili A.; Bica I.; Snydman D. R.; Hamer D. H. (2005) Daptomycin-resistant, methicillin-resistant Staphylococcus aureus bacteremia. Clin. Infect. Dis. 40, 1058–1060. [DOI] [PubMed] [Google Scholar]
- Tsiodras S.; Gold H. S.; Sakoulas G.; Eliopoulos G. M.; Wennersten C.; Venkataraman L.; Moellering R. C.; Ferraro M. J. (2001) Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 358, 207–208. [DOI] [PubMed] [Google Scholar]
- Lim D.; Strynadka N. C. (2002) Structural basis for the beta lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 9, 870–876. [DOI] [PubMed] [Google Scholar]
- Pinho M. G.; de Lencastre H.; Tomasz A. (2001) An acquired and a native penicillin-binding protein cooperate in building the cell wall of drug-resistant staphylococci. Proc. Natl. Acad. Sci. U.S.A. 98, 10886–10891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llarrull L. I.; Fisher J. F.; Mobashery S. (2009) Molecular basis and phenotype of methicillin resistance in Staphylococcus aureus and insights into new beta-lactams that meet the challenge. Antimicrob. Agents Chemother. 53, 4051–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk S.; Verlaine O.; Gerards T.; Zivec M.; Humljan J.; Sosic I.; Amoroso A.; Zervosen A.; Luxen A.; Joris B.; Gobec S. (2011) New noncovalent inhibitors of penicillin-binding proteins from penicillin-resistant bacteria. PLoS One 6, e19418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy D. Y.; Unal S. (2008) New antimicrobial agents for the treatment of Gram-positive bacterial infections. Clin. Microbiol. Infect. 14, 411–420. [DOI] [PubMed] [Google Scholar]
- Berger-Bachi B. (1999) Genetic basis of methicillin resistance in Staphylococcus aureus. Cell. Mol. Life Sci. 56, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lencastre H.; Tomasz A. (1994) Reassessment of the number of auxiliary genes essential for expression of high-level methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 38, 2590–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lencastre H.; Wu S. W.; Pinho M. G.; Ludovice A. M.; Filipe S.; Gardete S.; Sobral R.; Gill S.; Chung M.; Tomasz A. (1999) Antibiotic resistance as a stress response: complete sequencing of a large number of chromosomal loci in Staphylococcus aureus strain COL that impact on the expression of resistance to methicillin. Microb. Drug Resist. 5, 163–175. [DOI] [PubMed] [Google Scholar]
- Labischinski H.; Ehlert K.; Berger-Bachi B. (1998) The targeting of factors necessary for expression of methicillin resistance in staphylococci. J. Antimicrob. Chemother. 41, 581–584. [DOI] [PubMed] [Google Scholar]
- Memmi G.; Filipe S. R.; Pinho M. G.; Fu Z.; Cheung A. (2008) Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob. Agents Chemother. 52, 3955–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leski T. A.; Tomasz A. (2005) Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J. Bacteriol. 187, 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardete S.; Ludovice A. M.; Sobral R. G.; Filipe S. R.; de Lencastre H.; Tomasz A. (2004) Role of murE in the Expression of beta-lactam antibiotic resistance in Staphylococcus aureus. J. Bacteriol. 186, 1705–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobral R. G.; Ludovice A. M.; Gardete S.; Tabei K.; De Lencastre H.; Tomasz A. (2003) Normally functioning murF is essential for the optimal expression of methicillin resistance in Staphylococcus aureus. Microb. Drug Resist. 9, 231–241. [DOI] [PubMed] [Google Scholar]
- Berger-Bachi B.; Rohrer S. (2002) Factors influencing methicillin resistance in staphylococci. Arch. Microbiol. 178, 165–171. [DOI] [PubMed] [Google Scholar]
- Kuroda M.; Kuroda H.; Oshima T.; Takeuchi F.; Mori H.; Hiramatsu K. (2003) Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49, 807–821. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Jarantow L. W.; Wang H.; Sillaots S.; Cheng H.; Meredith T. C.; Thompson J.; Roemer T. (2011) Antagonism of chemical genetic interaction networks resensitize MRSA to beta-lactam antibiotics. Chem. Biol. 18, 1379–1389. [DOI] [PubMed] [Google Scholar]
- Tan C. M.; Therien A. G.; Lu J.; Lee S. H.; Caron A.; Gill C. J.; Lebeau-Jacob C.; Benton-Perdomo L.; Monteiro J. M.; Pereira P. M.; Elsen N. L.; Wu J.; Deschamps K.; Petcu M.; Wong S.; Daigneault E.; Kramer S.; Liang L.; Maxwell E.; Claveau D.; Vaillancourt J.; Skorey K.; Tam J.; Wang H.; Meredith T. C.; Sillaots S.; Wang-Jarantow L.; Ramtohul Y.; Langlois E.; Landry F.; Reid J. C.; Parthasarathy G.; Sharma S.; Baryshnikova A.; Lumb K. J.; Pinho M. G.; Soisson S. M.; Roemer T. (2012) Restoring methicillin-resistant Staphylococcus aureus Susceptibility to beta-lactam antibiotics. Sci. Transl. Med. 4, 126ra135. [DOI] [PubMed] [Google Scholar]
- Maki H.; Yamaguchi T.; Murakami K. (1994) Cloning and characterization of a gene affecting the methicillin resistance level and the autolysis rate in Staphylococcus aureus. J. Bacteriol. 176, 4993–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Elia M. A.; Pereira M. P.; Chung Y. S.; Zhao W.; Chau A.; Kenney T. J.; Sulavik M. C.; Black T. A.; Brown E. D. (2006) Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J. Bacteriol. 188, 4183–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavsar A. P.; Beveridge T. J.; Brown E. D. (2001) Precise deletion of tagD and controlled depletion of its product, glycerol 3-phosphate cytidylyltransferase, leads to irregular morphology and lysis of Bacillus subtilis grown at physiological temperature. J. Bacteriol. 183, 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J.; Singh A. K.; Santa Maria J. P. Jr.; Kim Y.; Brown S.; Swoboda J. G.; Mylonakis E.; Wilkinson B. J.; Walker S. (2011) Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem. Biol. 6, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Elia M. A.; Millar K. E.; Beveridge T. J.; Brown E. D. (2006) Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J. Bacteriol. 188, 8313–8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidenmaier C.; Peschel A. (2008) Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 6, 276–287. [DOI] [PubMed] [Google Scholar]
- Brown S.; Xia G.; Luhachack L. G.; Campbell J.; Meredith T. C.; Chen C.; Winstel V.; Gekeler C.; Irazoqui J. E.; Peschel A.; Walker S. (2012) Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc. Natl. Acad. Sci. U.S.A. 10.1073/pnas.1209126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price N. P.; Tsvetanova B. (2007) Biosynthesis of the tunicamycins: a review. J. Antibiot. (Tokyo) 60, 485–491. [DOI] [PubMed] [Google Scholar]
- D’Elia M. A.; Millar K. E.; Bhavsar A. P.; Tomljenovic A. M.; Hutter B.; Schaab C.; Moreno-Hagelsieb G.; Brown E. D. (2009) Probing teichoic acid genetics with bioactive molecules reveals new interactions among diverse processes in bacterial cell wall biogenesis. Chem. Biol. 16, 548–556. [DOI] [PubMed] [Google Scholar]
- Schlag M.; Biswas R.; Krismer B.; Kohler T.; Zoll S.; Yu W.; Schwarz H.; Peschel A.; Gotz F. (2010) Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 75, 864–873. [DOI] [PubMed] [Google Scholar]
- Atilano M. L.; Pereira P. M.; Yates J.; Reed P.; Veiga H.; Pinho M. G.; Filipe S. R. (2010) Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 107, 18991–18996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze U. U.; Berger-Bachi B. (1995) Staphylococcus aureus penicillin-binding protein 4 and intrinsic beta-lactam resistance. Antimicrob. Agents Chemother. 39, 2415–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong C. R.; Sullivan D. J. Jr. (2007) New uses for old drugs. Nature 448, 645–646. [DOI] [PubMed] [Google Scholar]
- Ejim L.; Farha M. A.; Falconer S. B.; Wildenhain J.; Coombes B. K.; Tyers M.; Brown E. D.; Wright G. D. (2010) Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 7, 348–350. [DOI] [PubMed] [Google Scholar]
- Odds F. C. (2003) Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52, 1. [DOI] [PubMed] [Google Scholar]
- Christianson S.; Golding G. R.; Campbell J.; Mulvey M. R. (2007) Comparative genomics of Canadian epidemic lineages of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 45, 1904–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W.; Chua K.; Davies J. K.; Newton H. J.; Seemann T.; Harrison P. F.; Holmes N. E.; Rhee H. W.; Hong J. I.; Hartland E. L.; Stinear T. P.; Howden B. P. (2010) Two novel point mutations in clinical Staphylococcus aureus reduce linezolid susceptibility and switch on the stringent response to promote persistent infection. PLoS Pathog. 6, e1000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves J.; Cockayne A.; Moody P. C.; Morrissey J. A. (2010) Comparison of the regulation, metabolic functions, and roles in virulence of the glyceraldehyde-3-phosphate dehydrogenase homologues gapA and gapB in Staphylococcus aureus. Infect. Immun. 78, 5223–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Elia M. A.; Henderson J. A.; Beveridge T. J.; Heinrichs D. E.; Brown E. D. (2009) The N-acetylmannosamine transferase catalyzes the first committed step of teichoic acid assembly in Bacillus subtilis and Staphylococcus aureus. J. Bacteriol. 191, 4030–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda J. G.; Meredith T. C.; Campbell J.; Brown S.; Suzuki T.; Bollenbach T.; Malhowski A. J.; Kishony R.; Gilmore M. S.; Walker S. (2009) Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus. ACS Chem. Biol. 4, 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera A.; Biswas R.; Herbert S.; Kulauzovic E.; Weidenmaier C.; Peschel A.; Gotz F. (2007) Influence of wall teichoic acid on lysozyme resistance in Staphylococcus aureus. J. Bacteriol. 189, 280–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S. R.; Cherubin C. E. (1978) Use of cefoxitin, new cephalosporin-like antibiotic, in the treatment of aerobic and anaerobic infections. Antimicrob. Agents Chemother. 14, 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipe S. R.; Tomasz A.; Ligoxygakis P. (2005) Requirements of peptidoglycan structure that allow detection by the Drosophila Toll pathway. EMBO Rep. 6, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavsar A. P.; Erdman L. K.; Schertzer J. W.; Brown E. D. (2004) Teichoic acid is an essential polymer in Bacillus subtilis that is functionally distinct from teichuronic acid. J. Bacteriol. 186, 7865–7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer C.; Wugeditsch T.; Messner P.; Whitfield C. (2002) Functional expression of enterobacterial O-polysaccharide biosynthesis enzymes in Bacillus subtilis. Appl. Environ. Microbiol. 68, 4722–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie E. S.; Lorenz L. L. (1952) Staphylococcal coagulase; mode of action and antigenicity. J. Gen. Microbiol. 6, 95–107. [DOI] [PubMed] [Google Scholar]
- Kreiswirth B. N.; Lofdahl S.; Betley M. J.; O’Reilly M.; Schlievert P. M.; Bergdoll M. S.; Novick R. P. (1983) The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.