

Abstract
The skin and its appendages constitute the largest organ of the body. Its stratified epithelia offer protection from environmental stresses such as dehydration, irradiation, mechanical trauma, and pathogenic infection, whereas its appendages, like hair and sebaceous glands, help regulate body temperature as well as influence animal interaction and social behavior through camouflage and sexual signaling. To respond to and function effectively in a dynamic external environment, the skin and its appendages possess a remarkable ability to regenerate in a carefully controlled fashion. When this finely tuned homeostatic process is disrupted, skin diseases such as cancers may result. At present, the molecular signals that orchestrate cell proliferation, differentiation, and patterning in the skin remain incompletely understood. It is increasingly apparent that many morphogenetic pathways with key roles in development are also important in regulating skin biology. Of these, Wnt signaling has emerged as the dominant pathway controlling the patterning of skin and influencing the decisions of embryonic and adult stem cells to adopt the various cell lineages of the skin and its appendages, as well as subsequently controlling the function of differentiated skin cells. Here we will review established concepts and present recent advances in our understanding of the diverse roles that Wnt signaling plays in skin development, homeostasis, and disease.
Wnt signaling influences embryonic and adult stem cells to adopt the various cell lineages of the skin and its appendages (e.g., hair). It also controls hair cycling and may manage skin and hair pigmentation.
WNT IN THE DEVELOPMENT OF SKIN AND ITS APPENDAGES (FIG. 1)
Figure 1.

Anatomy of mammalian skin. The skin is a complex composite of multiple miniorgans. The topmost layer is a stratified epidermis consisting of keratinocytes that differentiate as they progress from the basal layer to the suprabasal and eventually form dead enucleated squames that are sloughed off from the cornified layer. Multiple different appendages branch from the epidermal layer, including hair follicles (HFs), sebaceous glands, and sweat glands. The dermis, which lies below the epidermis and surrounds the HFs, is composed of fibroblasts of mesenchymal origin that reside in a fibrous matrix that they produce. Melanocytes are interspersed among the keratinocytes of the epidermis and HF (bulge and matrix), where they pigment the skin and hair. Below all these is a subcutaneous layer of adipocytes that serve to insulate the animal.
Wnt signaling is involved at the very earliest stages of skin development (Fuchs 2007). Following gastrulation, embryonic cells of the ectoderm and the mesoderm will differentiate to form the epidermis and dermis, respectively.
The ectoderm layer that emerges at the surface of the embryo is able to specify either the nervous system or the skin epithelium. The decision of which fate to adopt is strongly influenced by Wnt signaling, which inhibits the ectoderm’s responsiveness to fibroblast growth factors (FGFs). Without FGF signaling, the ectodermal cells are able to express bone morphogenetic proteins (BMPs), which block neural induction and direct the cells to differentiate into keratin-expressing cells (keratinocytes) that form the epidermis (Wilson and Hemmati-Brivanlou 1995; Wilson et al. 2001).
The mesoderm, on the other hand, segments into somites, which eventually give rise to the dermis. Wnt signals are important for somitogenesis, and also instruct the lateral plate mesoderm and somite-derived cells to form the mesenchymal cells populating the dermis (Atit et al. 2006; Ohtola et al. 2008).
When the embryonic epidermis and dermis are both in place, complex signaling begins between them that leads to fate changes in both tissue layers, ultimately resulting in epidermal patterning and the development of skin appendages like hair follicles (HFs) or feathers (Hardy 1992). Hair morphogenesis begins with an initial signal from the dermis (“first dermal message”) that initiates the aggregation of the epidermis at regularly spaced intervals, leading to the formation of epidermal placodes that eventually develop into hair. These placodes then emit an epidermal signal that instructs underlying mesenchymal cells to cluster and form “dermal condensates.” A “second dermal message” from the dermal condensates induces epidermal placode cells to rapidly divide downward and invade the dermis, enwrapping the dermal condensate, which becomes the HF dermal papilla. Further cross talk between epithelial cells of the developing hair leads to their proliferation and differentiation to form the inner root sheath and hair shaft of the mature HF.
Although the precise nature of the epidermal placode-inducing “first dermal message” remains poorly understood, several studies suggest that Wnts may constitute both the “first dermal message” and the upstream signal stimulating its expression (Fig. 2A,B). This is evidenced by the fact that Wnt/β-catenin signaling is first activated in the dermis and subsequently in the epidermis. In developing chick feathers, nuclear localization of β-catenin is observed transiently in the dense dermis below the feather tract 2 days before placode development (Noramly et al. 1999). Formation of this feather-inducing dermis is dependent on a signal emanating from the dorsal neural tube, likely to be Wnt1 (Olivera-Martinez et al. 2001). In mice, the LEF/TCF transcription factor family member LEF-1 is detected in the mesenchyme of the mouse whisker (vibrissa) pad before discernible whisker development and is required for subsequent vibrissa but not dorsal back skin (pelage) follicle formation (van Genderen et al. 1994; Kratochwil et al. 1996). Corroborating this observation of Wnt/β-catenin pathway activation first occurring in the dermis, in Axin2-lacZ transgenic Wnt-reporter mice (where a bacterial β-galactosidase [lacZ] reporter gene was knocked into the conductin/Axin2 locus), lacZ reporter activity is present in the dermis at E12.5, before placode development (Zhang et al. 2009). In both mouse and chick, Wnt/β-catenin signaling activity appears uniformly in the dermis (Noramly et al. 1999; Zhang et al. 2009), suggesting that the “first dermal message” is uniform rather than being localized in a periodic fashion (Millar 2002).
Figure 2.

Wnt signaling during skin development and hair follicle morphogenesis. Hair follicle (HF) morphogenesis proceeds in three phases: induction (A–C), organogenesis (D,E), and cytodifferentiation (F), which can be further separated into eight stages (for a detailed review of these stages see Paus et al. 1999 and Schneider et al. 2009). The morphology and molecular events occurring at each stage are depicted. Data for developmental timings are from Blanpain and Fuchs (2006) and Zhang et al. (2009). Expression data are from DasGupta and Fuchs (1999), Reddy et al. (2001, 2004), and Zhang et al. (2009). Induction: (A) Stage 0 (E12.5): Before any visible hair placode formation, dermal fibroblasts appear to uniformly receive an unidentified Wnt signal from an unknown source, resulting in nuclear localization of β-catenin. (B) Stage 0 (E13.5): Dermal fibroblasts then produce the first dermal signal, which is likely also a Wnt. (C) Stage 1 (E14.5): On receiving the first dermal signal, Wnt/β-catenin signaling becomes active in the epidermis and is especially elevated at placode-forming regions. Reaction–diffusion reactions between slower-diffusing activating Wnts and faster-diffusing inhibitory Dkks produce a regular array of these high-Wnt placode-forming regions that eventually result in patterned hair growth (see also Box 1). The nascent epidermal placode cells proliferate and accumulate and also start producing Wnt ligands like Wnt10b. Organogenesis: (D) Stage 2 (E15.5): As the placode develops into a visible hair germ, the Wnt ligands it produces induce underlying fibroblasts to form a dermal condensate that will eventually develop into a follicular dermal papilla. This process is also dependent on Shh pathway activity in the dermal condensates. (E) Stages 3–5 (E18.5): As the hair germ grows into a bulbous hair peg, differentiating epithelial layers become apparent. Wnt/β-catenin is evident in these layers. Cytodifferentiation: (F) Stages 6–8 (bulbous peg) (early postnatal): The morphology and biochemistry of the growing HF resembles that of a postnatal anagen follicle. Wnt/β-catenin signaling also seems to be involved in determining the specific follicular lineages that progenitor keratinocytes adopt, with strong and complex expression of Wnt ligands, receptors, and pathway reporters in the various follicular layers.
Various genetic studies further underscore the importance of Wnt/β-catenin signaling in the early dermis for subsequent placode initiation (Table 1). Ectopic epidermal expression of Dkk1, the diffusible inhibitor of Wnt/β-catenin signaling, results in complete loss of β-catenin and LEF-1 expression in the dermis and abrogation of subsequent placode formation (Andl et al. 2002). Similarly, Porcupine (Porcn) heterozygous-null embryonic mouse skins display a general loss of LEF-1-positive placodes, suggesting that fully functional Wnt ligand production and secretion is essential for placode formation (Barrott et al. 2011). Interestingly, a subset of these PorcnΔ/+ mice also displays large areas of dermal atrophy, such that the internal organs are visible through an epidermal monolayer.
Table 1.
Summary of mouse models for the functional study of Wnt/β-catenin signaling in the skin
| Hair phenotype | ||||||||
|---|---|---|---|---|---|---|---|---|
| Mutation | References | Description | Induction | Examination | Morphogenesis | Postnatal hair cycle | IFE phenotype | Notes |
| K14-ΔN87β-catenin | Gat et al. 1998 | Constitutively stable β-catenin (human K14 promoter; transgene) | E9.5 onward | P24, P90–120 | No defects described during embryogenesis | Misangled follicles | Thickening; cell morphology changes | K14 activity by E9.5; strong K14 transgene activity by E14.5 |
| ΔNB-catenin | Zhu and Watt 1999 | Constitutively stable β-catenin (in vitro) | Constitutive | NA | NA | NA | Increased proportion of stem cells | Stem cell population shows higher endogenous cytoplasmic β-catenin and TOPFLASH activity |
| K14-ΔNΔCβ-catenin | DasGupta et al. 2002 | Dominant-negative constitutively stable β-catenin without transcriptional effector domain | E9.5 onward | P21–28 | No defects described during embryogenesis | Formation of epidermal and sebaceous cysts; loss of TOPGAL reporter expression in bulge | Thickening; also, TOPGAL+ IFE resembling de novo hair germs (similar to Gat et al. 1998; Van Mater et al. 2003) | |
| K5-S33Y-β-catenin-ER | Van Mater et al. 2003 | Tamoxifen-inducible constitutively active β-catenin (bovine K5 promoter; transgene) | Starting from P50, either single 4OHT dose or daily administration for 1, 3, 7, 14 d | P51–64 | NA | Early induction of anagen; hyperplastic anagen HFs, hyperkeratosis | Little to no effect | 4OHT has no significant effect on progression to anagen in wild-type mice (Chanda et al. 2000) |
| K14-ΔNβ-catenin-ER | Lo Celso et al. 2004 | Tamoxifen-inducible constitutively active β-catenin (transgene) | Starting from P42, tamoxifen daily up to 14 d | P51–64 | NA | Early induction of anagen; abnormal HFs that progress into trichofolliculomas | Hyperproliferative epidermis (pelage and dorsal paw); ectopic hair follicle rudiments in otherwise hairless ventral paw skin | |
| K14-ΔNβ-catenin-ER | Silva-Vargas et al. 2005 | Tamoxifen-inducible constitutively active β-catenin (transgene) | Starting from P42–56, tamoxifen daily up to 21 d | P63–77 | NA | With increasing induction of ΔNβ-catenin, altered timing and location of ectopic HF formation | None reported | |
| K14-ΔN87β-catenin (hemizygous) | Lowry et al. 2005 | Constitutively stable β-catenin (human K14 promoter; transgene) | E9.5 onward | P28, P49, P55 | No defects described during embryogenesis | Increased proliferation during second telogen, and subsequent early anagen entry (by P55) | None reported | |
| K14-Cre(neo)/K14-Cre(ΔNeo);β-cateninΔex3-6 | Huelsken et al. 2001 | Inducible β-catenin loss-of-function; Cre-recombinase knocked into endogenous mouse K14 locus | K14-Cre(neo): strong expression by E15; K14-Cre(neo and Δneo): strong expression after birth | P8, P16, P30 | No defects described during embryogenesis | Shaggy fur and fewer zigzag HFs until P16, hair loss to baldness from P17 onward; bulge cells lose stem cell markers | Epidermal cysts expressing some bulge stem cell markers; increased proliferation in basal epidermal cells | |
| K14-Cre(neo);β-cateninΔex3-6 | Teulière et al. 2004 | Inducible β-catenin loss-of-function; Cre-recombinase knocked into endogenous mouse K14 locus | E15 onward | P30 | No defects described during embryogenesis | Presumably similar to that described above in Huelsken et al. (2001) | Thickening; proliferating cells in basal and suprabasal layers; differentiation defects | |
| K14-CreER;B-cateninΔex2-6 | Lowry et al. 2005 | Inducible β-catenin loss of function (human K14 promoter; transgene) | BrdU at P28 for 72 h; tamoxifen injection at P44 | P57 | NA | Loss of bulge label- retaining cells and CD34 expression, possibly converted to IFE fate | None reported | |
| LEF1−/− | van Genderen et al. 1994 | LEF-1 knockout | Constitutive | P16 | Follicle forms normally but in reduced numbers | At P3–9: follicles are short and rudimentary (poorly differentiated); at P16, lack of whiskers and bodily HFs | None reported; lacks dermal fat | Melanocytes present, although pigmentation lost |
| K14-LEF1 | Zhou et al. 1995 | LEF-1 overexpression (human K14 promoter fused to human LEF-1 cDNA; transgene) | E9.5 onward | P28 | Refer to postnatal hair section | Irregularly clustered, misoriented, and rudimentary follicles | None reported | Ectopic hair formation in oral epithelium |
| K14-ΔNLef1 | Merrill et al. 2001 | LEF-1 unable to interact with β-catenin (transgene) | E9.5 onward | 9 mo | No defects described during embryogenesis | Progressively sparse hair coat; hair germ cells adopt sebocyte fate and form sebaceous cysts | None reported | |
| K14-ΔNLef1 | Niemann et al. 2002 | Lef1 unable to interact with β-catenin (different deletion from Merrill et al. 2001; transgene) | E9.5 onward | P42–10 mo | No defects described during embryogenesis | Delayed entry into first postnatal hair cycle; progressive hair loss; hair loss complete only at 10 mo | Formation of epithelial cysts but no sebaceous differentiation (different from Merrill et al. 2001); light IFE thickening not confined to cyst regions; delayed but normal IFE wound healing; sebaceous adenoma/sebeomas and squamous papillomas/carcinomas observed at ∼3 mo | Transgene expression at 100× of endogenous LEF1; effects on hair only after first postnatal cycle, possibly because transgene not strongly expressed during development; LEF1 is not expressed in postnatal IFE |
| K14-Tcf3 | Merrill et al. 2001 | Overexpression of wild-type mouse Tcf3 (transgene) | E9.5 onward | P0 | Epidermal differentiation defect and impaired barrier function | No hair phenotype, although increased nuclear β-catenin in ORS and bulge | Late-stage epidermal differentiation defect; impaired barrier function | |
| K14-Cre;Tcf3-fl/fl | Nguyen et al. 2009 | Conditional Tcf3 knockout (K14-Cre transgene) | E9.5 onward | P0, adult | No defects | No defects | No defects | |
| K14-myc-ΔNTcf4 | Nguyen et al. 2009 | Overexpression of Tcf4 lacking β-catenin-interacting domain (transgene) | E9.5 onward | P0 | Epidermal differentiation defect and impaired barrier function | None reported | Epidermal differentiation defects similar to those stemming from K14-Tcf3 overexpression (Merrill et al. 2001) | |
| K14-Cre;Tcf3-fl/fl;Tcf4−/− | Nguyen et al. 2009 | Conditional Tcf3 and Tcf4 knockout (K14-Cre transgene) | E9.5 onward | P0 and grafted skin | Refer to postnatal hair and IFE sections | Lack whiskers at birth; HFs initiated, but defective downgrowth; Tcf3/4-null skin grafts are hairless | Thinner skin and flattened basal cells; grafted Tcf3/4-null skin shrank in overall area, although epidermis is slightly thickened; loss of clonogenic skin stem cells | |
| K14-Dkk1 | Andl et al. 2002 | Dkk1 overexpression (transgene) | E9.5 onward | E14.5, E15.5, E18.5, P1, P7, P13, P17, P28, P33, P52 | No or few primary, secondary, and vibrissae follicles develop | Loss of ventral HFs and thin ventral dermis; sparse dorsal guard HFs; reduced dermal fat | No differentiation or proliferation defects | |
| K5rtTA;tet-Wnt1 | Castilho et al. 2009 | Inducible mouse Wnt1 overexpression (transgene) | Fed Dox pellets since birth | P7, P14, P17, P21, and >1 yr | No defects | Premature entry into anagen and sustained anagen with increased follicular density, subsequently progressing into hair loss; increased proliferation in K5+ basal ORS cells | Thickening at P90 | Wnt1 overexpression may induce HF stem cell to exit from quiescence, causing proliferation and subsequent exhaustion of HF stem cell population |
| Wnt5a−/− | Hu et al. 2010 | Wnt5a knockout originally described in Yamaguchi et al. (1999) | Constitutive | Grafted skin | No defects described during embryogenesis | Follicular abnormalities in skin grafts; dermal papilla cells induce extensive cyst formation instead of hair | None reported | Wnt5a−/− mice die at birth (Yamaguchi et al. 1999) |
| ROR2−/− | Hu et al. 2010 | ROR2-knockout originally described in Takeuchi et al. (2000) | Constitutive | Grafted skin | No defects described during embryogenesis | Altered biochemistry in grafted follicles | None reported | ROR2−/− mice die at birth (Takeuchi et al. 2000) |
| Sox2-Cre; Porcn−/+ (females) | Barrott et al. 2011 | Porcupine knockout, gene deleted during epiblast stage | Epiblast | E15.5–E18.5 | Refer to postnatal hair and IFE sections | Loss of HFs and Lef1 expression in nascent HFs | Epidermal monolayer, dermal atrophy but otherwise apparently normal keratinocyte differentiation; loss of Lef1 expression in basal epidermis | |
All K14- and K5-promoter-driven transgenes are overexpressed in the basal epidermal and follicular bulge and ORS cells.
IFE, interfollicular epidermis; ORS, outer root sheath; ER, estrogen receptor; HF, hair follicle.
However, even though all this evidence strongly suggests that the first dermal message and its upstream initiating signal are Wnt ligands, the precise source and identity of these Wnt ligands remains unknown.
In response to the first dermal signal (Fig. 2B), Wnt/β-catenin signaling is activated in the epidermis to promote placode fate (Fig. 2C). Both Wnt ligands and receptors are expressed in the E14.5 mouse epidermis, including Wnts 3, 4, 6, 7a, 7b, 10a, 10b, and 16 (Reddy et al. 2001) and Frizzleds 1, 6, and 10 (Reddy et al. 2004). By one day following the appearance of nuclear β-catenin in the dense chick dermis, nuclear β-catenin becomes apparent in the entire epidermis and is specifically up-regulated in regions that will form the placodes (Noramly et al. 1999), whereas in mice, nuclear β-catenin is observed in dermal condensates (Zhang et al. 2009). Further corroborating these observations, Wnt-reporter mouse lines like Axin2-lacZ and TOPGAL (containing the lacZ reporter gene under the control of lymphoid enhancer factor/T-cell factor (LEF/TCF) consensus binding motifs and a minimal c-fos promoter) (DasGupta and Fuchs 1999) also display up-regulated and restricted reporter activity within developing placodes (Zhang et al. 2009).
When stabilized β-catenin (Gat et al. 1998; Noramly et al. 1999) or a Wnt ligand (Wnt7a) (Widelitz et al. 1999) is overexpressed in mouse and chick epidermis, ectopic HFs and feather buds form, demonstrating that activation of the Wnt/β-catenin signaling pathway is sufficient to direct epidermal cells toward an appendage fate. In contrast, mouse epidermis carrying a loss-of-function mutation in β-catenin failed to develop placodes (Huelsken et al. 2001), and a LEF-1-null mutation in mouse epidermis resulted in arrested development and loss of vibrissae as well as two-thirds of the pelage follicles (van Genderen et al. 1994; Kratochwil et al. 1996), suggesting that Wnt/β-catenin is necessary for placode formation.
After the placodes have formed, epithelial placode cells signal to underlying mesenchymal cells to form the dermal condensate that ultimately develops into the dermal papilla. Again, Wnt/β-catenin signaling is important for these processes (Fig. 2D). Dermal condensates do not develop if epithelial β-catenin is not present (Huelsken et al. 2001), and the presence of Axin2-lacZ and TOPGAL reporter activity in the dermal condensates suggests that Wnt/β-catenin signaling is taking place there (DasGupta and Fuchs 1999; Zhang et al. 2009). Additionally, knocking out Sonic hedgehog (Shh) in embryos results in the loss of Wnt5a expression in dermal condensates and the inhibition of dermal papilla maturation (Reddy et al. 2001). From this, it may seem that Wnt5a plays a role in dermal papilla development. However, the deletion of Wnt5a in the skin appears to have no effects on HF morphogenesis (Hu et al. 2010).
Epidermal keratinocytes surrounding the placodes form stretches of hairless interfollicular epidermis (IFE) that evenly space hair follicles (HFs) apart into regular arrays (Fig. 2C, Box 1). This array structure suggests the presence of global spatial patterning mechanisms that either promote or inhibit HF fate. The periodicity of placode formation and subsequent formation of a regular, patterned array of HFs is believed to arise via a Turing model of reaction–diffusion involving competition between diffusible placode-promoting and placode-inhibiting morphogens (Jiang et al. 2004; Schmidt-Ullrich and Paus 2005). Such a reaction–diffusion system might result from differences in the rates of diffusion between placode promoters and inhibitors, setting up a “short-range activation, long-range inhibition” phenomenon that produces the diffusion-driven instability needed to produce Turing patterns (Maini et al. 2006). Both Wnt ligands such as Wnt10a and Wnt10b (Reddy et al. 2001) and inhibitors such as Dkk4 are expressed in the placodes and are secreted into the extracellular space, and can hence potentially participate in a reaction–diffusion system (Sick et al. 2006). Because it is smaller in molecular size, Dkk4 could potentially traverse a larger distance than the Wnt ligands and diffuse beyond the placode, reducing Wnt/β-catenin signaling in the IFE adjacent to the placodes and refining the placode boundaries (Zhang et al. 2009). Conversely, the larger and hydrophobic Wnt ligands do not move freely in an aqueous extracellular environment and hence remain within the placode and exert short-range effects to focally up-regulate Wnt/β-catenin signaling in the placode. These differences in the levels of Wnt/β-catenin signaling could thus result in the emergence of a periodic array of placodes (Box 1) (Sick et al. 2006). In further support of the notion that optimal levels of Wnt/β-catenin signaling exist that are required for proper placode patterning, constitutive expression of LEF1 or stabilized β-catenin in the basal epidermis causes irregular spacing and tight clustering of hair placodes and follicles (Zhou et al. 1995; Närhi et al. 2008).
BOX 1. FORMATION OF PLACODE ARRAY.
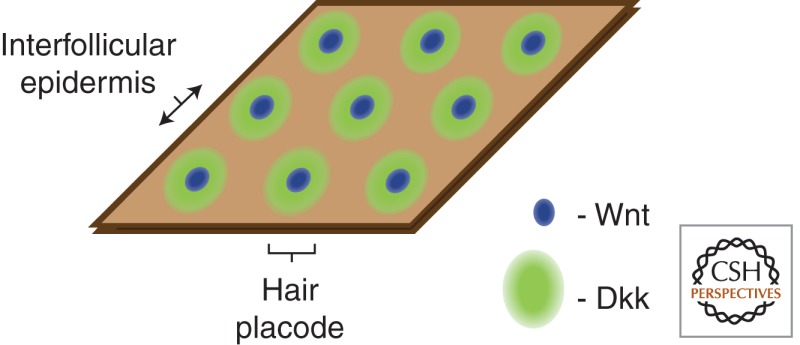
Reaction–diffusion reactions between a slowly diffusing Wnt and a faster diffusing Wnt inhibitor Dkk1 set up a regular array of nodes of elevated Wnt signaling that are the sites of placode formation.
Although the Turing model is elegant and produces remarkably accurate predictions of HF array patterns, it is based on the premise of freely diffusible and interacting chemicals. Because Wnt and Dkk are not freely diffusible in the extracellular milieu, and they do not directly interact but instead bind to cells, it is unclear if using reaction–diffusion models to describe their interaction is really representative of actual in vivo events. Additionally, it seems likely that mechanical forces may interact with these diffusible morphogens to influence spatial patterning, although this possibility remains relatively unexplored in the mammalian skin (Morelli et al. 2012).
WNT IN HF
In the postnatal skin, mature HFs undergo repeated cycles of growth (anagen), death/regression (catagen), and rest (telogen) in distinct morphological states (reviewed in Schneider et al. 2009). Similar to HF morphogenesis, the initiation of a new growth phase and the resulting proliferation, differentiation, and progressive downgrowth of the follicle is likely to involve extensive reciprocal interactions between dermal and epidermal cells (Oliver and Jahoda 1988; Schmidt-Ullrich and Paus 2005). Numerous molecular studies suggest that the same signaling pathways that are active during HF morphogenesis are reused in cycling postnatal hair. In particular, Wnt/β-catenin signaling is known to play a critical role at multiple stages of the hair cycle, from the earliest stages at the transition from rest to growth, to influencing lineage decisions during HF differentiation (Fig. 3).
Figure 3.

Wnt signaling during hair follicle cycling. Postnatal hair follicles (HFs) cycle repeatedly through rest (telogen), growth (anagen), and death/regression (catagen). The morphology and molecular events taking place at each stage of the hair cycle are depicted (for a comprehensive histological review of hair cycle stages see Müller-Röver et al. 2001). Expression data are from DasGupta and Fuchs (1999), Reddy et al. (2001, 2004), Blanpain et al. (2004), Morris et al. (2004), Tumbar et al. (2004), and Zhang et al. (2009). (A) During refractory telogen (BMP-high, Wnt-low) (Plikus et al. 2008), BMP is secreted from adipocytes and dermal fibroblasts, as well as the dermal papilla, to maintain the HF bulge in a low-growth, quiescent state. The bulge is thought to also be in a Wnt-inhibited state, by expressing various Wnt pathway inhibitors. (B) In the competent telogen phase (BMP-low, Wnt-high), BMP expression in adipocytes and dermal cells is reduced, allowing bulge cells to respond to anagen-inducing cues. One such cue is likely to involve an as-yet-unidentified Wnt. At this time, Wnt/β-catenin signaling becomes up-regulated in bulge and secondary hair germ cells, and they produce Wnt ligands such as Wnt10b. (C) As the follicle transitions from telogen to anagen and starts growing, bulge/secondary hair germ keratinocytes proliferate and migrate along the developing outer root sheath (ORS) to enwrap the dermal papilla and push it downward, away from the bulge. Wnt/β-catenin signaling continues to be active in these keratinocytes that form the developing matrix and the precortex. (D) The HF continues to grow downward in anagen. Distinct, differentiated layers (sheaths) are observed, each expressing different Wnt ligands and receptors. The significance of these complex expression patterns remains unknown. Lgr5+ follicle stem cells are found throughout the ORS and likely migrate to the matrix, where they differentiate into keratinocytes populating the various lineages of the anagen HF. Wnt/β-catenin signaling is also active in the matrix and is thought to be important for specifying these lineages. It is unclear if particular Wnts are responsible for specifying particular anagen lineages. (E) As the follicle exits anagen and enters catagen, it stops growing and starts dying. The follicle regresses and forms an epithelial strand that slowly pulls the dermal papilla upward to the level of the bulge, a process that is dependent on β-catenin (van Genderen et al. 1994).
During the telogen phase of hair cycle, the resting HF is ensheathed by layers of epithelial keratinocytes in a structure called the bulge (Fig. 3A). Numerous lineage tracing and functional genetic studies have elegantly shown that follicular stem cells are keratinocytes that reside in the outermost bulge layer, expressing cytokeratin-14 (K14) (Vasioukhin et al. 1999) and -15 (K15) (Morris et al. 2004), Shh (Levy et al. 2005), and the transcription factors Sox9 (Vidal et al. 2005; Sick et al. 2006) and Lhx2 (Rhee et al. 2006). DNA and histone label-retaining assays further suggest that many bulge cells cycle slowly (Cotsarelis et al. 1990; Tumbar et al. 2004), and remain largely quiescent during telogen, not taking up any BrdU label or expressing proliferation markers (Greco et al. 2009).
It is conventionally believed that little or no Wnt/β-catenin signaling takes place during most of telogen (Fig. 3A), and that Wnt/β-catenin signaling is up-regulated only at the end of telogen, promoting entry into anagen (Fig. 3B). Wnt ligand expression is not detected in telogen follicles, and Wnt10a and 10b only become expressed at anagen onset in the dermal papilla and secondary hair germ, respectively (Reddy et al. 2001). This is supported by the observation that β-catenin is generally confined to the membranes of bulge cells through most of telogen, and nuclear β-catenin only becomes apparent in hair germ cells just before the follicle enters anagen (Fig. 3B) (Lowry et al. 2005; Greco et al. 2009). The TOPGAL Wnt/β-catenin signaling reporter is generally inactive in telogen follicles (DasGupta and Fuchs 1999; Plikus et al. 2008), although TOPGAL-positive bulge cells are sometimes detected (DasGupta and Fuchs 1999; Merrill et al. 2001). In further support of the notion that the bulge is a Wnt-inhibited environment, microarray gene expression analyses show that prospectively isolated telogen follicular bulge cells and slow-cycling cells express secreted repressors of Wnt/β-catenin signaling such as Sfrp1, Wif1, and Dkk3 (Blanpain et al. 2004; Morris et al. 2004; Tumbar et al. 2004), as well as Tcf3, which is believed to repress Wnt/β-catenin signaling in the absence of nuclear β-catenin (Nguyen et al. 2006, 2009). Interestingly, isolated human bulge cells also express slightly elevated levels of Wif1 and Dkk3, supporting the findings in mice (Ohyama et al. 2006).
Loss- and gain-of-function mutations in β-catenin provide further genetic support for Wnt/β-catenin signaling as an inducer of anagen onset (Table 1). Expression of constitutively stable β-catenin in basal epidermal keratinocytes leads to hyperactive Wnt/β-catenin signaling and is sufficient to induce anagen onset (Van Mater et al. 2003) and ectopic HF formation (Gat et al. 1998; Lo Celso et al. 2004), whereas loss-of-functional β-catenin blocked entry into anagen when induced postnatally (Huelsken et al. 2001).
The decision to exit telogen and enter anagen appears to result from the antagonistic interplay between BMP, TGF-β, and Wnt/β-catenin signaling in follicular stem cells. Although Wnt/β-catenin signaling and TGF-β (Oshimori and Fuchs 2012) promote anagen onset, BMP signaling is thought to inhibit it. This growth inhibitory role of BMP is supported by studies in mice where BMP receptor 1a was conditionally ablated in basal keratinocytes, leading to continuous activation and aberrant proliferation of follicular stem cells and the eventual loss of slow-cycling cells from the bulge (Kobielak et al. 2003, 2007; Andl et al. 2004; Yuhki et al. 2004). BMPR1a loss in bulge cells also results in increased levels of Lef1 and stabilized β-catenin, providing evidence that BMP signaling may inhibit anagen onset by preventing the up-regulation of Wnt/β-catenin signaling in follicular stem cells. This mutual antagonism divides telogen into two phases: refractory, which is BMP-high and Wnt-low, and competent telogen, which is BMP-low and Wnt-high (Plikus et al. 2008). During the refractory phase, dermal cells such as fibroblasts, adipocytes, and dermal papillae express high levels of BMPs 2 and 4, inhibiting the ability of follicular epithelial stem cells to respond to growth stimuli (Plikus et al. 2008). In the competent phase, BMP expression in the dermal macroenvironment is reduced (Plikus et al. 2008) and the dermal papilla secretes noggin (Botchkarev et al. 2001; Zhang et al. 2006), turning BMP signaling off in bulge cells and allowing them to respond to anagen-inducing cues and up-regulate Wnt signaling.
Although most studies place the role of Wnt/β-catenin signaling in the activation of stem cells to transit from telogen to anagen, some evidence exists for the presence of active Wnt/β-catenin signaling throughout telogen. Both human and mouse bulge cells may be Wnt-responsive, expressing Frizzled receptors (Reddy et al. 2004; Tumbar et al. 2004; Ohyama et al. 2006) as well as Wnt-signaling-related transcription factors (Tumbar et al. 2004; Nguyen et al. 2006, 2009). They also express Wnt target genes such as the leucine-rich G-protein-coupled receptor 5 (Lgr5) (Jaks et al. 2008), initially identified as a β-catenin/TCF4 target gene in colon cancer (van de Wetering et al. 2002) and one of the most highly expressed genes in follicular bulge cells (Morris et al. 2004). If Wnt/β-catenin signaling is indeed present during the telogen phase, then the initiation of anagen could be the result of an increase in relative levels of Wnt/β-catenin signaling, rather than activation from a state of no signaling. The manner in which quantitative differences in Wnt signaling levels are interpreted to produce distinct cell fates remains an unanswered question of fundamental importance.
Despite the well-established role for Wnt/β-catenin signaling as an anagen inducer, the cellular source and precise Wnt ligand(s) involved remain unknown. As detected by in situ hybridization, some of the earliest Wnt ligands expressed during the telogen–anagen transition include Wnt5a and Wnt10a in the dermal papilla, and Wnt10b in the secondary hair germ (Reddy et al. 2001). Examination of Shh-knockout skin showed that Wnt10a and 10b expression were unaffected, whereas Wnt5a expression was lost, placing Wnt10a and 10b as upstream of Shh, which in turn regulates Wnt5a expression (Reddy et al. 2001). Although in vitro and skin reconstitution and grafting experiments suggest that Wnt10b is sufficient to promote hair development and growth (Ouji et al. 2006, 2008), Wnt10b-null mice are viable and show no reported skin phenotypes (Stevens et al. 2010). There are no known Wnt10a-null mice, although Wnt10a mutations are associated with human follicular hyperkeratosis and abnormal hair patterning (Adaimy et al. 2007; Bohring et al. 2009; Nawaz et al. 2009). Although Wnt5a is downstream from Wnts10a and 10b, its expression is necessary for normal progression of anagen, as ablation of Wnt5a in the dermal papilla results in impaired HF induction and the formation of epithelial cysts (Hu et al. 2010). Interestingly, continued Wnt/β-catenin signaling within the dermal papilla is also required for normal progression through anagen, possibly by regulating the secretion of anagen-inducing signals, or by influencing the response to those inductive signals or follicular maturation following anagen onset (Enshell-Seijffers et al. 2010).
As the follicle enters the growth (anagen) phase, BMP signaling is inhibited and Wnt/β-catenin signaling is ramped up (Fig. 3B). Nuclear β-catenin accumulates in secondary hair germ cells, concomitant with large-scale alterations in histone modifications and the derepression and subsequent expression of cell cycling, extracellular matrix-remodeling, and HF fate specification genes (Lowry et al. 2005; Greco et al. 2009; Lien et al. 2011). Bulge cells also begin to proliferate, resulting in growth of the follicle and migration of bulge/hair germ cells along the growing ORS (Fig. 3C) (Greco et al. 2009; Hsu et al. 2011). As the follicle continues to grow downward, the bulge becomes more distant from the dermal papilla (DP), which becomes surrounded by the concentric layers of epithelial keratinocytes that form the outer root sheath (ORS), inner root sheath (IRS), and matrix (Mx) and the medulla, cortex, and cuticle of the hair shaft.
Many Wnt ligands (Reddy et al. 2001), Frizzled receptors (Reddy et al. 2004), and the effector Dishevelled (Millar et al. 1999) are expressed in overlapping patterns throughout the anagen follicle, suggesting that Wnt/β-catenin signaling continues to be active throughout the progression of anagen (Fig. 3D). However, in most anagen follicular compartments, the precise role of Wnt signaling in orchestrating the follicular differentiation program remains unclear. The most well-examined role for Wnt/β-catenin signaling is in the anagen matrix, where it likely plays a critical role in influencing the differentiation decisions of matrix cells along the various hair lineages. Matrix cells express LEF1 (Zhou et al. 1995; Merrill et al. 2001) and accumulate nuclear β-catenin (Rabbani et al. 2011), suggesting that active Wnt/β-catenin signaling is taking place. Additionally, Wnt-reporter genes like TOPGAL (DasGupta and Fuchs 1999) turn on in the apical region of the matrix known as the precortex, consisting of cells about to differentiate into the cortex of the hair shaft. As follicular keratinocytes differentiate to form the hair shaft, they express multiple keratin genes, which have TCF/LEF binding sites in their promoters and are thought to be Wnt target genes (Zhou et al. 1995; Merrill et al. 2001; Fuchs 2007). Additional Wnt target genes such as Sonic Hedgehog (Gat et al. 1998; Lo Celso et al. 2004; Silva-Vargas et al. 2005) and the Notch ligand Jagged1 (Estrach et al. 2006) are also expressed in the anagen matrix and function to promote keratinocyte proliferation and differentiation. Within the anagen DP, Wnt/β-catenin signaling continues to be required to maintain matrix cell proliferation and differentiation, possibly by influencing the secretion of various secreted growth factors like FGFs 7 and 10, and insulinlike growth factor (IGF) (Enshell-Seijffers et al. 2010).
Quantitative differences in the levels of Wnt/β-catenin signaling influence the decisions of follicular cells to adopt particular lineages. This is evident from transgenic mouse experiments involving epidermal loss and gain of function of β-catenin signaling. For instance, expression of constitutively stable β-catenin promotes differentiation toward the hair lineage (Gat et al. 1998; Lo Celso et al. 2004), whereas inhibition of Wnt/β-catenin signaling in the basal epidermis directs epidermal keratinocytes toward the interfollicular and sebocyte lineages, resulting in progressive hair loss (Merrill et al. 2001; Niemann et al. 2002). Interestingly, K14-mediated ectopic expression of Wnt3 in the ORS results in the formation of fragile hair shafts and up-regulated expression of hair shaft intermediate filament proteins, suggesting that modulating the levels of Wnt signaling can result in more subtle differences in cellular phenotypes (Millar et al. 1999).
Much less is known about the involvement of Wnt/β-catenin signaling during catagen (Fig. 3E). There are no known published studies on the expression of various Wnt pathway components, and most studies on Wnt pathway mutations do not describe catagen defects (for example, see Millar et al. 1999), although there is some evidence that β-catenin is required for the normal regression of the HF and expression of catagen-stage genes (Huelsken et al. 2001).
Apart from controlling the hair cycle telogen–anagen transition and follicular lineage decisions, Wnt/β-catenin signaling may also influence the direction in which hairs grow. HFs emerge at an angle to the skin surface, and in mice, they point in an anterior to posterior direction (reviewed in Millar 2002). The polarity of follicular orientation may be regulated by the Wnt target gene Shh, which is asymmetrically expressed at only one side of the hair matrix (Gat et al. 1998), and has been shown to have a functional role in regulating feather bud orientation in embryonic chicken skin (Ting-Berreth and Chuong 1996a,b). Up-regulated Wnt/β-catenin signaling can disrupt the normal polar orientation of follicles, as observed in embryonic chick (Noramly et al. 1999; Widelitz et al. 1999) and mouse skin (Zhou et al. 1995; Gat et al. 1998). This may be the result of hyperactive β-catenin signaling inducing symmetrical expression of Shh in both sides of the follicle matrix (Gat et al. 1998). Additionally, the Wnt receptor and planar cell polarity gene Frizzled 6 is also asymmetrically polarized in epidermal keratinocytes to orient developing HFs, a process that is independent of placode formation or epidermal stratification (Devenport and Fuchs 2008; Devenport et al. 2011). It remains unclear if or how Wnt ligands are actually involved in the process of setting up epidermal tissue polarity.
Sebocytes are specialized skin cells that produce and accumulate lipids (sebum), eventually releasing these lipids to the skin surface. Sebocytes exist either in sebaceous glands associated with a HF (forming the pilosebaceous unit), or independently in specialized glands (Schneider and Paus 2010).
Numerous studies suggest that inhibition of Wnt/β-catenin signaling may be necessary for specification of the sebocyte lineage (reviewed in Fuchs and Horsley 2008). In mice, K14-driven expression of dominant–negative LEF-1 (Niemann et al. 2002) results in progressive hair loss and concomitant induction of hair-follicle-derived sebaceous tumors. Corroborating this, one-third of human sebaceous tumors were found to contain mutations in LEF-1 that result in similar impairment of β-catenin binding (Takeda et al. 2006). Additionally, the balance between Wnt and Hedgehog signaling is critical for proper sebocyte fate determination (Lo Celso et al. 2008).
Although most studies largely support a role for repression of Wnt/β-catenin signaling in adoption of the sebocyte lineage, some deviations from this paradigm may exist. K14-driven overexpression of TCF-3 induced a Wnt-inhibited state but also resulted in repression of proadipogenic genes like peroxisome proliferator-activated receptor-γ (PPAR-γ) and loss of sebaceous glands (Nguyen et al. 2006). These findings underscore the complexity of the cross talk among signaling pathways in the regulation of adipogenesis and sebaceous gland development.
WNT IN INTERFOLLICULAR EPIDERMIS
Conflicting evidence exists as to whether Wnt/β-catenin signaling is involved in IFE development and adult IFE maintenance. As discussed above, the constitutive attenuation of Wnt/β-catenin signaling in the epidermis throughout development as well as postnatally, either by loss-of-function mutations in β-catenin (Huelsken et al. 2001), LEF1 (Merrill et al. 2001; Niemann et al. 2002), or Porcupine (Porcn) (Barrott et al. 2011), generally results in the loss of hair morphogenesis or cycling. In contrast, the IFE in these mice appears mostly normal.
However, many Wnt/β-catenin signaling mutations do result in IFE phenotypes (Table 1). For instance, mice overexpressing stabilized β-catenin or Wnt1 in the epidermis displayed hyperthickening together with altered HF structures (Lo Celso et al. 2004; Castilho et al. 2009). Mice with β-catenin null mutations in skin display IFE thickening in young adult mice (Huelsken et al. 2001; Teulière et al. 2004), and when β-catenin null skins were grafted onto nude mouse recipients, the grafts also showed epidermal hyperthickening (Nguyen et al. 2009). Constitutive overexpression of Tcf-3 in the skin resulted in embryonic lethality with defective late-stage epidermal differentiation and impaired skin barrier function (Merrill et al. 2001). These observations suggest that an optimal level of β-catenin signaling is somehow required for IFE maintenance, by either affecting progenitor proliferation or differentiation into more mature keratinocytes.
Consistent with this, experiments with human keratinocytes suggest that Wnt/β-catenin signaling may indeed play a role in maintaining the IFE stem cell pool and promoting adoption of the IFE lineage. In cultured human keratinocytes, putative IFE stem cells showed increased clone-forming ability and expressed higher levels of noncadherin-associated, cytoplasmic β-catenin than putative transit-amplifying cells (Zhu and Watt 1999). Retroviral transduction of constitutively stable β-catenin increased the proportion of putative stem cells to ∼90% of the proliferative subpopulation with no effect on terminal differentiation or alterations to cell-cycle kinetics. Conversely, induced expression of a dominant–negative form of β-catenin stimulated cells to exit from the stem cell compartment (Zhu and Watt 1999). These experiments provide evidence for a role of Wnt/β-catenin signaling in maintaining the IFE stem cell pool, while also working coordinately with other signals to influence subsequent differentiation decisions into terminal IFE, hair, or sebocytic lineages.
If Wnt/β-catenin signaling is indeed involved in IFE homeostasis, then what is the precise level of Wnt signaling needed? This remains to be addressed in detail, although closer examination of some of the loss-of-function studies above reveals that Wnt/β-catenin signaling may not have been completely suppressed, potentially allowing a low level of Wnt/β-catenin signaling to continue that might suffice to maintain the IFE. For example, Andl and colleagues examined only mice moderately overexpressing Dkk1 or that were mosaic for transgene expression (which showed sparse hair or bald patches with apparently normal IFE when analyzed up to 60 days postnatally) because mice strongly expressing the K14-Dkk1 transgene were not viable, precluding analysis of the effects of strong Dkk1 overexpression on postnatal IFE maintenance (Andl et al. 2002). The expression levels of Dkk1 in these surviving transgenic animals may not have completely abrogated Wnt/β-catenin signaling in all IFE cells, leaving a low level of Wnt signaling or a population of unaffected IFE progenitors that is sufficient for IFE maintenance. Similarly, Huelsken and colleagues managed to mostly but not completely delete the wild-type β-catenin allele (Fig. 2) in the skin (Huelsken et al. 2001). Any small amount of wild-type β-catenin remaining might provide enough signaling activity to maintain the IFE, accounting for a relatively normal IFE in older animals (Teulière et al. 2004). Consistent with this idea that some β-catenin signaling might remain, hair loss only occurred in patches, and the mice generally developed hair normally up till postnatal day 16. Other plausible explanations for a lack of IFE phenotype in Wnt/β-catenin signaling mutants might include the presence of other unidentified proteins redundant with the mutated signaling components that could substitute for their function in the IFE but not hair keratinocytes.
Many other signaling pathways intersect with Wnt/β-catenin signaling to regulate the adult IFE, sometimes with seemingly contradictory effects. One example is the protooncogene c-Myc (Watt et al. 2008), a known Wnt/β-catenin signaling targeting gene and the key effector of β-catenin/TCF-induced intestinal proliferation (Klaus and Birchmeier 2008). In the skin, Myc overexpression results in epidermal hyperplasia (Waikel et al. 2001) similar to that observed in epidermis overexpressing Wnt1 (Castilho et al. 2009). At the same time, however, Myc and β-catenin appear to exert rather opposite effects. Constitutively high levels of β-catenin signaling induce ectopic HF formation, whereas constitutive expression of Myc promotes differentiation into IFE and sebaceous gland (reviewed in Watt et al. 2008). Clearly, whether or not Myc is indeed a β-catenin/TCF target gene in the skin remains to be addressed.
Taken together, these experiments provide evidence for a role of Wnt/β-catenin signaling in maintaining the IFE stem cell pool. Based on the studies above, it is tempting to speculate that Wnt/β-catenin signaling is ubiquitous in the epidermis, and it is the precise amount of Wnt/β-catenin signaling, coupled with other signaling inputs, that may influence the specific epidermal lineage decisions. In this scenario, relatively lower (but present) levels of Wnt/β-catenin signaling may maintain IFE stem cells and promote IFE fate, intermediate levels promote sebocyte differentiation, whereas high levels direct IFE cells to form hair (as first proposed in Niemann and Watt 2002; Niemann et al. 2002). In essence, any epidermal stem cell can adopt any of the epidermal lineages, given the right signals at the appropriate levels and at the right time (Silva-Vargas et al. 2005). Consistent with this, following a full-thickness skin injury in postnatal skin, a Wnt-dependent wound signal can induce the de novo formation of HFs in the healed skin through a process that closely resembles that of embryonic follicular morphogenesis (Ito et al. 2007), underscoring the multipotency of interfollicular keratinocytes and the importance of high-Wnt signaling in promoting adoption of the HF lineage.
WNT IN MELANOCYTES
Skin pigmentation plays important physiological and evolutionary roles, by protecting animals against mutagenic UV radiation and allowing camouflage, mimicry, and social communication. Pigmentation results from the deposition of melanin pigment from melanin-producing melanocytes into recipient skin cells. Melanocytes arise from neural crest-derived melanoblasts, and are maintained in adulthood by melanocyte stem cells. The embryonic development and tissue regeneration of melanocytes share many common cellular and genetic events. Among them, Wnt/β-catenin signaling is known to govern many aspects of melanocyte biology, from developmental specification of the melanocyte lineage, to subsequent maintenance of the adult melanocyte regenerative unit (comprising adult stem and progenitor cells) and even melanoma pathogenesis.
During embryonic development, multipotent trunk neural crest cells migrate from the neural plate to the epidermis and dermis, undergoing gradual lineage restriction to form committed precursor melanoblasts, which subsequently produce the adult melanocyte stem and progenitor cells and their differentiated, pigmented melanocyte progeny (Fig. 4) (Rawles 1947), reviewed in White and Zon (2008). Dopachrome tautomerase (DCT, also known as Tyrosinase-related protein 2, TRP-2) -expressing melanoblasts are not present in mice mutant for both Wnt-1 and Wnt-3a, providing one of the earliest indications that Wnt/β-catenin signaling might be important for melanoblast specification (Fig. 4A) (Ikeya et al. 1997). Subsequent studies in zebrafish showed that activated β-catenin signaling directed neural crest cells toward pigment–cell fates, at the expense of neuronal and glial cell fates (Dorsky et al. 1998). Transgenic overexpression of Wnt1 in neural crest explant cultures was sufficient to expand the number of melanoblasts and increase differentiation to pigmented melanocytes, showing in vitro that Wnt signaling can direct melanocyte lineage specification and differentiation decisions (Dunn et al. 2000).
Figure 4.

Wnt signaling during melanocyte development and homeostasis. (A) As the embryo develops, multipotent neural crest cells receive Wnt signals that instruct them to adopt the melanocyte lineage and form melanoblasts. These melanoblasts migrate and populate the embryonic epidermis and developing hair follicles (HFs). (B) Embryonic melanoblasts eventually give rise to adult melanocyte stem cells, which reside in the telogen bulge in close proximity to follicular stem cells. Follicular and melanocyte stem cells regulate each other and coordinately activate Wnt/β-catenin signaling at the onset of anagen (Rabbani et al. 2011). (C) The activation of Wnt signaling in melanocyte stem cells causes them to proliferate and differentiate into melanocytes, which then migrate along the outer root sheath (ORS) to enter the matrix. In the matrix, they potentially receive additional Wnt signals that cause them to transfer pigment to differentiating cortical keratinocytes, producing a pigmented hair shaft.
Much mechanistic insight into the way Wnt/β-catenin signaling drives the melanocyte lineage has emerged from studies of melanocyte-specific microphthalmia-associated transcription factor (MITF), a gene that regulates melanocyte differentiation and causes auditory–pigmentary diseases like Waardenburg syndrome type 2 (WS2) when mutated in humans (Saito et al. 2003). A promoter/enhancer region of nacre, the zebrafish homolog of MITF, is bound by LEF1 in vitro and contains TCF/LEF binding sites that are required for MITF promoter–reporter gene expression (Dorsky et al. 2000). By adding Wnt-3a to cultured melanocytes, Takeda and colleagues (2000) found that exogenous Wnt recruited β-catenin and LEF-1 to the MITF promoter. Together, these studies provide strong evidence that Wnt/β-catenin signaling directly regulates the transcription of melanocyte-specific genes like MITF. The MITF protein itself directly interacts with LEF-1, and MITF and LEF-1 function synergistically to up-regulate DCT expression. Thus, by functioning as both a Wnt target gene as well as a nuclear effector of the Wnt pathway, MITF participates in a feed-forward loop to efficiently propagate Wnt signals in melanocytes (Yasumoto et al. 2002).
How is the sensitive balance between self-renewal and differentiation of lineage-restricted melanocyte stem cells controlled? Studies in mouse HFs and cell culture suggest that Wnt signaling interacts with Pax3 and MITF to mediate stem cell self-renewal versus differentiation decisions (Lang et al. 2005). In HF bulge melanocyte stem cells, Pax3 cooperates with Sox10 to promote MITF expression, hence directing these cells toward a melanocyte fate. Interestingly, however, Pax3 simultaneously prevents further differentiation of these cells, by associating with Groucho corepressors and competing with MITF for binding to an enhancer element in the DCT promoter, causing strong repression of DCT transcription. This results in a buildup of Pax3-expressing melanocyte precursors that accumulate MITF and so are primed to express further differentiation genes when the right signals are received. Activated nuclear β-catenin displaces Pax3 and the Groucho corepressors from the enhancer, allowing MITF binding and activating DCT expression. In this way, low levels of Wnt/β-catenin signaling maintain melanocyte stem cells in an undifferentiated state, whereas increased Wnt/β-catenin signaling promotes terminal differentiation to mature melanocytes.
If Wnt/β-catenin signaling is required for proper differentiation of melanocytes, where does the Wnt signal come from? Unpigmented DCT-expressing HF melanocyte stem cells are present in the secondary hair germ near the follicular bulge (Nishimura et al. 2002), which incidentally is also where K15+ and Lgr5+ follicular stem cells reside (Fig. 4B). Because of the proximity of these two stem cell populations and the fact that they act coordinately to regenerate pigmented hair with each hair cycle, it has been proposed that follicular and melanocyte stem cells may regulate each other. Indeed, several recent studies show that follicular stem cells provide adhesion (Tanimura et al. 2011) and extrinsic signals, including Wnt, necessary for melanocyte stem cell maintenance (Nishimura et al. 2010; Rabbani et al. 2011). At the onset of anagen, secondary hair germ keratinocytes stabilize β-catenin and subsequently express Wnt10b, activating Wnt/β-catenin signaling in adjacent melanocyte stem cells and promoting their proliferation and differentiation to melanocytes (Fig. 4B,C). Strikingly, hyperactive Wnt/β-catenin signaling in melanocyte stem cells by DCT-mediated expression of a constitutively stabilized β-catenin resulted in exhaustion of the stem cell compartment and premature hair graying (Rabbani et al. 2011).
Wnt/β-catenin signaling may even be involved in melanocyte function. LEF-1 knockout mouse HFs lack melanin even though melanocytes are still present, suggesting that LEF-1 may be involved in regulating the ability of melanocytes to synthesize or transfer pigment to adjacent hair keratinocytes (van Genderen et al. 1994). Wnt5a mutant mice also show reduced pigment deposition in the hair cortex, possibly by decreasing FoxN1 expression (Hu et al. 2010). As such, it is tempting to speculate that Wnt signals in the matrix and precortex instruct the melanocytes there to deposit pigment in differentiating cortical keratinocytes, resulting in pigmented hair shafts.
Wnt/β-catenin signaling may also have a role in controlling different relative pigmentation of the same skin. In humans, palmoplantar epidermis contains much fewer melanocytes and is subsequently much less pigmented than nonpalmoplantar epidermis. Compared to nonpalmoplantar dermal fibroblasts, palmoplantar dermal fibroblasts secrete higher levels of the Wnt-antagonist Dkk1, which acts on cultured melanocytes to decrease β-catenin expression and suppress transcription of MITF (Yamaguchi et al. 2004). As such, reduced Wnt/β-catenin signaling in palmoplantar skin may account for the reduced numbers of melanocytes there.
What is the role of Wnt/β-catenin signaling in the pathogenesis of melanocyte diseases? Hyperactive β-catenin signaling seems to initiate melanoma genesis, as evidenced by the fact that coexpression of stabilized nuclear β-catenin (bcatsta) with constitutively active N-ras in mouse melanocytes leads to enhanced melanoma formation in vivo and increased immortalization of melanocytes in vitro (Delmas et al. 2007). Consistent with this, as observed in multiple immunohistochemistry studies of human melanomas, benign melanocytic neoplasms (nevi) and many primary melanoma tumors have up-regulated levels of nuclear β-catenin. Interestingly, however, the loss of Wnt/β-catenin signaling may aid the progression of melanoma. As the melanomas progress to become more malignant and metastatic, nuclear β-catenin levels appear to decrease (Kageshita et al. 2001; Maelandsmo et al. 2003; Bachmann et al. 2005; Chien et al. 2009). In fact, loss of nuclear β-catenin in malignant tumors was associated with decreased patient survival (Kageshita et al. 2001; Maelandsmo et al. 2003; Bachmann et al. 2005; Chien et al. 2009). The suppression of Wnt/β-catenin signaling may be achieved through the BRAF oncogene, which is constitutively activated by the BRAFV600E mutation in the majority of melanomas (Davies et al. 2002; Biechele et al. 2012). Knocking down Axin1, a negative regulator of Wnt/β-catenin signaling, synergized with inhibition of BRAFV600E to induce apoptosis in previously resistant melanoma cell lines, suggesting that a combination of enhanced Wnt/β-catenin signaling with other therapies may be a valuable strategy in the treatment of melanoma (Biechele et al. 2012).
WNT IN EPIDERMAL AND HAIR DISEASES
Given the importance of Wnt/β-catenin signaling in skin development and homeostasis, it is not surprising that perturbations in signaling pathway activity can lead to developmental defects and diseases like cancer (reviewed in Watt and Collins 2008; Ambler and Määttä 2009; Blanpain and Fuchs 2009).
Prolonged activation of Wnt/β-catenin signaling leads to a variety of tumors in transgenic mice (Owens and Watt 2003; Klaus and Birchmeier 2008). In the hair, constitutive expression of stabilized β-catenin leads to the formation of pilomatricomas, which are densely packed benign tumors with a center of hair shaft cells surrounded by matrix cells, or trichofolliculomas displaying multilineage differentiation (Gat et al. 1998) that regress when the pathway activation is stopped (Lo Celso et al. 2004). Strikingly, many human pilomatricomas also harbor β-catenin-stabilizing mutations, confirming the relevance of the mouse findings (Chan et al. 1999). Conversely, when Wnt/β-catenin signaling is reduced via expression of the K14-ΔNLef1 transgene in mice, sebaceous gland tumors develop with high frequency, consistent with the sebocyte-promoting property of the transgene (Niemann et al. 2002). This occurs in human tumors as well, as examination of human sebaceous tumors revealed that one-third of them contained mutations in the amino terminus of LEF-1 that similarly blocked β-catenin binding (Takeda et al. 2006). Taken together, these studies suggest that an optimal level of Wnt/β-catenin signaling exists that is required for skin homeostasis, and that either hyperactivation or attenuation of signaling can lead to tumor formation.
As stem cells are long-lived residents in tissue, they are able to accumulate many mutations that might confer tumor-initiating properties, and are hence attractive candidates for tumor-initiating cells or cancer stem cells. It is plausible that some of these mutations may affect Wnt signaling. In a two-step chemical carcinogenesis model of squamous cell carcinoma, tumor-propagating cells expressing the follicular bulge cell marker CD34 also showed nuclear β-catenin, suggesting the presence of active Wnt signaling. The tumors were functionally dependent on β-catenin signaling for their maintenance, as loss of β-catenin resulted in complete regression of the tumors (Malanchi et al. 2008). However, the source of the Wnt ligands for these tumors remains unknown. It is tempting to speculate that a candidate source of Wnt signals might be fibroblasts residing in the tumor stroma, which are known to produce Wnt ligands to support tumors of the mammary epithelia (Bhowmick et al. 2004; Kalluri and Zeisberg 2006). In a genetic mouse model of basal cell carcinoma (BCC), constitutive Hedgehog signaling in a small subset of BCC tumor cells induces Wnt/β-catenin signaling in the BCC tumor cells and surrounding stromal cells, as evidenced by their expression of various Wnt ligands and target genes (Yang et al. 2008). Blocking Wnt/β-catenin signaling via conditional and concomitant expression of Dkk1 resulted in suppression of tumor formation (Yang et al. 2008). Consistent with these animal model observations, human BCC samples also show nuclear accumulation of β-catenin (Salto-Tellez et al. 2006, and references therein), supporting a role for Wnt/β-catenin signaling in BCC etiology. Together, these studies show the potentially crucial function of Wnt/β-catenin signaling in development and progression of various skin cancers.
In concert with other signaling pathways, altered Wnt/β-catenin signaling may determine the specific types of tumors formed (Watt and Collins 2008). In wild-type skin, dimethyl benzanthracene/tetradecanoyl-phorbol acetate (DMBA/TPA) chemical carcinogenesis induces papillomas possessing Ha-Ras mutations that progress into squamous cell carcinomas. In K14-ΔNLef1 mice, the same DMBA/TPA treatment protocol results in sebaceous gland tumors (Niemann et al. 2007). Expression of constitutively active β-catenin in the absence of vitamin D receptor (VDR) leads to the formation of BCC-like lesions with up-regulated Ptch1 expression rather than pilomatricomas (Pálmer et al. 2008). Human trichofolliculoma cells have high levels of β-catenin and VDR, whereas infiltrative human BCCs have high β-catenin levels and low VDR levels (Pálmer et al. 2008). Clearly, altered Wnt/β-catenin signaling with concomitant mutations in other signaling pathways can yield very different tumor types.
Some human skin diseases are known to result from mutations in Wnt genes themselves. Wnt10a missense mutations have been associated with a rare form of ectodermal dysplasia called odonto-onycho-dermal (OOD) dysplasia, which results in dry hair and skin that may result from reduced numbers of sebaceous glands, lesions, and excessive sweating of palms and soles, as well as abnormal thickening of the cornified layer of the epidermis (Adaimy et al. 2007; Bohring et al. 2009; Nawaz et al. 2009). Patients with heterozygotic mutations also presented with some of the OOD phenotype, suggesting that the precise levels of nonmutated Wnt10a are crucial for normal skin homeostasis (Bohring et al. 2009).
CONCLUSIONS AND PERSPECTIVES
From the large body of evidence reviewed here, it is clear that Wnt signaling is important right from the earliest stages of skin development by initiating, patterning, and propagating hair morphogenesis, and continues to be involved postnatally by controlling hair cycling and possibly IFE maintenance, and even managing hair and skin pigmentation.
The fate of skin cells appears to be exquisitely sensitive to the precise levels of Wnt/β-catenin signaling: High-Wnt signaling instructs keratinocytes to form hair, whereas intermediate to low levels of Wnt signaling specify the sebocyte and interfollicular epidermis lineages. When this delicate balance of Wnt/β-catenin signaling is upset, skin cells are driven excessively to form particular lineages, leading to developmental defects or cancer.
Although much has been discovered, several outstanding questions remain.
What is the Wnt ligand that initiates hair morphogenesis, and where does it come from?
If Wnts and their inhibitors are not freely diffusible in the milieu of the embryonic skin, how exactly is an array of foci of up-regulated Wnt signaling established to form hair placodes? What are the identities of the patterning Wnts and their inhibitors? Are they distinct from the morphogenesis-initiating Wnts, and where do they come from?
How exactly are the levels of Wnt/β-catenin signaling modulated to specify the various skin lineages?
Do either or both follicular stem cells and their niche cells require Wnt signaling for their function?
Which Wnt ligands and receptors expressed in the anagen follicle specify which different anagen follicular keratinocyte fates?
Does Wnt/β-catenin signaling have a broader role in the progression of catagen?
How precisely does Wnt/β-catenin signaling influence the polarity of HFs, and how is this polarity set up in the first place?
What exactly is the role that Wnt/β-catenin signaling plays in IFE homeostasis?
As work continues at the intersection of skin and Wnt biology, it seems likely that these and other questions will be addressed, yielding exciting, interesting, and perhaps unexpected new insights into the roles that Wnt/β-catenin signaling plays in the development, homeostasis, and diseases of the skin.
ACKNOWLEDGMENTS
We thank the Agency for Science, Technology and Research (A*STAR), Singapore, and the Howard Hughes Medical Institute (HHMI) for financial support. X.L. is a recipient of a National Science Scholarship from A*STAR, and R.N. is an Investigator of the HHMI.
Footnotes
Editors: Roel Nusse, Xi He, and Renee van Amerongen
Additional Perspectives on Wnt Signaling available at www.cshperspectives.org
REFERENCES
- Adaimy L, Chouery E, Megarbane H, Mroueh S, Delague V, Nicolas E, Belguith H, de Mazancourt P, Megarbane A 2007. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am J Hum Genet 81: 821–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambler CA, Määttä A 2009. Epidermal stem cells: Location, potential and contribution to cancer. J Pathol 217: 206–216 [DOI] [PubMed] [Google Scholar]
- Andl T, Reddy ST, Gaddapara T, Millar SE 2002. WNT signals are required for the initiation of hair follicle development. Dev Cell 2: 643–653 [DOI] [PubMed] [Google Scholar]
- Andl T, Ahn K, Kairo A, Chu EY, Wine-Lee L, Reddy ST, Croft NJ, Cebra-Thomas JA, Metzger D, Chambon P, et al. 2004. Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development 131: 2257–2268 [DOI] [PubMed] [Google Scholar]
- Atit R, Sgaier SK, Mohamed OA, Taketo MM, Dufort D, Joyner AL, Niswander L, Conlon RA 2006. β-Catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev Biol 296: 164–176 [DOI] [PubMed] [Google Scholar]
- Bachmann IM, Straume O, Puntervoll HE, Kalvenes MB, Akslen LA 2005. Importance of P-cadherin, β-catenin, and Wnt5a/frizzled for progression of melanocytic tumors and prognosis in cutaneous melanoma. Clin Cancer Res 11: 8606–8614 [DOI] [PubMed] [Google Scholar]
- Barrott JJ, Cash GM, Smith AP, Barrow JR, Murtaugh LC 2011. Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc Natl Acad Sci 108: 12752–12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Neilson EG, Moses HL 2004. Stromal fibroblasts in cancer initiation and progression. Nature 432: 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biechele TL, Kulikauskas RM, Toroni RA, Lucero OM, Swift RD, James RG, Robin NC, Dawson DW, Moon RT, Chien AJ 2012. Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant Kinase BRAFV600E in human melanoma. Sci Signal 5: ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E 2006. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol 22: 339–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E 2009. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat Rev Mol Cell Biol 10: 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E 2004. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 118: 635–648 [DOI] [PubMed] [Google Scholar]
- Bohring A, Stamm T, Spaich C, Haase C, Spree K, Hehr U, Hoffmann M, Ledig S, Sel S, Wieacker P, et al. 2009. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet 85: 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botchkarev VA, Botchkareva NV, Nakamura M, Huber O, Funa K, Lauster R, Paus R, Gilchrest BA 2001. Noggin is required for induction of the hair follicle growth phase in postnatal skin. FASEB J 15: 2205–2214 [DOI] [PubMed] [Google Scholar]
- Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS 2009. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell 5: 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EF, Gat U, McNiff JM, Fuchs E 1999. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet 21: 410–413 [DOI] [PubMed] [Google Scholar]
- Chanda S, Robinette CL, Couse JF, Smart RC 2000. 17β-Estradiol and ICI-182780 regulate the hair follicle cycle in mice through an estrogen receptor-α pathway. Am J Physiol Endocrinol Metab 278: E202–E210 [DOI] [PubMed] [Google Scholar]
- Chien AJ, Moore EC, Lonsdorf AS, Kulikauskas RM, Rothberg BG, Berger AJ, Major MB, Hwang ST, Rimm DL, Moon RT 2009. Activated Wnt/β-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci 106: 1193–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotsarelis G, Sun T-T, Lavker RM 1990. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 61: 1329–1337 [DOI] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E 1999. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development 126: 4557–4568 [DOI] [PubMed] [Google Scholar]
- DasGupta R, Rhee H, Fuchs E 2002. A developmental conundrum: A stabilized form of β-catenin lacking the transcriptional activation domain triggers features of hair cell fate in epidermal cells and epidermal cell fate in hair follicle cells. J Cell Biol 158: 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. 2002. Mutations of the BRAF gene in human cancer. Nature 417: 949–954 [DOI] [PubMed] [Google Scholar]
- Delmas V, Beermann F, Martinozzi S, Carreira S, Ackermann J, Kumasaka M, Denat L, Goodall J, Luciani F, Viros A, et al. 2007. β-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev 21: 2923–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devenport D, Fuchs E 2008. Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat Cell Biol 10: 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devenport D, Oristian D, Heller E, Fuchs E 2011. Mitotic internalization of planar cell polarity proteins preserves tissue polarity. Nat Cell Biol 13: 893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsky RI, Moon RT, Raible DW 1998. Control of neural crest cell fate by the Wnt signalling pathway. Nature 396: 370–373 [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Raible DW, Moon RT 2000. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev 14: 158–162 [PMC free article] [PubMed] [Google Scholar]
- Dunn KJ, Williams BO, Li Y, Pavan WJ 2000. Neural crest-directed gene transfer demonstrates Wnt1 role in melanocyte expansion and differentiation during mouse development. Proc Natl Acad Sci 97: 10050–10055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enshell-Seijffers D, Lindon C, Kashiwagi M, Morgan BA 2010. β-catenin activity in the dermal papilla regulates morphogenesis and regeneration of hair. Dev Cell 18: 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrach S, Ambler CA, Lo Celso C, Hozumi K, Watt FM 2006. Jagged 1 is a β-catenin target gene required for ectopic hair follicle formation in adult epidermis. Development 133: 4427–4438 [DOI] [PubMed] [Google Scholar]
- Fuchs E 2007. Scratching the surface of skin development. Nature 445: 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E, Horsley V 2008. More than one way to skin. Genes Dev 22: 976–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gat U, DasGupta R, Degenstein L, Fuchs E 1998. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated β-catenin in skin. Cell 95: 605–614 [DOI] [PubMed] [Google Scholar]
- Greco V, Chen T, Rendl M, Schober M, Pasolli HA, Stokes N, Cruz-Racelis Dela J, Fuchs E 2009. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell 4: 155–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy MH 1992. The secret life of the hair follicle. Trends Genet 8: 55–61 [DOI] [PubMed] [Google Scholar]
- Hsu Y-C, Pasolli HA, Fuchs E 2011. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell 144: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Lefort K, Qiu W, Nguyen BC, Rajaram RD, Castillo E, He F, Chen Y, Angel P, Brisken C, et al. 2010. Control of hair follicle cell fate by underlying mesenchyme through a CSL-Wnt5a-FoxN1 regulatory axis. Genes Dev 24: 1519–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W 2001. β-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 105: 533–545 [DOI] [PubMed] [Google Scholar]
- Ikeya M, Lee SM, Johnson JE, McMahon AP, Takada S 1997. Wnt signalling required for expansion of neural crest and CNS progenitors. Nature 389: 966–970 [DOI] [PubMed] [Google Scholar]
- Ito M, Yang Z, Andl T, Cui C, Kim N, Millar SE, Cotsarelis G 2007. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 447: 316–320 [DOI] [PubMed] [Google Scholar]
- Jaks V, Barker N, Kasper M, van Es JH, Snippert HJ, Clevers H, Toftgård R 2008. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat Genet 40: 1291–1299 [DOI] [PubMed] [Google Scholar]
- Jiang T-X, Widelitz RB, Shen W-M, Will P, Wu D-Y, Lin C-M, Jung H-S, Chuong C-M 2004. Integument pattern formation involves genetic and epigenetic controls: Feather arrays simulated by digital hormone models. Int J Dev Biol 48: 117–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageshita T, Hamby CV, Ishihara T, Matsumoto K, Saida T, Ono T 2001. Loss of β-catenin expression associated with disease progression in malignant melanoma. Br J Dermatol 145: 210–216 [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M 2006. Fibroblasts in cancer. Nat Rev Cancer 6: 392–401 [DOI] [PubMed] [Google Scholar]
- Klaus A, Birchmeier W 2008. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 8: 387–398. [DOI] [PubMed] [Google Scholar]
- Kobielak K, Pasolli HA, Alonso L, Polak L, Fuchs E 2003. Defining BMP functions in the hair follicle by conditional ablation of BMP receptor IA. J Cell Biol 163: 609–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak K, Stokes N, la Cruz de J, Polak L, Fuchs E 2007. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc Natl Acad Sci 104: 10063–10068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochwil K, Dull M, Farinas I, Galceran J, Grosschedl R 1996. Lef1 expression is activated by BMP-4 and regulates inductive tissue interactions in tooth and hair development. Genes Dev 10: 1382–1394 [DOI] [PubMed] [Google Scholar]
- Lang D, Lu M-M, Huang L, Engleka KA, Zhang M, Chu EY, Lipner S, Skoultchi A, Millar SE, Epstein JA 2005. Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature 433: 884–887 [DOI] [PubMed] [Google Scholar]
- Levy V, Lindon C, Harfe BD, Morgan BA 2005. Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev Cell 9: 855–861 [DOI] [PubMed] [Google Scholar]
- Lien W-H, Guo X, Polak L, Lawton LN, Young RA, Zheng D, Fuchs E 2011. Genome-wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell Stem Cell 9: 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Celso C, Prowse DM, Watt FM 2004. Transient activation of β-catenin signalling in adult mouse epidermis is sufficient to induce new hair follicles but continuous activation is required to maintain hair follicle tumours. Development 131: 1787–1799 [DOI] [PubMed] [Google Scholar]
- Lo Celso C, Berta MA, Braun KM, Frye M, Lyle S, Zouboulis CC, Watt FM 2008. Characterization of bipotential epidermal progenitors derived from human sebaceous gland: Contrasting roles of c-Myc and β-catenin. Stem Cells 26: 1241–1252 [DOI] [PubMed] [Google Scholar]
- Lowry WE, Blanpain C, Nowak JA, Guasch G, Lewis L, Fuchs E 2005. Defining the impact of β-catenin/Tcf transactivation on epithelial stem cells. Genes Dev 19: 1596–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelandsmo GM, Holm R, Nesland JM, Fodstad Ø, Flørenes VA 2003. Reduced β-catenin expression in the cytoplasm of advanced-stage superficial spreading malignant melanoma. Clin Cancer Res 9: 3383–3388 [PubMed] [Google Scholar]
- Maini PK, Baker RE, Chuong C-M 2006. Developmental biology. The Turing model comes of molecular age. Science 314: 1397–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, et al. 2008. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 452: 650–653 [DOI] [PubMed] [Google Scholar]
- Merrill BJ, Gat U, DasGupta R, Fuchs E 2001. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev 15: 1688–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar SE 2002. Molecular mechanisms regulating hair follicle development. J Invest Dermatol 118: 216. [DOI] [PubMed] [Google Scholar]
- Millar SE, Willert K, Salinas PC, Roelink H, Nusse R, Sussman DJ, Barsh GS 1999. WNT signaling in the control of hair growth and structure. Dev Biol 207: 133–149 [DOI] [PubMed] [Google Scholar]
- Morelli LG, Uriu K, Ares S, Oates AC 2012. Computational approaches to developmental patterning. Science 336: 187–191 [DOI] [PubMed] [Google Scholar]
- Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, Lin JS, Sawicki JA, Cotsarelis G 2004. Capturing and profiling adult hair follicle stem cells. Nat Biotechnol 22: 411–417 [DOI] [PubMed] [Google Scholar]
- Müller-Röver S, Handjiski B, van der Veen C, Eichmüller S, Foitzik K, McKay IA, Stenn KS, Paus R 2001. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J Invest Dermatol 117: 3–15 [DOI] [PubMed] [Google Scholar]
- Närhi K, Järvinen E, Birchmeier W, Taketo MM, Mikkola ML, Thesleff I 2008. Sustained epithelial β-catenin activity induces precocious hair development but disrupts hair follicle down-growth and hair shaft formation. Development 135: 1019–1028 [DOI] [PubMed] [Google Scholar]
- Nawaz S, Klar J, Wajid M, Aslam M, Tariq M, Schuster J, Baig SM, Dahl N 2009. WNT10A missense mutation associated with a complete Odonto-Onycho-Dermal Dysplasia syndrome. Eur J Hum Genet 17: 1600–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Rendl M, Fuchs E 2006. Tcf3 governs stem cell features and represses cell fate determination in skin. Cell 127: 171–183 [DOI] [PubMed] [Google Scholar]
- Nguyen H, Merrill BJ, Polak L, Nikolova M, Rendl M, Shaver TM, Pasolli HA, Fuchs E 2009. Tcf3 and Tcf4 are essential for long-term homeostasis of skin epithelia. Nat Genet 41: 1068–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann C, Watt FM 2002. Designer skin: Lineage commitment in postnatal epidermis. Trends Cell Biol 12: 185–192 [DOI] [PubMed] [Google Scholar]
- Niemann C, Owens DM, Hülsken J, Birchmeier W, Watt FM 2002. Expression of ΔNLef1 in mouse epidermis results in differentiation of hair follicles into squamous epidermal cysts and formation of skin tumours. Development 129: 95–109 [DOI] [PubMed] [Google Scholar]
- Niemann C, Owens DM, Schettina P, Watt FM 2007. Dual role of inactivating Lef1 mutations in epidermis: Tumor promotion and specification of tumor type. Cancer Res 67: 2916–2921 [DOI] [PubMed] [Google Scholar]
- Nishimura EK, Jordan SA, Oshima H, Yoshida H, Osawa M, Moriyama M, Jackson IJ, Barrandon Y, Miyachi Y, Nishikawa S-I 2002. Dominant role of the niche in melanocyte stem-cell fate determination. Nature 416: 854–860 [DOI] [PubMed] [Google Scholar]
- Nishimura EK, Suzuki M, Igras V, Du J, Lonning S, Miyachi Y, Roes J, Beermann F, Fisher DE 2010. Key roles for transforming growth factor β in melanocyte stem cell maintenance. Cell Stem Cell 6: 130–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noramly S, Freeman A, Morgan BA 1999. β-catenin signaling can initiate feather bud development. Development 126: 3509–3521 [DOI] [PubMed] [Google Scholar]
- Ohtola J, Myers J, Akhtar-Zaidi B, Zuzindlak D, Sandesara P, Yeh K, Mackem S, Atit R 2008. β-catenin has sequential roles in the survival and specification of ventral dermis. Development 135: 2321–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama M, Terunuma A, Tock CL, Radonovich MF, Pise-Masison CA, Hopping SB, Brady JN, Udey MC, Vogel JC 2006. Characterization and isolation of stem cell-enriched human hair follicle bulge cells. J Clin Invest 116: 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver RF, Jahoda CA 1988. Dermal-epidermal interactions. Clin Dermatol 6: 74–82 [DOI] [PubMed] [Google Scholar]
- Olivera-Martinez I, Thélu J, Teillet MA, Dhouailly D 2001. Dorsal dermis development depends on a signal from the dorsal neural tube, which can be substituted by Wnt-1. Mech Dev 100: 233–244 [DOI] [PubMed] [Google Scholar]
- Oshimori N, Fuchs E 2012. Paracrine TGF-β signaling counterbalances BMP-mediated repression in hair follicle stem cell activation. Cell Stem Cell 10: 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouji Y, Yoshikawa M, Shiroi A, Ishizaka S 2006. Promotion of hair follicle development and trichogenesis by Wnt-10b in cultured embryonic skin and in reconstituted skin. Biochem Biophys Res Commun 345: 581–587 [DOI] [PubMed] [Google Scholar]
- Ouji Y, Yoshikawa M, Moriya K, Nishiofuku M, Matsuda R, Ishizaka S 2008. Wnt-10b, uniquely among Wnts, promotes epithelial differentiation and shaft growth. Biochem Biophys Res Commun 367: 299–304 [DOI] [PubMed] [Google Scholar]
- Owens DM, Watt FM 2003. Contribution of stem cells and differentiated cells to epidermal tumours. Nat Rev Cancer 3: 444–451 [DOI] [PubMed] [Google Scholar]
- Pálmer HG, Anjos-Afonso F, Carmeliet G, Takeda H, Watt FM 2008. The vitamin D receptor is a Wnt effector that controls hair follicle differentiation and specifies tumor type in adult epidermis. PLoS ONE 3: e1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus R, Müller-Röver S, van der Veen C, Maurer M, Eichmüller S, Ling G, Hofmann U, Foitzik K, Mecklenburg L, Handjiski B 1999. A comprehensive guide for the recognition and classification of distinct stages of hair follicle morphogenesis. J Invest Dermatol 113: 523–532 [DOI] [PubMed] [Google Scholar]
- Plikus MV, Mayer JA, la Cruz de D, Baker RE, Maini PK, Maxson R, Chuong C-M 2008. Cyclic dermal BMP signalling regulates stem cell activation during hair regeneration. Nature 451: 340–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbani P, Takeo M, Chou W, Myung P, Bosenberg M, Chin L, Taketo MM, Ito M 2011. Coordinated activation of Wnt in epithelial and melanocyte stem cells initiates pigmented hair regeneration. Cell 145: 941–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawles ME 1947. Origin of pigment cells from the neural crest in the mouse embryo. Physiol Zool 20: 248–266 [DOI] [PubMed] [Google Scholar]
- Reddy S, Andl T, Bagasra A, Lu MM, Epstein DJ, Morrisey EE, Millar SE 2001. Characterization of Wnt gene expression in developing and postnatal hair follicles and identification of Wnt5a as a target of Sonic hedgehog in hair follicle morphogenesis. Mech Dev 107: 69–82 [DOI] [PubMed] [Google Scholar]
- Reddy ST, Andl T, Lu M-M, Morrisey EE, Millar SE 2004. Expression of Frizzled genes in developing and postnatal hair follicles. J Invest Dermatol 123: 275–282 [DOI] [PubMed] [Google Scholar]
- Rhee H, Polak L, Fuchs E 2006. Lhx2 maintains stem cell character in hair follicles. Science 312: 1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, Yasumoto K-I, Takeda K, Takahashi K, Yamamoto H, Shibahara S 2003. Microphthalmia-associated transcription factor in the Wnt signaling pathway. Pigment Cell Res 16: 261–265 [DOI] [PubMed] [Google Scholar]
- Salto-Tellez M, Peh BK, Ito K, Tan SH, Chong PY, Han HC, Tada K, Ong WY, Soong R, Voon DC, et al. 2006. RUNX3 protein is overexpressed in human basal cell carcinomas. Oncogene 25: 7646–7649 [DOI] [PubMed] [Google Scholar]
- Schmidt-Ullrich R, Paus R 2005. Molecular principles of hair follicle induction and morphogenesis. Bioessays 27: 247–261 [DOI] [PubMed] [Google Scholar]
- Schneider MR, Paus R 2010. Sebocytes, multifaceted epithelial cells: Lipid production and holocrine secretion. Int J Biochem Cell Biol 42: 181–185 [DOI] [PubMed] [Google Scholar]
- Schneider MR, Schmidt-Ullrich R, Paus R 2009. The hair follicle as a dynamic miniorgan. Curr Biol 19: R132–R142 [DOI] [PubMed] [Google Scholar]
- Sick S, Reinker S, Timmer J, Schlake T 2006. WNT and DKK determine hair follicle spacing through a reaction-diffusion mechanism. Science 314: 1447. [DOI] [PubMed] [Google Scholar]
- Silva-Vargas V, Lo Celso C, Giangreco A, Ofstad T, Prowse DM, Braun KM, Watt FM 2005. β-catenin and Hedgehog signal strength can specify number and location of hair follicles in adult epidermis without recruitment of bulge stem cells. Dev Cell 9: 121–131 [DOI] [PubMed] [Google Scholar]
- Stevens JR, Miranda-Carboni GA, Singer MA, Brugger SM, Lyons KM, Lane TF 2010. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J Bone Miner Res 25: 2138–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Yasumoto K, Takada R, Takada S, Watanabe K, Udono T, Saito H, Takahashi K, Shibahara S 2000. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J Biol Chem 275: 14013–14016 [DOI] [PubMed] [Google Scholar]
- Takeda H, Lyle S, Lazar AJF, Zouboulis CC, Smyth I, Watt FM 2006. Human sebaceous tumors harbor inactivating mutations in LEF1. Nat Med 12: 395–397 [DOI] [PubMed] [Google Scholar]
- Takeuchi S, Takeda K, Oishi I, Nomi M, Ikeya M, Itoh K, Tamura S, Ueda T, Hatta T, Otani H, et al. 2000. Mouse Ror2 receptor tyrosine kinase is required for the heart development and limb formation. Genes Cells 5: 71–78 [DOI] [PubMed] [Google Scholar]
- Tanimura S, Tadokoro Y, Inomata K, Binh NT, Nishie W, Yamazaki S, Nakauchi H, Tanaka Y, McMillan JR, Sawamura D, et al. 2011. Hair follicle stem cells provide a functional niche for melanocyte stem cells. Cell Stem Cell 8: 177–187 [DOI] [PubMed] [Google Scholar]
- Teulière J, Faraldo M, Shtutman M, Birchmeier W, Huelsken J, Thiery J, Glukhova M 2004. β-Catenin-dependent and -independent effects of ΔN-plakoglobin on epidermal growth and differentiation. Mol Cell Biol 24: 8649–8661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting-Berreth SA, Chuong CM 1996a. Local delivery of TGF β2 can substitute for placode epithelium to induce mesenchymal condensation during skin appendage morphogenesis. Dev Biol 179: 347–359 [DOI] [PubMed] [Google Scholar]
- Ting-Berreth SA, Chuong CM 1996b. Sonic Hedgehog in feather morphogenesis: Induction of mesenchymal condensation and association with cell death. Dev Dyn 207: 157–170 [DOI] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E 2004. Defining the epithelial stem cell niche in skin. Science 303: 359–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, et al. 2002. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111: 241–250 [DOI] [PubMed] [Google Scholar]
- van Genderen C, Okamura RM, Farinas I, Quo RG, Parslow TG, Bruhn L, Grosschedl R 1994. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev 8: 2691–2703 [DOI] [PubMed] [Google Scholar]
- Van Mater D, Kolligs FT, Andrzej A Dlugosz, Fearon ER 2003. Transient activation of β-catenin signaling in cutaneous keratinocytes is sufficient to trigger the active growth phase of the hair cycle in mice. Genes Dev 17: 1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin V, Degenstein L, Wise B, Fuchs E 1999. The magical touch: Genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci 96: 8551–8556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal VPI, Chaboissier M-C, Lützkendorf S, Cotsarelis G, Mill P, Hui C-C, Ortonne N, Ortonne J-P, Schedl A 2005. Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr Biol 15: 1340–1351 [DOI] [PubMed] [Google Scholar]
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR 2001. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet 28: 165–168 [DOI] [PubMed] [Google Scholar]
- Watt FM, Collins CA 2008. Role of β-catenin in epidermal stem cell expansion, lineage selection, and cancer. Cold Spring Harbor Symp Quant Biol 73: 503–512 [DOI] [PubMed] [Google Scholar]
- Watt FM, Frye M, Benitah SA 2008. MYC in mammalian epidermis: How can an oncogene stimulate differentiation? Nat Rev Cancer 8: 234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RM, Zon LI 2008. Melanocytes in development, regeneration, and cancer. Cell Stem Cell 3: 242–252 [DOI] [PubMed] [Google Scholar]
- Widelitz RB, Jiang TX, Chen CW, Stott NS, Jung HS, Chuong CM 1999. Wnt-7a in feather morphogenesis: Involvement of anterior-posterior asymmetry and proximal-distal elongation demonstrated with an in vitro reconstitution model. Development 126: 2577–2587 [DOI] [PubMed] [Google Scholar]
- Wilson PA, Hemmati-Brivanlou A 1995. Induction of epidermis and inhibition of neural fate by Bmp-4. Nature 376: 331–333 [DOI] [PubMed] [Google Scholar]
- Wilson SI, Rydström A, Trimborn T, Willert K, Nusse R, Jessell TM, Edlund T 2001. The status of Wnt signalling regulates neural and epidermal fates in the chick embryo. Nature 411: 325–330 [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Bradley A, McMahon AP, Jones S 1999. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 126: 1211–1223 [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Itami S, Watabe H, Yasumoto K-I, Abdel-Malek ZA, Kubo T, Rouzaud F, Tanemura A, Yoshikawa K, Hearing VJ 2004. Mesenchymal-epithelial interactions in the skin: Increased expression of dickkopf1 by palmoplantar fibroblasts inhibits melanocyte growth and differentiation. J Cell Biol 165: 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Andl T, Grachtchouk V, Wang A, Liu J, Syu L-J, Ferris J, Wang TS, Glick AB, Millar SE, et al. 2008. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/β3-catenin signaling. Nat Genet 40: 1130–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasumoto K-I, Takeda K, Saito H, Watanabe K-I, Takahashi K, Shibahara S 2002. Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. EMBO J 21: 2703–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuhki M, Yamada M, Kawano M, Iwasato T, Itohara S, Yoshida H, Ogawa M, Mishina Y 2004. BMPR1A signaling is necessary for hair follicle cycling and hair shaft differentiation in mice. Development 131: 1825–1833 [DOI] [PubMed] [Google Scholar]
- Zhang J, He XC, Tong W-G, Johnson T, Wiedemann LM, Mishina Y, Feng JQ, Li L 2006. Bone morphogenetic protein signaling inhibits hair follicle anagen induction by restricting epithelial stem/progenitor cell activation and expansion. Stem Cells 24: 2826–2839 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, Birchmeier W, Paus R, Piccolo S, Mikkola ML, et al. 2009. Reciprocal requirements for EDA/EDAR/NF-κB and Wnt/β-catenin signaling pathways in hair follicle induction. Dev Cell 17: 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Byrne C, Jacobs J, Fuchs E 1995. Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes Dev 9: 700–713 [DOI] [PubMed] [Google Scholar]
- Zhu AJ, Watt FM 1999. β-catenin signalling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development 126: 2285–2298 [DOI] [PubMed] [Google Scholar]