

Summary
OLE RNAs are large, noncoding transcripts characterized by their ornate secondary structure and presence predominantly in Gram-positive, extremophilic bacteria. A gene for an OLE-associated protein (OAP) is almost always located immediately downstream of the OLE gene. OAP has no extensive homology to other proteins and is predicted to form multiple transmembrane domains. We show that this protein forms a ribonucleoprotein complex with OLE RNA using at least 2:1 protein:RNA stoichiometry. A series of truncated OLE RNA constructs was used to establish that most of the RNA can be deleted without eliminating protein binding. Two primary binding sites are present within the RNA, although additional binding determinants exist and extensive structural stabilization is induced by OAP. RNA fluorescence in situ hybridization was used in Escherichia coli to demonstrate that ribonucleoprotein complex formation localizes the RNA near cell membranes of this heterologous system. Therefore the majority of the complex structure formed by OLE RNA may perform a biochemical function that requires membrane localization.
Keywords: anaerobe, extremophile, noncoding RNA, ribonucleoprotein, ribozyme
Introduction
OLE (ornate, large, extremophilic) RNA representatives are unusually complex and large noncoding RNAs that are distributed predominantly in extremophiles (Puerta-Fernandez et al., 2006). This RNA class was first identified by using comparative sequence analysis methods (Corbino et al., 2005), which can readily identify even rare noncoding RNAs that exhibit extensive sequence and secondary structure conservation. While much is known about small RNAs, from the microRNAs of eukaryotes (Bartel and Chen, 2004, Zamore and Haley, 2005) to the various riboregulators in bacteria (Waters and Storz, 2009, Gottesman, 2005), far less is understood about the many large RNAs that have been identified in both bacteria and eukaryotes. Recently, analyses of chromatin signatures in eukaryotic cells revealed that many long, noncoding RNAs are produced (Khalil et al., 2009, Guttman et al., 2009). Initial structural and functional analyses of some RNAs from this collection indicate that they function as regulators of gene expression (Gupta et al., 2010, Guttman et al., 2009). However, the functions of many of these RNAs have yet to be characterized, and there are likely many more yet to be discovered.
In bacteria, several large, noncoding RNAs were recently identified by using comparative sequence analysis (Weinberg et al., 2009), and their ornate secondary structures are consistent with complex functions. It is interesting to note that some of the most studied large and complex-folded RNAs, such as group II self-splicing introns (Pyle, 2010) and RNase P ribozymes (Kazantsev and Pace, 2006), have catalytic function. Many of these RNAs exist as RNP complexes, suggesting that while protein components may not be directly involved in catalysis, they commonly play an important role in ribozyme folding. Whether newly identified long, noncoding RNAs also have catalytic function remains to be determined.
Because of the phylogenetic distribution of OLE RNA amongst anaerobic Gram-positive bacteria that typically live in extreme environments, we are particularly interested in the possibility that OLE RNA allows these bacteria to adapt to their extreme surroundings. The previous identification of a protein of unknown function that tracks phylogenetically with OLE RNA (Puerta-Fernandez et al., 2006) suggested the possibility that the molecules form a ribonucleoprotein (RNP) complex. In the following studies, we observe that the RNA is highly abundant during exponential growth in Bacillus halodurans. Additionally, we find that OLE RNA and the conserved protein form an RNP complex in vitro. This RNP formation in vivo leads to localization of OLE RNA to cell membranes, a rare characteristic of noncoding RNAs that suggests a novel function for this RNA.
Results and Discussion
Expansion of the number of OLE RNA representatives
OLE RNA was initially identified through comparative sequence analysis in 15 species of Firmicutes, and an additional 20 examples of OLE RNA were isolated by amplifying homologs from a hypersaline microbial mat (Puerta-Fernandez et al., 2006). In the current study, we conducted sequence homology searches to further expand the number of OLE RNAs in sequenced organisms to 48, and we have identified more than 200 further examples in a human gut metagenome (Qin et al., 2010). Many of the species identified in the gut metagenome belong to the class Clostridia (Qin et al., 2010) and OLE RNAs are frequently encountered in sequenced genomes of species within this class. At least some of the species encountered within the gut microenvironment are anaerobic, a characteristic of all known OLE-carrying species. By combining bioinformatics algorithms that examine conserved secondary structures of noncoding RNAs (Nawrocki et al., 2009) and structural probing of the full-length RNA, we have refined the original consensus sequence and secondary structure model for OLE RNA (Fig. 1).
Fig. 1.

Revised consensus sequence and secondary structure model for OLE RNAs. Phylogenetic analysis of 78 examples from sequenced genomes, metagenomic data, and community PCR were used to define the consensus model. Dashed lines indicate highly conserved regions implicated in protein binding.
Strikingly, almost one-third of the ~600 nucleotides of OLE RNAs remain more than 90% conserved among all OLE RNAs identified. However, the representatives identified in human gut metagenomic data were not included in this analysis because the method of sequencing produced several single-nucleotide insertions that hindered accurate sequence and structure comparisons. Many of the most highly conserved nucleotides are clustered in regions predicted to reside in loops or bulges, which is typical of functional RNAs that form complex tertiary structures. Nucleotide covariation that retains predicted base complementarity is observed within every proposed base-paired region, which is indicative of extensive conservation of numerous secondary substructures. This extensive conservation of sequence and structure places OLE RNAs among the top five most complex noncoding RNAs currently known to exist in bacteria (Weinberg et al., 2009).
OLE RNA expression and abundance
With very few exceptions, the gene for OLE RNA is located in the middle of a large operon of genes whose functions are diverse (Puerta-Fernandez et al., 2006). Several nearby genes code for proteins that are involved in isoprenoid biosynthesis or in DNA repair, while the gene immediately downstream has no previously known function (see below). Previously, RT-PCR analysis (Puerta-Fernandez et al., 2006) was used to establish that OLE RNA can be produced as part of extended transcripts that carry protein-coding regions on either flank. However, a reporter gene fused downstream of the noncoding portion of the genome carrying the OLE gene gives substantial expression (Puerta-Fernandez et al., 2006), suggesting that OLE RNA could also be expressed from its own promoter. Moreover, a transcriptional promoter is predicted by sequence analysis to reside immediately upstream of the sequence encoding OLE (Ko and Altman, 2007). Therefore primary transcripts of OLE RNA may not be uniform.
We conducted a transcriptome analysis to assess the relative amount of OLE RNA in B. halodurans. Parallel sequencing of cDNAs generated from B. halodurans RNAs extracted during exponential growth phase revealed that OLE RNA is among the most abundant transcripts (Fig. 2A), with RNA copy number exceeding more than 99.8% of mRNAs (Table S1). Indeed, the abundance of OLE RNA is approximately one tenth that of the RNA component of the signal recognition particle (SRP), which is among the most abundant RNA transcripts in bacterial cells. OLE abundance was confirmed by quantitative reverse transcription and PCR (qRT-PCR), which indicates there is 40-fold more SRP RNA than OLE RNA. SRP RNA is present at roughly 2% the levels of tRNA (Hsu et al., 1984) and 4% the levels of ribosomes in Escherichia coli (Jensen and Pedersen, 1994). Furthermore, the abundance of Bacillus subtilis SRP RNA is similar to that of E. coli (Struck et al., 1988), and therefore we suspect that the abundance of OLE RNA is roughly 0.1 to 0.4% of ribosomes in B. halodurans.
Fig. 2.
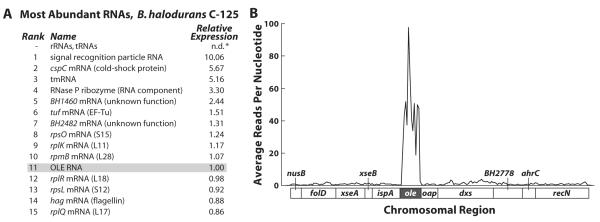
Transcriptome deep sequencing reveals that OLE RNA is highly expressed in B. halodurans. (A) Listed are the fifteen most common RNAs in B. halodurans C-125 other than ribosomal and transfer RNAs. Both rRNAs and tRNAs are likely more abundant than those RNAs listed but were eliminated from the RNA sample before cDNA generation. Relative abundance was determined by calculating RPKM scores (Reads Per Kilobase per Million reads (Mortazavi et al., 2008)) for each RNA before normalizing to OLE RNA. (B) The average reads per nucleotide using a 50-base-pair window are graphed for the genomic region (bottom of graph) corresponding to the OLE RNA-containing operon as defined previously (Puerta-Fernandez et al., 2006) (see Table S1 for raw data).
Interestingly, OLE RNA is roughly 100-fold more abundant than the transcripts for genes on its immediate flanks (Fig. 2B). Also, the abrupt boundaries in abundance of sequence reads near the beginning and the end of the conserved portion of OLE RNA suggests that independent transcripts of OLE RNA are produced, or that there is precise processing of longer precursor transcripts, or both. Previous 5′ RACE data revealed processing of OLE RNA at the 5′ base of the P1 stem (Puerta-Fernandez et al., 2006), perhaps suggesting that OLE RNAs are selectively stabilized compared to the surrounding transcript regions. However, the putative promoter located immediately upstream of the OLE RNA gene also appears functional and induces high levels of expression (unpublished observations), indicating that some of the disproportionately high amount of OLE RNA originates from its own promoter. Regardless of its cause, the abundance of this RNA during exponential growth suggests that it may serve its function under these conditions.
Conserved features of OLE-Associated Protein
We previously reported that a protein of unknown function is conserved only in species that carry OLE RNA (Puerta-Fernandez et al., 2006). Moreover, the gene for this protein is almost exclusively located immediately downstream of the OLE RNA gene even among the representatives on the expanded list reported in the current study. This protein has been renamed OLE-Associated Protein (OAP) due to its syntenic association with OLE RNA.
Inspection of the amino acid sequences of this expanded collection of OAP representatives (Fig. 3) revealed only short stretches of similarity to other known proteins. Interestingly, considerably less of the sequence of OAP is conserved compared to that of OLE RNA, suggesting that the protein may be subject to fewer evolutionary constraints than the RNA. Moreover, the different levels of conservation of the protein and RNA indicate that the two molecules do not coexist simply due to recent horizontal transfers of this block of genes. Rather, most pairs of OLE RNA and OAP loci appear to have persisted as co-localized genes for long periods despite much evolutionary change to their RNA or amino acid sequences. It is common for the products of bacterial genes that are co-localized to be functionally linked as partners in metabolic pathways or in the formation of complexes. Clustering of related protein coding genes in bacteria can even be exploited to predict functions for new genes (Galperin and Koonin, 2000, Overbeek et al., 1999). The genomic organization of functionally linked molecules proximal to each other also occurs with RNA-protein complexes, as is observed for smpB and tmRNA (Karzai et al., 1999) as well as the CRISPR/cas protein system (Karginov and Hannon, 2010). Therefore, the location of OAP next to OLE in all but two examples strongly suggests the two gene products are functionally linked.
Fig. 3.
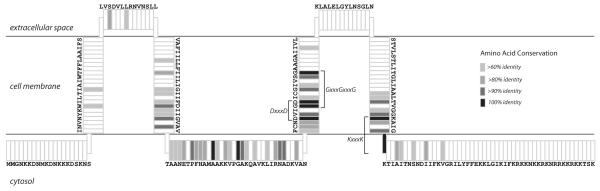
Consensus and topology of OLE-Associated Proteins. The consensus sequence and predicted topology of OAP are shown with transmembrane domains depicted as vertical segments and extracellular or cytosolic domains shown as horizontal segments. Orientation with respect to the cytosol and extracellular space is indicated. Shading depicts conservation of primary sequence, with black positions representing 100% amino acid conservation across all known examples.
An algorithm to predict membrane-spanning domains (Krogh et al., 2001) was used to reveal four putative conserved transmembrane domains (Fig. 3). A fifth transmembrane domain is predicted only for some representatives, which is partly responsible for the lack of extensive conservation of the C-terminus. Consistent with the prediction of transmembrane helices, initial efforts to isolate a recombinant OAP in E. coli revealed that the protein exists in the membrane fraction (Puerta-Fernandez et al., 2006). Numerous conserved amino acids are concentrated in the second, third, and fourth putative transmembrane domains, with additional conserved residues in the proposed central cytosolic domain. Notably, within the third predicted transmembrane domain reside three highly conserved glycine residues that form consecutive GxxxG motifs (i.e. GxxxGxxxG). This motif has been predicted to participate in oligomerization of transmembrane helices (Senes et al., 2000). A DxxxD motif at the cytosolic base of putative transmembrane domain 3 is strictly conserved (Fig. 3). The DxxxD motif has been shown to coordinate Mg2+ in the recognition and cleavage of pyrophosphate moieties in prenyldiphosphate-converting enzymes (Brandt et al., 2009). Although we cannot conclude that OAP similarly functions in modification of isoprenoids due to the presence of this motif, a role for OAP in isoprenoid biology is plausible given the association of the OAP locus with other isoprenoid biosynthetic genes (Puerta-Fernandez et al., 2006).
Likewise, a KxxxK motif is conserved at the cytosolic base of the predicted transmembrane domain 4. The KxxxK motif carries RNA-binding capacity in the eukaryotic LysRS (Francin and Mirande, 2003). Whether this motif, which may be buried in the membrane, can directly interact with RNA is unknown. Also, similarly spaced basic residues are found throughout the proposed central cytosolic domain and could also participate in RNA binding. Finally, we observe a large number of basic amino acids distributed throughout the C-terminal domain that could contribute to RNA binding, although exact positioning of these amino acids is far less conserved than in other regions of OAP. Thus there are several possible RNA-binding regions within OAP.
OLE RNA forms an RNP with OAP
Because the genes encoding OLE and OAP are syntenic, and OAP has several motifs consistent with RNA-binding function, we investigated the possibility that OLE and OAP form an RNP complex. Recombinant B. halodurans OAP fused to an N-terminal hexahistidine tag was expressed in E. coli, a heterologous system that has successfully yielded other functionally active transmembrane spanning proteins from Gram-positive organisms (e.g. Baliarda et al., 2003, Kappes et al., 1996). The bulk of the protein was localized to membrane rather than cytoplasmic cell fractions, as previously observed (Puerta-Fernandez et al., 2006), and therefore membrane preparations were used to isolate OAP for subsequent use in RNA binding assays. Purification of OAP from membranes resulted in a single protein species of the correct size as determined by SDS-PAGE (Fig. S1). Electrophoretic mobility shift assays (EMSA) were performed first with a 5′ 32P-labeled B. halodurans OLE RNA containing nucleotides 1 through 637 (OLE1-637), which encompasses the entire conserved motif. With increasing concentrations of OAP, OLE1-637 shifts completely from unbound RNA to a less-mobile form we attribute to RNP formation (Fig. 4A). By contrast, heat-denatured OAP does not induce a shift in RNA mobility (Fig. S2). Most 5′ 32P-labeled RNA fragments (less than 5% of the total labeled RNA) resulting from partial degradation of OLE do not undergo mobility shift on addition of OAP (Fig. 4A). This suggests that specific sequences or structural motifs of OLE RNA necessary for RNP formation are lost or are malformed in these RNA fragments.
Fig. 4.
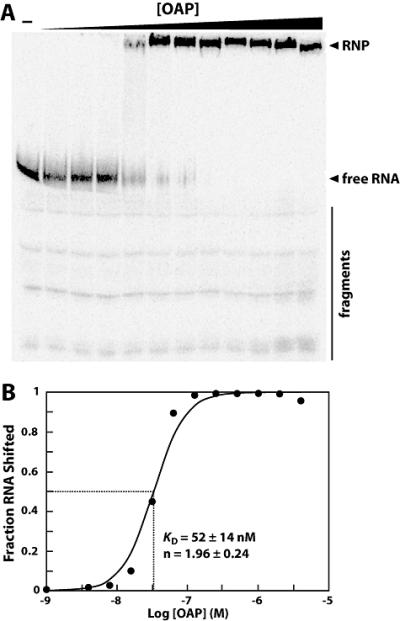
OLE and OAP form a ribonucleoprotein complex. (A) 5′ 32P-labeled OLE1-637 RNA was subjected to electrophoretic mobility shift assays with increasing concentrations of N-terminal histidine-tagged OAP to assess ribonucleoprotein complex formation. Arrowheads denote free, full-length RNA and shifted RNA (RNP) as noted. A bar highlights residual 3′ degradation products (fragments). These degraded RNAs do not shift in mobility in response to OAP. (B) To determine the binding characteristics of OAP, the fraction OLE RNA shifted was plotted versus the logarithm of the concentration of OAP. The data depicted is a representative and plots data calculated from the gel shown in part A. The KD and Hill coefficient (n) were calculated from four separate experiments, and the average and standard error are listed on the graph.
To examine the binding characteristics of the OLE-OAP complex, the fraction of OLE RNA shifted was plotted relative to the concentration of OAP (Fig. 4B), and a dissociation constant (KD) of approximately 50 nM was estimated for the full-length RNA. The affinity of OAP for OLE RNA is comparable to a number of other RNA-protein interactions. For example, components of the 30S and 50S ribosomal subunits in E. coli bind 16S rRNA and 5S rRNA, respectively, with KD values ranging from 4 to 625 nM (Schwarzbauer and Craven, 1981, Spierer et al., 1978). However, unlike the binding curves derived from the interaction between OLE RNA and OAP, those plotted for nearly all ribosomal proteins follow what would be expected for a one-to-one interaction. By contrast, the binding of OLE RNA by OAP displays a far sharper transition from free RNA to RNP (Fig. 4). The binding curve displays a Hill coefficient of 1.96, indicating cooperative binding, with more than one molecule of OAP for each molecule of OLE RNA.
A series of truncations of the OLE molecule were made to determine what regions of the RNA were responsible for RNP formation. Preliminary truncation studies suggested that the central region of the molecule, from P7 to P11 (Fig. 5A) contains protein binding sites (data not shown). We subsequently sought to map the location of binding with greater resolution. From the various truncated RNAs, two regions appeared to be necessary for RNP formation. These regions are separated by approximately 140 nucleotides and encompass the P9 stem as well as the central P2, P3, P3.1, and P10 structures (Fig. 5B). For each construct, a KD and Hill coefficient were determined (see below). Most of the truncated RNAs vary only slightly from each other in KD, and they additionally show less than an order of magnitude decrease in affinity from wild type. However, altered Hill coefficient values close to 1 for some of the truncated constructs imply disruption of RNA-protein contacts.
Fig. 5.
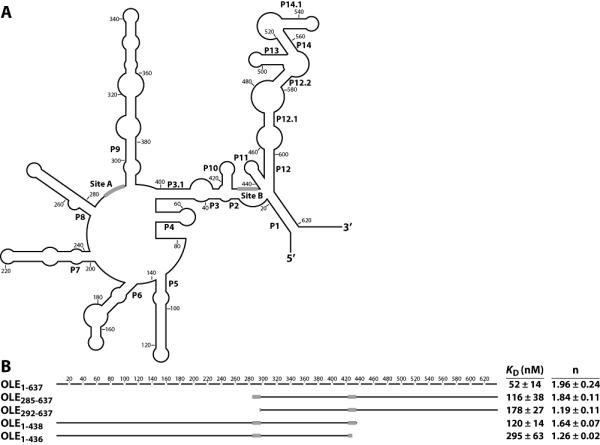
Identification of nucleotides necessary for formation of the OLE-OAP complex. (A) Secondary structure of the B. halodurans OLE RNA sequence is depicted in outline. Black bars indicate the predicted OAP binding sites as determined in B. (B) Full-length OLE RNA is depicted as a broken line, with each line segment corresponding to 20 nucleotides. Solid lines correspond to the indicated truncated constructs, with approximate location depicted graphically. The KD and Hill coefficient for each construct were calculated from at least four experiments and are given with the standard error. From these truncations, predicted OAP-binding sites were identified and depicted as black boxes on both the truncated RNAs and in part A. No binding (n.b.) was observed for the construct OLE285-438.
Removal of the first 284 nucleotides (OLE285-637) yields a construct with a nearly unperturbed Hill coefficient of 1.84. However, further removal of seven nucleotides in the construct OLE292-637 results in a Hill coefficient decrease to 1.19 (Fig. 5B), suggesting that the removed nucleotides include important protein contacts or help form an RNA structure necessary for normal binding. This region, termed Site A, consists of some of the most highly conserved nucleotides in the RNA, fitting a consensus of UAGCCUG. The high degree of conservation across this stretch of nucleotides suggested the possibility that this region is important for establishing contact with OAP. A second site (Site B) located between stems P10 and P11 has the sequence UAGRCUG, which is near identical to Site A. Removal of the last 199 nucleotides of OLE RNA to generate the construct OLE1-438 resulted in a Hill coefficient of 1.64. However, disrupting Site B by removal of just two more nucleotides (OLE1-436) further impairs the RNA-protein interaction, as indicated by a Hill coefficient of 1.26 (Fig. 5B).
Interestingly, an antisense OLE RNA (OLE AS, Table S2) displays similar binding characteristics to that of the full-length sense molecule. This could be explained if the OAP can bind the both the sense and antisense version of these sites, which conform to the consensus YAGNCUR. Other RNAs unrelated to OLE but that carry YAGNCUR sequence elements exhibit mixed results in gel mobility shift assays. For example, a large non-coding RNA of unknown function called HEARO (Weinberg et al., 2009), derived from the bacterium Arthrospira maxima, is completely shifted by OAP with a K of 59 nM, although with reduced cooperativity (Hill coefficient of 1.38). In contrast, RNase PD RNA from B. halodurans has two perfect matches for the YAGNCUR consensus, but only 25% of the RNA can be shifted by OAP with a KD of 216 nM and a reduced Hill coefficient of 1.2. Additionally, some truncated OLE RNAs that contain the YAGNCUR sequence do not interact at all with OAP (see below). Thus, while OAP binds some RNAs other than OLE, the protein does not bind RNAs indiscriminately.
To determine if specific nucleotides within the proposed binding sites are important for RNP formation, mutations were introduced into Site A of the construct OLE285-637. Every position was mutated individually to a U, or to an A if the wild-type position was U. Of the seven constructs tested, A287U, C289U, and G292U exhibit Hill coefficients of approximately 1 (Table S2), indicating that these changes perturb cooperative binding. By contrast, mutants U286A, G288U, and U291A are near wild-type in function, and the C290U mutant exhibits a slightly diminished Hill coefficient of 1.40. These data indicate that the nucleotide identities of only some positions in the YAGNCUR consensus are important for cooperative binding. However, most truncated RNA constructs tested yield only ~50% RNP formation (data not shown). To avoid complications due to misfolding, we examined select mutations in the context of the full-length RNA construct.
We also tested mutations to either Site A or Site B in the context of full-length OLE RNAs. Single mutations in these regions of the B. halodurans OLE RNA displayed only moderate effects on the KD or Hill coefficient values (Table S2). This suggests that while nucleotides in these regions may be important for establishing contact with OAP, there are likely other determinants that participate in RNP formation and can compensate for single nucleotide mutations. To account for the fact that the antisense molecule and unrelated RNAs can be bound by OAP to different extents, the sequences and structures necessary for RNP formation are likely to be relatively simple.
Several smaller OLE-derived constructs were examined to provide further evidence for selective binding by OAP. For example, OAP fails to bind OLE285-438 despite the fact that this construct carries Site A and Site B at its 5′ and 3′ termini, respectively (Table S2). Shorter construct carrying either Site A alone, or both Site A and Site B separated by nine nucleotides also fail to bind. The latter RNA gives two distinct bands after non-denaturing polyacrylamide gel electrophoresis, which is consistent with hairpin formation due to the palindromic nature of Site A and Site B. Therefore, OAP is unlikely to recognize a helical structure formed between these sites in the full RNA construct, unless perhaps additional recognition sites are present.
The full length OLE RNA was subjected to structural probing, via a Pb2+-mediated RNA cleavage assay, to assess RNA structure changes induced by OAP binding. In the presence of OAP, several regions become less susceptible to Pb2+-induced cleavage in the presence of the protein (Fig. 6). Importantly, the internucleotide linkages encompassing Site A, as well as a few linkages downstream, undergoes less cleavage in the presence of OAP. The linkages joining nucleotides 35-49, which form parts of helices P3 and P3.1, also undergo less cleavage. Other sites of reduced cleavage are located in the loop of P4, throughout P5, the bulge separating P6.1 and P6.2, and the distal part of P7. These reductions in strand scission are indicative of larger structural changes induced by protein binding.
Fig. 6.
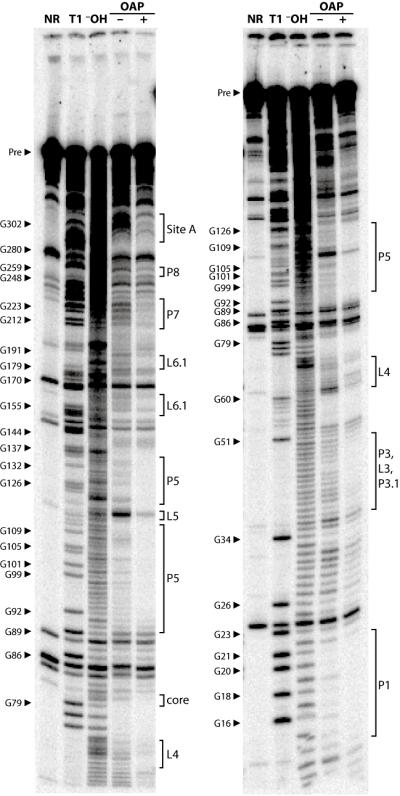
Probing of OLE RNA with OAP reveals structural modulation in response to protein binding. 5′ 32P-labeled OLE1-637 RNA was subjected to Pb2+ -catalyzed cleavage in the presence (+) or absence (□) of OAP to identify regions of OAP-mediated structural modulation. Other lane assignments are as follows: NR, no reaction; T1, partial digestion with RNase T1 under denaturing conditions; □OH, partial digestion with alkali. Select bands corresponding to RNase T1 digestion products (cleavage after G residues) are labeled. The locations of structural elements as defined in Fig. 1 are indicated. Bands in the no reaction lanes are fragments produced by spontaneous cleavage during the long elution times needed to recover precursor (Pre) RNAs from purification gels.
OAP localizes OLE RNA to membranes
Although OLE and OAP interact in vitro, we sought to determine if complex formation occurs in vivo. OAP exists as a membrane-associated protein, and hence RNP formation in vivo should localize the RNA to cell membranes. We used digoxigenin-labeled antisense OLE RNA and immunofluorescence microscopy to determine the location of OLE RNA expressed in E. coli either with or without co-expression of OAP. Membrane stain FM 4-64 FX was used to visualize the membranes.
Co-expression of OLE RNA and OAP results in a strong signal at the periphery of the cells (Fig. 7A). Merging of images recording RNA staining and membrane staining reveals an extensive overlap, suggesting that the RNA is localized near membranes when RNA and protein are co-expressed. By contrast, OLE RNA appears diffuse throughout the cell when expressed alone (Fig. 7B), suggesting that membrane localization of OLE is dependent on OAP and not simply a byproduct of overexpression. Levels of OLE and oap RNAs produced in these cells are estimated to be only one to two orders of magnitude less than the highly abundant 16s rRNA. Furthermore, a similar strain expressing OAP for protein preparations contained no less than 100 μM OAP. Thus, the concentration of OAP in the strain used for microscopy is likely to be above the KD measured for OLE-OAP complex formation in vitro.
Fig. 7.
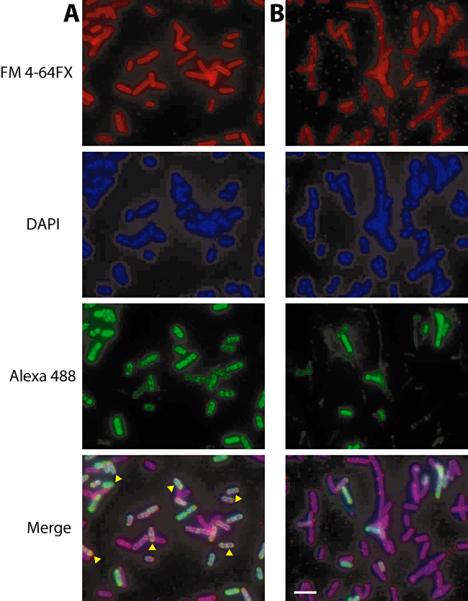
Subcellular localization of exogenously expressed OLE RNA when co-expressed with OAP (A) or expressed alone (B). E. coli cells expressing OLE with or without OAP were probed for cellular localization of OLE RNA using fluorescence in situ hybridization (RNA FISH). Fluorescence signal from the membrane stain (FM 4-64 FX, top), nucleic acids (DAPI, second), antisense RNA probe (Alexa 488, third) and merge (bottom) are shown. Yellow arrowheads indicate cells with overlap between RNA localization and the membrane stain. The scale bar in the lower right panel denotes 5 μm.
Examination of cell morphology in E. coli strains expressing OLE RNA, either alone or with OAP, did not reveal significant differences from wild-type cells (data not shown), indicating that OLE RNA does not alter gross morphology when overexpressed to this extent and under the growth conditions tested. However, because OLE RNA normally exists within a small subset of species whose environments are categorized by extreme characteristics, we cannot exclude the possibility that expression and localization of OLE RNA could alter morphology under these extreme environmental pressures.
We used similar methods to probe E. coli cells overexpressing the E. coli SRP RNA (4.5S RNA, Fig. S3) for comparison with localization of OLE RNA. As expected for this known membrane-associated RNA, our FISH data confirm cell membrane localization of SRP RNA. Importantly, the localization pattern of SRP RNA is similar to the pattern observed with OLE RNA when co-expressed with OAP. We additionally expressed in E. coli the B. halodurans copy of ispA, which is the gene immediately upstream of OLE RNA in its native context, and similarlyexamined mRNA localization. Similarly to the staining pattern of OLE RNA when overexpressed alone, ispA mRNA appears to lack membrane localization (Fig. S1), albeit with granular staining, indicating that the membrane localization is specific for the OLE-OAP complex.
Conclusions
OLE RNAs are widely distributed among a subset of anaerobic Firmicutes, many of which are extremophilic. OLE is more highly abundant in B. halodurans during exponential growth than over 99.8% of transcripts, including almost 100-fold excess over the oap mRNA. This difference in RNA levels for a noncoding RNA and protein that interact is similar to that observed for other noncoding RNAs and the transcripts of the protein partners with which they interact (Table S1). For example, both tmRNA and SRP RNA appear in E. coli total RNA in significant excess over the mRNAs of their corresponding protein partners (Lee et al., 1978), likely owing to the ability of a single mRNA transcript to generate many protein copies. Regardless, the over-representation of OLE RNA in our transcriptome sequencing analysis suggests that it is unlikely to be involved strictly in the regulation of nearby genes. Rather, given the nature of similarly abundant RNAs in B. halodurans, the function of OLE RNA may be catalytic, structural, or required for global regulation of gene expression. Regardless, we currently have two lines of support for membrane localization of OLE RNA. First, we know that OAP localizes to membranes because protein preparations reveal that the membrane fractions carry the bulk of OAP. This, coupled with the observation that OAP and OLE form tight complexes suggests that OLE may be co-localized. Second, imaging data in a surrogate organism is consistent with membrane localization of the RNA.
Our binding data indicate that the OLE-OAP complex consists of at least two molecules of OAP per molecule of OLE RNA. Analyses of truncated RNAs indicate that only some regions of OLE RNA are necessary for establishing contacts with OAP, and binding of both the antisense OLE RNA construct and HEARO RNA by OAP suggests that a simple and common structural motif serves as a recognition determinant. Our data do not rule out the possibility that an even larger complex exists, in which OAP, OLE, or both can serve as scaffolds to bring together other components to form a functional complex.
Most well-characterized RNA binding proteins use large domains for RNA sequence recognition (Perez-Canadillas and Varani, 2001). Given its small size and relatively simple topology, OAP may not recognize RNA sequence alone. The influenza viral matrix protein M1 binds RNA nonspecifically, and its simple binding site involves several basic amino acids aligned on one face of an α-helix (Wakefield and Brownlee, 1989), likely through electrostatic interactions (Rodriguez et al., 2004). Despite the similarity between the M1 binding domain and the proposed RNA binding domains in OAP, OAP does not bind RNA completely indiscriminately (Fig. 3 and Table S2). The RNA binding promiscuity of OAP may be overcome in part because of the abundance of OLE RNA in cells. Moreover, recent findings indicate that some bacterial RNA transcripts do not disperse freely, but rather are localized to the site of transcription (Llopis et al., 2010). Given the proximity of OLE and oap RNA production, the effective concentration of the partner molecules may be even higher when being synthesized.
Localization of mRNAs in eukaryotes has long been understood as important for cellular function (Holt and Bullock, 2009, Meignin and Davis, 2010). Similarly, recent work in bacteria suggests parallel implications of subcellular localization of mRNA in their function. Uneven distribution of the nifH mRNA was observed in Klebsiella oxytoca (Pilhofer et al., 2009), perhaps mirroring eukaryotes in targeting protein production to specific subcellular loci. Examination of localization of noncoding RNAs, both in bacteria and eukaryotes, could shed additional light on the importance of RNA localization for function. Recently, tmRNA was observed with specific distribution in bacteria, localizing to helical structures in Caulobacter crescentus (Russell and Keiler, 2009). How this localization of tmRNA may be important for its role in trans-translation remains to be determined. Likewise, our data regarding OLE RNA localization is intriguing because of the implied involvement in cell membrane function and the need to strengthen cell membranes in various extremophilic environments.
The association of RNAs with membranes is rare in nature and in molecular engineering (Khvorova et al., 1999, Vlassov et al., 2001). Some membrane-localizing RNAs likely have a passive role such as those in viral RNP particles that interact with the lipid bilayer (Wakefield and Brownlee, 1989, Sha and Luo, 1997). Others require membrane localization as part of their function, such as the small RNA SgrS from E. coli, which requires membrane association to downregulate translation of the ptsG mRNA encoding a glucose transporter (Kawamoto et al., 2005). An exceptionally common membrane-associated RNA is the SRP RNA, which functions as part of a larger RNP complex to target the mRNAs of secreted or membrane-bound proteins. Hence, some membrane-associated RNAs perform critical functions that require precise localization, which might suggest a similarly important role for OLE RNA at cell membranes.
Given the precedence for RNA localization at cell membranes and our data demonstrating membrane localization of OLE RNA, it is likely that additional RNAs exist at the periphery of cells where they serve regulatory or other unknown functions. Conservation of the DxxxD motif in OAP might indicate a function for the RNP complex in lipid biochemistry. This function would also be consistent with the near-universal placement of the genes for OLE and OAP between isoprenoid biosynthetic genes. While the biochemical functions for OLE RNA remain unresolved, there is precedence for large RNAs functioning as ribozymes (Weinberg et al., 2009). Regardless of its precise function, understanding the role of the OLE-OAP complex will likely shed light on one mechanism by which bacteria adapt to their anaerobic or extreme environments.
Experimental procedures
Bioinformatics
The previously published alignment of OLE RNA (Puerta-Fernandez et al., 2006) was updated using the Infernal 1.0 algorithm (Nawrocki et al., 2009) and version 38 of the RefSeq database (Pruitt et al., 2005). Additional examples of OLE RNA were identified by BLASTN and added to the alignment manually using RALEE (Griffiths-Jones, 2005). Conservation of nucleotide identity and covariation were determined as described previously (Weinberg et al., 2007).
BLASTP was used to identify examples of OAP. The sequences of unique representatives were aligned and compared using the CLUSTALW algorithm (Larkin et al., 2007). Consensus sequence and amino acid conservation was determined using Jalview (Waterhouse et al., 2009) and weighted to adjust for highly similar sequences resulting when multiple strains of the same species are included in the analysis. Topology was predicted using TMHMM (Krogh et al., 2001).
Transcriptome Sequencing and Analysis
B. halodurans C-125 cells were grown to exponential phase in LB (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl) adjusted to pH ~10.5 with 1% (w/v) Na2CO3, resuspended in TE (10 mM Tris-HCl [pH 7.5 at 23°C], 1 mM EDTA [pH 8.0 at 23°C]) with 3 mg/mL lysozyme, and subjected to three freeze-thaw cycles to lyse the cells. Total RNA was extracted with TRIzol reagent (Invitrogen), and approximately 20 μg RNA was treated with 5 U of RQ1 DNase (Promega) for 40 minutes at 37°C. RNA was precipitated by adding 0.1 volumes of 3 M NaOAc (pH 5.2 at 23°C) and 2.5 volumes of 100% ethanol at -20°C. Precipitate was recovered by centrifugation and resuspended in water. Following precipitation, a RiboMinus Transcriptome Isolation kit (Invitrogen) was used to remove rRNAs from 10 μg of total RNA. The remaining RNA was fragmented by incubation at 70°C for 5 minutes using Ambion RNA fragmentation buffer, and precipitated with ethanol as described above after adding 10 μg glycogen (Roche) to facilitate precipitation. qRT-PCR was performed as previously described (Block et al., 2010) using this fragmented RNA as template.
Synthesis and preparation of cDNA for sequencing was performed using a protocol similar to that described previously (Nagalakshmi et al., 2010). First-strand cDNA synthesis was performed using an Invitrogen Superscript II reverse transcriptase kit with 3 μg random hexamers (Invitrogen) in a total volume of 20 μL. Second-strand cDNA was generated by incubating the first-strand cDNA at 16°C for 2.5 h with 300 mM dNTP, 2 U RNase H (Invitrogen), and 50 U E. coli DNA polymerase I (NEB) in buffer containing 50 mM Tris-HCl (pH 7.8 at 23°C), 5 mM MgCl2 and 1 mM DTT The reaction was purified using a QIAquick PCR purification kit (Qiagen). Eluted DNA was end-repaired . in a reaction with 400 μM dNTP, 15 U T4 DNA polymerase, 5 U Klenow DNA polymerase, and 50 U T4 polynucleotide kinase (PNK) in T4 DNA ligase buffer (all reagents from NEB). Reactions were incubated at 20°C for 30 min and the products were purified using a QIAquick PCR purification kit. 5′-A overhangs were added by combining the eluted DNA in a reaction mixture with 200 mM dATP and 15 U Klenow Fragment (3′ to 5′ exo-) in the supplied buffer (NEB) at 37°C for 30 min, then purified with a MinElute PCR purification kit (Qiagen). Illumina sequencing adaptors were added with Quick Ligase (NEB) according to the manufacturer’s protocol. Reactions were purified on an agarose gel, amplified by PCR, and purified again as described previously (Nagalakshmi et al., 2010).
Shotgun sequencing and mapping of reads to the B. halodurans genome were conducted by the Yale Center for Genomics and Proteomics on an Illumina GAII sequencer, resulting in 3.99 million mappable reads of 34 bp. Relative abundance was computed by using in-house Perl scripts to convert mapped reads to Reads Per Kilobase per Million Reads (RPKM) scores (Mortazavi et al., 2008) for all genes in the B. halodurans genome. Gene boundaries were taken from Genbank (accession BA000004.3). Boundaries for unannotated genes such as OLE RNA or the RNase P RNA component were entered manually.
Purification of OAP
The gene encoding B. halodurans OAP (BH2780; accession BAB06499) was amplified from genomic DNA by PCR using primers OLE1 and OLE2 (for a complete list of primers, see Table S3) and ligated into the KpnI/BamHI sites of a pET-HT vector (gift of Dr. Lynne Regan and Dr. Aitziber L. Cortajarena), which fuses an N-terminal hexahistidine-tag to the protein of interest. The plasmid was transformed into E. coli BL21 cells (Novagen) immediately before each round of purification. A single colony was inoculated overnight at 37°C in 5 mL LB containing 50 μg/mL carbenicillin.
Overnight cultures were diluted 1:500 in 1 L LB containing 50 μg/mL carbenicillin and grown to an OD600 of 0.4-0.6, at which point 1 mM IPTG (Sigma-Aldrich) was added to induce expression of OAP. To avoid inclusion bodies, cells were grown at 28°C approximately 3-4 h until the OD600 approached 1. Cells were pelleted at 6500 rpm in a JLA 10.500 rotor (Beckman Coulter) for 6 min. Cells were washed once each in 50 mM K2HPO4 (pH 7.2), 5 mM MgSO4 and in 50 mM K2HPO4. Pellets were frozen overnight before proceeding with cell lysis.
Frozen pellets were thawed on ice and resuspended in a solution containing 50 mM K2HPO4 (pH 7.2 at 23°C), 5 mM MgSO4, 10 mM β-mercaptoethanol, and 30 μg/mL DNase (Roche) and subjected to lysis in a microfluidizer (Micrufluidics, model 110EH). Immediately after lysis, 1 mM PMSF (Sigma-Aldrich) was added, and the cell lysate was centrifuged at 12,000 rpm in a JA-30.50 Ti rotor (Beckman Coulter) for 20 min to collect cell debris. Cleared lysate was then centrifuged at 43,000 rpm in a Ti45 rotor (Beckman Coulter) for 3 h, and membranes were resuspended in 20 mM HEPES (pH 7.5 at 23°C), 200 mM NaCl. Membranes were snap-frozen in liquid nitrogen if long-term storage was necessary.
A Bradford assay was performed to determine the total amount of protein in the membrane preparation using the Quick Start Bovine Serum Albumin standards (Bio-Rad) and Bradford Reagent (Sigma-Aldrich). Membranes were then diluted to approximately 2-3 mg/mL total protein for extraction. NaCl was added to a final concentration of 500 mM along with 2% LysoFos Choline 14 (Anatrace), and membranes were stirred gently at 4°C for 1 h to solubilize all proteins. The extract was centrifuged for 30 min at 60,000 rpm in a Ti70.1 rotor (Beckman Coulter) to pellet insoluble material. 20 mM HEPES (pH 7.5 at 23°C) was added to the extract to bring the total NaCl concentration to 200 mM for column purification. The extract was applied to HIS-Select Nickel Affinity gel (Sigma-Aldrich) that had been washed in water and equilibrated in 20 mM HEPES (pH 7.5 at 23°C), 200 mM NaCl, 0.01% n-Dodecyl-β-D-maltopyranoside (DDM; Anatrace), 5 mM imidazole. Extract and beads were agitated at 4°C for 2 hours and applied to a gravity-flow column. Beads were washed twice with 20 mM HEPES (pH 7.5 at 23°C), 200 mM NaCl, 0.01% DDM, 10 mM imidazole, and eluted with 20 mM HEPES (pH 7.5 at 23°C), 0.01% DDM, 200 mM imidazole (pH 8.0 at 23°C). Fractions were collected and examined by SDS-PAGE to ensure retention of OAP on the beads until elution.
The elution fraction was applied to a Vivaspin 10 kDa MWCO spin filter to concentrate OAP. The concentration of OAP was determined by Bradford assay. 5% glycerol was added to the final stock, and 2 μL aliquots were stored at -80°C for later use.
Binding Assays
To generate the full-length OLE RNA template (OLE1-637), primers OLE3 and OLE4 were used to amplify B. halodurans genomic DNA with incorporation of a T7 promoter for subsequent in vitro transcription. The template was cloned into the TOPO-TA vector (Invitrogen), and the DNA sequence was confirmed (W.M. Keck Foundation Biotechnology Resource Laboratory, Yale University). All truncated constructs were generated by PCR amplification from this full-length template. Internal point mutations were made by a two-stage PCR process in which the first stage generated two portions of the construct with mutations incorporated at the sites of the internal primers. A second stage of PCR generated the full-length construct with the mutations present. Mutations were also confirmed by DNA sequencing.
RNAs were generated by in vitro transcription in reactions containing 80 mM HEPES (pH 7.5 at 23°C), 24 mM MgCl2, 2 mM spermidine, 40 mM DTT, 4 mM of each NTP, and 5 U/μL T7 RNA polymerase. Transcription products were separated by denaturing (8 M urea) 6% polyacrylamide gel electrophoresis (PAGE; National Diagnostics) and the desired products were eluted from the gel in a buffer containing 200 mM NaCl, 10 mM Tris-HCl (pH 7.5 at 23°C) and 1 mM EDTA (pH 8.0 at 23°C). Eluted RNAs were subsequently precipitated with ethanol as described above. To 5′-end label the RNA, 10 pmol RNA was used in a dephosphorylation reaction using the Roche rAPid alkaline phosphatase and a subsequent 5′ 32P-labeling reaction using T4 PNK (NEB). Radiolabeled RNA was purified by PAGE and isolated as described above.
Trace amounts of RNA (approximately 1 nM) were used for binding assays. Frozen aliquots of OAP were thawed on ice and diluted in 20 mM HEPES (pH 7.5 at 23°C), 0.01% DDM to bring the final imidazole concentration of the working stock protein to 5 mM. Subsequent serial dilutions (1:2) were made to the working stock to generate assay samples. Binding reactions were performed for 5 min at room temperature in a solution containing 10 mM HEPES (pH 7.5 at 23°C), 3 mM MgCl2, 40 mM KCl, 5% Glycerol, 1 mM DTT and 0.15 mg/mL tRNA. Longer incubation times did not improve binding and were detrimental to RNA stability. Reactions were terminated by the addition of stop buffer (final concentration 5% glycerol, 10 mM EDTA [pH 8.0 at 23°C], 0.1% SDS, 0.01% bromophenol blue, and 0.01% xylene cyanol) and placed on ice. Samples were loaded on a nondenaturing 6% polyacrylamide gel (19:1 acrylamide:bisacrylamide; Bio-Rad). Gels were run at 300 V for 4 h at 4°C in 90 mM Tris base, 90 mM Boric acid, and 5 mM Mg(OAc)2 (pH 8.0 at 23°C). After electrophoresis, gels were dried, visualized using a Phosphorimager (Storm 820. GE Healthcare), and quantified using ImageQuant software (GE Healthcare). For KD and Hill coefficient calculations, a Hill plot was generated, and a linear regression was fitted to the plot. All reactions were replicated in at least four assays.
RNA Probing
Trace amounts of 5′ 32P labeled OLE1-637 were prepared and incubated with 500 nM OAP or buffer only under the binding assay conditions described above. After completion of the binding reaction, 1 mM Pb(OAc)2 was added to the reaction and incubated for 2 min at 23°C. To stop cleavage, 6 M urea and 10 mM EDTA (pH 8.0 at 23°C) was added, and the reactions were subjected to phenol-chloroform extraction to remove protein, followed by precipitation with ethanol. Reaction products were separated on a denaturing 10% polyacrylamide gel at 4°C in 90 mM Tris base, 90 mM boric acid, and 1 mM EDTA (pH 8.0 at 23°C). After electrophoresis, gels were dried and visualized on a Phosphoimager.
Fluorescence In Situ Hybridization
Digoxigenin (DIG)-labeled RNA probes were transcribed using DNA templates amplified from B. halodurans ATCC strain 21591 (OLE RNA, ispA) or E. coli BL21 (4.5S RNA) genomic DNA. Transcriptions incorporated DIG-UTP (Roche) at a ratio of 3.5:6.5 with unlabeled UTP and were purified on a microspin G-25 column (GE Healthcare). Of the 50 μL transcription reaction, 5 μL was used for subsequent RNA FISH.
E. coli BL21 cells containing the appropriate pET-11a vectors were grown to an OD600 0.5-0.7 in LB containing 1 mM IPTG. 2 mL cells were stained using 5 μg/mL freshly made FM 4-64 FX (Invitrogen) for 5 min and were fixed in 7.5% formaldehyde, 90 mM Na2HPO4 (pH 7.5 at 23°C) for 1 h at room temperature. Cells were pelleted, washed three times with 1 mL phosphate buffered saline (PBS; American Bioanalytical), and resuspended in 100 μL GTE (50 mM glucose, 20 mM Tris-HCl [pH 7.5 at 23°C], 10 mM EDTA) with 2 mM vanadyl-ribonucleoside complex (VRC; Sigma-Aldrich). Cells were partially lysed by adding 4 μL 20 μg/mL lysozyme in GTE to 12 μL and incubating at room temperature for 20 min. 12 μL partially lysed cell mixtures were spotted to slides pretreated with poly-L-lysine (Sigma-Aldrich) and incubated for 10 min. Slides were allowed to air-dry and further fixed in -20°C methanol for 10 min and -20°C acetone for 30 sec.
For detection, dried slides were rehydrated for 60 min in a solution containing 2XSSC (1XSSC: 300 mM NaCl, 30 mM sodium citrate [pH 7.0 at 23°C]) and 50% formamide. Cells were hybridized overnight at 42°C in 40 μL hybridization buffer containing 10% dextran sulfate, 2 mM VRC, 0.02% acetylated BSA (Sigma-Aldrich), 40 μg tRNA (Roche), 50% formamide and 5 μL DIG-labeled antisense probe in 2X SSC. Slides were washed twice for 30 min in pre-warmed 0.1XSSC with 50% formamide at 50°C and twice for 10 min in PBS at room temperature. Slides were blocked in 2XSSC, 8% formamide, 2 mM VRC, and 2% acetylated BSA for 2 h at 37°C. Slides were incubated with mouse anti-DIG (1:100, Jackson Labs), washed in a solution containing 2XSSC and 8% formamide, and stained with 1:200 Alexa488-conjugated donkey anti-mouse IgG (Molecular Probes) for 1 h. Slides were mounted in SlowFade Gold antifade reagent media (Molecular Probes) containing 1 μg/mL DAPI. Slides were examined on a Nikon E1000 microscope fitted with a Hamamatsu Orca-ER LCD camera. Images were taken with Metamorph software and analyzed using ImageJ.
Supplementary Material
Acknowledgements
The authors thank Dr. José Luis Vázquez-Ibar for providing protocols for membrane isolation and protein purification and Dr. Vincenz Unger and Krasimir Spasov for advice and technical assistance in protein purification. We also thank Drs. Christine Jacobs-Wagner and Audrey Jackson for FISH protocols, Dr. Peter Angelastro for technical assistance with the microscope, and members of the Michael Snyder laboratory for Illumina sequencing protocols and reagents. Additionally, we thank the members of the Breaker laboratory for helpful discussions.
This work was supported by funding to R.R.B from the NIH (GM022778) and from the Howard Hughes Medical Institute. E.P.-F. was supported by a postdoctoral fellowship from the Spanish Ministerio de Educación y Ciencia.
Footnotes
Author contributions: All authors participated in experimental design and K.F.B., J.G.W., and E.P.-F. performed experiments. K.F.B., J.G.W., and R.R.B. wrote the manuscript.
Conflict of interest The authors declare that they have no conflict of interest.
References
- Baliarda A, Robert H, Jebbar M, Blanco C, Le Marrec C. Isolation and characterization of ButA, a secondary glycine betaine transport system operating in Tetragenococcus halophila. Curr Microbiol. 2003;47:347–351. doi: 10.1007/s00284-002-4017-x. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Block KF, Hammond MC, Breaker RR. Evidence for widespread gene control function by the ydaO riboswitch candidate. J Bacteriol. 2010;192:3983–3989. doi: 10.1128/JB.00450-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt W, Brauer L, Gunnewich N, Kufka J, Rausch F, Schulze D, et al. Molecular and structural basis of metabolic diversity mediated by prenyldiphosphate converting enzymes. Phytochemistry. 2009;70:1758–1775. doi: 10.1016/j.phytochem.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Corbino KA, Barrick JE, Lim J, Welz R, Tucker BJ, Puskarz I, et al. Evidence for a second class of S-adenosylmethionine riboswitches and other regulatory RNA motifs in alpha-proteobacteria. Genome Biol. 2005;6 doi: 10.1186/gb-2005-6-8-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francin M, Mirande M. Functional dissection of the eukaryotic-specific tRNA-interacting factor of lysyl-tRNA synthetase. J Biol Chem. 2003;278:1472–1479. doi: 10.1074/jbc.M208802200. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. Who’s your neighbor? New computational approaches for functional genomics. Nat Biotechnol. 2000;18:609–613. doi: 10.1038/76443. [DOI] [PubMed] [Google Scholar]
- Gottesman S. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet. 2005;21:399–404. doi: 10.1016/j.tig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S. RALEE--RNA ALignment editor in Emacs. Bioinformatics. 2005;21:257–259. doi: 10.1093/bioinformatics/bth489. [DOI] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Bullock SL. Subcellular mRNA Localization in Animal Cells and Why It Matters. Science. 2009;326:1212–1216. doi: 10.1126/science.1176488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LM, Zagorski J, Fournier MJ. Cloning and sequence-analysis of the Escherichia-coli 4.5 S RNA gene. J Mol Biol. 1984;178:509–531. doi: 10.1016/0022-2836(84)90236-5. [DOI] [PubMed] [Google Scholar]
- Jensen CG, Pedersen S. Concentrations of 4.5S RNA and Ffh protein in Escherichia coli: the stability of Ffh protein is dependent on the concentration of 4.5S RNA. J Bacteriol. 1994;176:7148–7154. doi: 10.1128/jb.176.23.7148-7154.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappes RM, Kempf B, Bremer E. Three transport systems for the osmoprotectant glycine betaine operate in Bacilluss subtilis: Characterization of OpuD. J Bacteriol. 1996;178:5071–5079. doi: 10.1128/jb.178.17.5071-5079.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol Cell. 2010;37:7–19. doi: 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karzai AW, Susskind MM, Sauer RT. SmpB, a unique RNA-binding protein essential for the peptide-tagging activity of SsrA (tmRNA) EMBO J. 1999;18:3793–3799. doi: 10.1093/emboj/18.13.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto H, Morita T, Shimizu A, Inada T, Aiba H. Implication of membrane localization of target mRNA in the action of a small RNA: mechanism of post-transcriptional regulation of glucose transporter in Escherichia coli. Genes Dev. 2005;19:328–338. doi: 10.1101/gad.1270605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev AV, Pace NR. Bacterial RNase P: a new view of an ancient enzyme. Nat Rev Microbiol. 2006;4:729–740. doi: 10.1038/nrmicro1491. [DOI] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, Kwak YG, Tamkun M, Majerfeld I, Yarus M. RNAs that bind and change the permeability of phospholipid membranes. Proc Natl Acad Sci. 1999;96:10649–10654. doi: 10.1073/pnas.96.19.10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lee SY, Bailey SC, Apirion D. Small stable RNAs from Escherichia coli: evidence for the existence of new molecules and for a new ribonucleoprotein particle containing 6S RNA. J Bacteriol. 1978;133:1015–1023. doi: 10.1128/jb.133.2.1015-1023.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopis PM, Jackson AF, Sliusarenko O, Surovtsev I, Heinritz J, Emonet T, Jacobs-Wagner C. Spatial organization of the flow of genetic information in bacteria. Nature. 2010;466:77–U90. doi: 10.1038/nature09152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meignin C, Davis I. Transmitting the message: intracellular mRNA localization. Curr Opin Cell Biol. 2010;22:112–119. doi: 10.1016/j.ceb.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Waern K, Snyder M. RNA-Seq: a method for comprehensive transcriptome analysis. Curr Protoc Mol Biol. 2010;89:4.11.11–14.11.13. doi: 10.1002/0471142727.mb0411s89. [DOI] [PubMed] [Google Scholar]
- Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D’Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Canadillas JM, Varani G. Recent advances in RNA-protein recognition. Curr Opin Struct Biol. 2001;11:53–58. doi: 10.1016/s0959-440x(00)00164-0. [DOI] [PubMed] [Google Scholar]
- Pilhofer M, Pavlekovic M, Lee NM, Ludwig W, Schleifer K-H. Fluorescence in situ hybridization for intracellular localization of nifH mRNA. Syst Appl Microbiol. 2009;32:186–192. doi: 10.1016/j.syapm.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puerta-Fernandez E, Barrick JE, Roth A, Breaker RR. Identification of a large noncoding RNA in extremophilic eubacteria. Proc Natl Acad Sci. 2006;103:19490–19495. doi: 10.1073/pnas.0607493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle AM. The tertiary structure of group II introns: implications for biological function and evolution. Crit Rev Biochem Mol Biol. 2010;45:215–232. doi: 10.3109/10409231003796523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez L, Cuesta I, Asenjo A, Villanueva N. Human respiratory syncytial virus matrix protein is an RNA-binding protein: binding properties, location and identity of the RNA contact residues. J Gen Virol. 2004;85:709–719. doi: 10.1099/vir.0.19707-0. [DOI] [PubMed] [Google Scholar]
- Russell JH, Keiler KC. Subcellular localization of a bacterial regulatory RNA. Proc Natl Acad Sci. 2009;106:16405–16409. doi: 10.1073/pnas.0904904106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbauer J, Craven GR. Apparent association constants for Escherichia-coli ribosomal-proteins S4, S7, S8, S15, S17 and S20 binding to 16S RNA. Nucleic Acids Res. 1981;9:2223–2237. doi: 10.1093/nar/9.9.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- Sha BD, Luo M. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nat Struct Biol. 1997;4:239–244. doi: 10.1038/nsb0397-239. [DOI] [PubMed] [Google Scholar]
- Spierer P, Bogdanov AA, Zimmermann RA. Parameters for the interaction of ribosomal proteins L5, L18, and L25 with 5S RNA from Escherichia coli. Biochemistry. 1978;17:5394–5398. doi: 10.1021/bi00618a012. [DOI] [PubMed] [Google Scholar]
- Struck JCR, Vogel DW, Ulbrich N, Erdmann VA. The Bacillus-subtilis scRNA is related to the 4.5S RNA from Escherichia-coli. Nucleic Acids Res. 1988;16:2719–2719. doi: 10.1093/nar/16.6.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassov A, Khvorova A, Yarus M. Binding and disruption of phospholipid bilayers by supramolecular RNA complexes. Proc Natl Acad Sci. 2001;98:7706–7711. doi: 10.1073/pnas.141041098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield L, Brownlee GG. RNA-binding properties of Influenza-A virus matrix protein M1. Nucleic Acids Res. 1989;17:8569–8580. doi: 10.1093/nar/17.21.8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Storz G. Regulatory RNAs in Bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, et al. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 2007;35:4809–4819. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Perreault J, Meyer MM, Breaker RR. Exceptional structured noncoding RNAs revealed by bacterial metagenome analysis. Nature. 2009;462:656–659. doi: 10.1038/nature08586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Haley B. Ribo-gnome: The Big World of Small RNAs. Science. 2005;309:1519–1524. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.