

Abstract
Background
Low dose aspirin reduces the secondary incidence of myocardial infarction and stroke. Drug resistance to aspirin might result in treatment failure. Despite this concern, no clear definition of “aspirin resistance” has emerged and estimates of its incidence have varied remarkably. We aimed to determine the commonality of a mechanistically consistent, stable and specific phenotype of true pharmacological resistance to aspirin – such as might be explained by genetic causes.
Methods and Results
Healthy volunteers (n=400) were screened for their response to a single oral dose of 325 mg immediate release or enteric coated aspirin. Response parameters reflected the activity of aspirin's molecular target, cyclooxygenase-1. Individuals who appeared “aspirin resistant” on one occasion underwent repeat testing and if still “resistant” were exposed to low dose enteric coated aspirin (81 mg) and clopidogrel (75 mg) for one week each. Variable absorption caused a high frequency of apparent resistance to a single dose of 325 mg enteric coated aspirin (up to 49%) but not to immediate release aspirin (0%). All individuals responded to aspirin upon repeated exposure, extension of the post dosing interval or addition of aspirin to their platelets ex vivo.
Conclusions
Pharmacological resistance to aspirin is rare; this study failed to identify a single case of true drug resistance. Pseudoresistance, reflecting delayed and reduced drug absorption, complicates enteric coated but not immediate release aspirin administration.
Clinical Trial Registration Information
clinicaltrials.gov. Identifier: NCT00948987.
Keywords: aspirin, pharmacology, platelets, thromboxane
Introduction
The efficacy of low dose aspirin in the secondary prevention of important vascular events, such as stroke and myocardial infarction, is well established while its relative benefit versus the risk of gastrointestinal bleeding in primary prevention is still debated.1–4 The mechanism by which this benefit is accrued is explained sufficiently by the irreversible acetylation of Ser530 in the enzyme prostaglandin G/H synthase-1 (commonly termed cyclooxygenase [COX]-1) and the consequent suppression of thromboxane (Tx) A2 formation. 3, 5 TxA2 is synthesized from arachidonic acid in activated platelets and released as a local signal which amplifies activation and recruits additional platelets to the site of clot formation. 6, 7 However, TxA2 is but one of several endogenous agonists that result in platelet activation and vasoconstriction, both pertinent to clinically important vascular occlusive events. 3, 7 Thus, inhibition of TxA2 signaling limits substantially the aggregation response, but it does not prevent completely clot formation. Given this capacity for redundancy, the magnitude of aspirin's clinical impact is perhaps surprising; for example, it reduces both death and myocardial infarction by 50% in placebo controlled trials of patients with unstable angina. 8, 9 What is unsurprising is that treatment failures occur on aspirin, as they do in patients taking other cardiovascular drugs. Many reasons for this have been invoked, from patients failing to take their medication to aspirin insensitive mechanisms of platelet activation to drug-drug interactions. 10–14 Despite this, the concept of “aspirin resistance” has emerged,15 prompting the development of point-of-care diagnostic approaches to assessing the inhibition of platelet aggregation in patients taking aspirin. 16 Estimates of the incidence of such a “resistant” phenotype have varied widely 16–20 and the reliance on an assay of platelet aggregability as a surrogate for enzyme acetylation and suppression of platelet TxA2 formation21 and for clinical outcomes 15 has been criticized. Similarly, reliance on post hoc analysis of the major urinary Tx metabolites 22 as a biomarker of resistance in large scale trials 23, 24 has been questioned. 15
True drug resistance to aspirin would result from (i) a failure to reach its molecular target, (ii) a failure to acetylate COX-1 despite sufficient concentrations in the platelet or (iii) a failure of COX-1 acetylation to suppress thromboxane formation. In the present study, we sought to parse the variance in response to aspirin and to estimate the incidence of an internally consistent phenotype of true drug resistance, reconciling biochemical and functional assays of aspirin action that are stable over time and specific for the effect of aspirin on platelets. Healthy volunteers (n=400) were screened for their response to a single oral dose of 325 mg regular “immediate release” aspirin or enteric coated aspirin, which was designed to reduce local damage of the gastric mucosa. Platelet aggregation induced by arachidonic acid, serum thromboxane formation and urinary excretion of a Tx metabolite (TxM) – all reflecting the activity of aspirin's molecular target, COX-1 – were measured before and after dosing. Individuals who appeared to be “aspirin resistant” underwent repeat testing. Individuals who failed to respond to aspirin twice were exposed to low dose enteric coated aspirin (81 mg) and clopidogrel (75 mg) for one week each in a cross over design.
While the distinct estimates of aspirin action were congruent and apparent “resistance” based on platelet aggregation inhibition at a single time point was common, this reflected variable drug exposure due to enteric coating of aspirin and was either inconstant over time or could be overcome by addition of aspirin ex vivo. Thus, we failed to find a single case of true drug resistance in this study of 400 healthy volunteers.
Methods
Study Design
The study protocol was approved by the Institutional Review Board of the University of Pennsylvania and conducted in University's Clinical Translational Research Center (CTRC). 13 Healthy, non-smoking volunteers (18–55 years) who provided written informed consent were enrolled and abstained from all medications and nutritional supplements for the duration of the study.
In Phase 1 fasted healthy volunteers (n=400) received a single oral dose of 325 mg aspirin and drug response parameters were assessed before and after dosing. Two distinct aspirin formulations were administered with 250 ml water, followed by a light standardized meal 2 hours later. Drug responsiveness was assessed at 4 or 8 hours after administration to create three groups (Figure 1). Group 1 (n=40) received regular, immediate release aspirin (Genuine Bayer Aspirin, Bayer Health Care, Morristown, NJ) and their response was assessed 8 hours after dosing. Group 2 (n=210) received enteric coated aspirin (Safety Coated Bayer Aspirin) and their response was measured 8 hours after dosing. Group 3 (n=150) received enteric coated aspirin and their response was assessed at 4 hours. Enteric coating retards aspirin absorption markedly and renders this process much more variable.25, 26 Thus, Groups 2 and 3 were characterized by decreasingly stringent conditions for the identification of variation in the response to aspirin. This would be expected to increase the likelihood of identifying host factors (e.g. genetics, nutritional habits, the composition of the gut microbiome) which might influence reproducibly the response to aspirin. Decisions regarding inclusion of subjects into the next study phases were made based on a “point-of-care” assessment, the platelet aggregation response to arachidonic acid, which was performed immediately following the blood draw. Individuals who showed less than 60% inhibition of their maximal arachidonic acid induced platelet aggregation (comparing post vs. pre dose aggregation) were classified as “non-responders”, who might potentially be resistant to aspirin and entered Phase 2 of the study. Additionally, a subset of individuals who showed an adequate inhibition of 60% or more was invited to participate in Phase 2 as “responder” controls.
Figure 1.
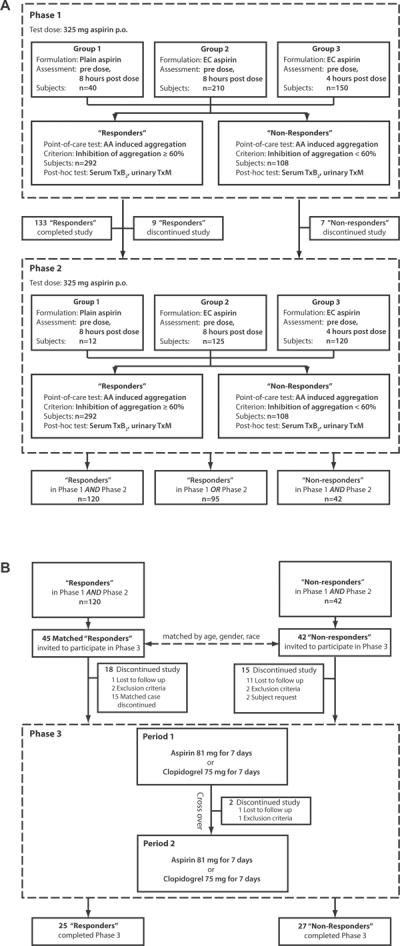
Study design (A) Phase 1 and 2; (B) Phase 3.
Phase 2 addressed the stability of the phenotype of aspirin resistance. Following a washout period of at least 14 days, the two cohorts identified in Phase 1 (108 “non-responders” and 149 “responders”) underwent repeat testing with a single dose of aspirin. Again, individuals showing less than 60% inhibition of platelet aggregation using arachidonic acid as a stimulus were classified as “non-responders”. Those who were “non-responders” in Phase 1 AND Phase 2 were invited to proceed to Phase 3. They were matched by gender, age (± 2.5 years) and race with subjects who were “responders” in Phase 1 AND Phase 2.
Phase 3 determined whether resistance persisted when the dose of aspirin was altered and multiple doses were administered. It also assessed whether the phenotype was specific for aspirin or extended to another platelet inhibitor, clopidogrel. Two cohorts (27 “non-responders” and 25 “responders”, who completed the study) received 81 mg enteric coated aspirin and 75 mg clopidogrel for 7 days, in a crossover design randomized by order and separated by a washout period of at least 14 days (Figure 1). Fifteen of the 42 volunteers, who were classified “nonresponders” in both Phase 1 and Phase 2, were unavailable for re-testing in Phase 3 mostly due to geographical mobility. However, the “non-responder' cohort included all individuals who had appeared “resistant” in Phase 2 even when aspirin was added to their platelets ex vivo. Outcome measurements were obtained prior to treatment begin, 4 hours after the first dose, just prior to the last dose and 4 hours after the last dose. Aspirin response status was assessed based on the last inhibition detected in the last measurement relative to baseline. Those who were responsive to clopidogrel but showed an impaired response only in the COX-1 pathway as determined by arachidonic acid induced platelet aggregation and ex vivo whole blood TxB2 release in all three study phases were considered pharmacologically “aspirin resistant”.
Efficacy Assessments
These included (i) platelet function tests ex vivo involving traditional optical platelet aggregation testing of four pathways of platelet activation (500 μg/mL arachidonic acid, 20 μmol/L ADP, 2 μg/mL collagen, 100 μmol/L the protease receptor-1 activating peptide [PAR-1AP]). 13 The sensitivity of platelets to aspirin added ex vivo was determined by addition of freshly prepared aspirin (dissolved in ethanol) at final concentrations of 30 and 100 μmol/L to PRP, 15 minutes before platelets were stimulated. (ii) The capacity of platelets to form Tx during clotting was assessed as an index of COX-1 enzymatic activity ex vivo. Serum TxB2 was detected by enzyme immunoassay (Cayman Chemicals, Ann Arbor, Michigan). 27 (iii) The urinary excretion of the Tx metabolite, 11-dehydro TxB2 (TxM) – an index of platelet COX-1 activity in vivo – was quantified as its methoxyamine derivative by liquid chromatography tandem mass spectrometry in spot urine samples that were collected 30 minutes after voiding. 28
Statistical considerations
Empirical cumulative frequency distributions were graphed in R (cran.r-project.org). The effect of aspirin exposure on the outcome variables (post vs. pre dose comparison) was assessed with the Wilcoxon Matched-Pairs Signed Ranks Test as implemented in R. A p-value below 0.05 was considered statistically significant.
Responder status to aspirin in the arachidonic acid induced aggregation and the serum TxB2 assays was reported using thresholds that were derived from data sets generated previously in our laboratory. 13, 29 Inhibition of aggregation and TxB2 formation relative to baseline (prior to aspirin administration) was calculated. Failure of aspirin to inhibit arachidonic acid induced aggregation more than 60% or serum Tx formation more than 95% was classified as “non-response”. These threshold values were chosen before the study was conducted. “Responders”/”non-responders” did not exhibit any differences in baseline AA aggregation measures before the administration of aspirin.
For all other outcome measures (maximal aggregation, maximal slope of aggregation, absolute serum TxB2 concentration, urinary TxM excretion), a data driven classification threshold for “responder” vs. “non-responder” status was identified using a modified Receiver Operating Characteristics (ROC) analyses with the inhibition of serum TxB2 formation as the gold standard. Thus, all values of a given outcome measure (e.g. maximal arachidonic acid induced aggregation) were considered as hypothetical cutoffs to predict class membership. For each hypothetical cutoff all instances of an outcome measurement were assigned a hypothetical class label. However, instead of assigning the “true” class labels based on an arbitrary threshold of serum TxB2 inhibition we allowed the best fitting random forest model 30 of serum TxB2 inhibition to define “true” class membership. The random forest classifier as implemented in the R package “randomForest” was run in classification mode for each hypothetical threshold. Each random forest model, consisting of 4000 trees, generated a confusion matrix with the classes predicted by the hypothetical cutoff and the “true” classes derived from the random forest fit of the serum TxB2 inhibition distribution. Graphs of “true” positive and “true” negative rates were used to evaluate the performance of the hypothetical thresholds. The random forest algorithm also generates an error estimate of the model fit, which is the error rate we report here together with the classification accuracy.
Results
Parsing variability in the response to aspirin
We studied a population of relatively young and healthy individuals (Table 1) to assess the inter- and intra-individual variability in the pharmacological response to aspirin without “environmental” variability contributed by disease. High inter- and low intra individual variability would suggest that host factors which predict the response to aspirin might be identified even in a much more heterogeneous population with cardiovascular disease. All 40 subjects administered immediate release aspirin (Group 1) in Phase 1 responded with a reduction of platelet aggregation induced by direct stimulation of the COX-1 pathway by arachidonic acid by more than 60% comparing pre and post dose measurements (Table 2). By contrast, 17% qualified as “non-responders” 8 hours after dosing with enteric coated aspirin (Group 2), increasing to 49% 4 hours after the dose (Group 3).
Table 1.
Demographics of the study population
| Group 1 | Group 2 | Group 3 | Total | |
|---|---|---|---|---|
| Group characteristics | ||||
| Aspirin formulation | Plain | EC | EC | |
| Assessment time | 8 hours | 8 hours | 4 hours | |
|
| ||||
| Ethnicity / race (n) | ||||
| American Indian or Alaskan Native | 1 | 1 | ||
| Asian | 2 | 27 | 18 | 47 |
| Native Hawaiian or Pacific Islander | 1 | 1 | ||
| Black (Non-Hispanic) | 13 | 23 | 26 | 62 |
| Hispanic | 1 | 10 | 8 | 19 |
| White (Non-Hispanic) | 23 | 144 | 96 | 263 |
| Other or more than one race | 1 | 5 | 1 | 7 |
| Totals | 40 | 210 | 150 | 400 |
|
| ||||
| Gender (%) | ||||
| Female | 65 | 52 | 60 | 57 |
|
| ||||
| Age (y) | ||||
| Median | 28 | 25 | 27 | 26 |
| 1. Quartile | 23 | 23 | 23 | 23 |
| 3. Quartile | 34 | 30 | 35 | 31 |
|
| ||||
| BMI (kg/m2) | ||||
| Median | 24.0 | 23.4 | 24.0 | 23·5 |
| 1. Quartile | 21.1 | 21.2 | 21.7 | 21·5 |
| 3. Quartile | 28.8 | 26.0 | 26.2 | 26·3 |
Table 2.
“Non-response” to administration of 325 mg aspirin in study Phases 1 and 2 as assessed by arachidonic acid induced platelet aggregation. Regular or enteric coated (EC) aspirin was administered and the response assessed either 8 or 4 hours post dosing. Platelet function was also measured in platelet rich plasma that was treated with 100 micromolar aspirin ex vivo for 15 minutes. "Non-responders" were arbitrarily defined as individuals who had an inhibition of platelet aggregation induced by arachidonic acid of less than 60% when comparing to pre dose assessments.
| % (N) | Group 1 | Group 2 | Group 3 | |||
|---|---|---|---|---|---|---|
| Plain aspirin, 8 hrs post drug | EC aspirin, 8 hrs post drug | EC aspirin, 4 hrs post drug | ||||
| N=40 | N=210 | N=150 | ||||
| Aspirin ex vivo | − | + | − | + | − | + |
|
| ||||||
| “Non-responders” in Phase 1 | 0 | 0 | 17 | 0 | 49 | 12 |
| (0/40) | (0/40) | (35/210) | (0/210) | (73/150) | (18/150) | |
|
| ||||||
| “Non-responders” in Phase 1 AND Phase 2 | 0 | 0 | 6 | 0 | 20 | 4 |
| (0/40) | (0/40) | (12/209) | (0/210) | (30/150) | (6/150) | |
If platelets are not inhibited by ingestion of aspirin, but can be inhibited by addition of aspirin ex vivo, this points to a pharmacokinetic, rather than a pharmacodynamic explanation for such a “non-responder” status. The addition of aspirin ex vivo dose dependently “corrected” non-responder status in the two groups of volunteers administered enteric coated aspirin, falling from 17 % to 0% 8 hours (Group 2) and from 49% to 12% 4 hours post dosing (Group 3) (Table 2).
Assessment of the response to aspirin by quantitation of serum TxB2, which is considered a reliable biochemical measure of platelet COX-1 function, but is not practical as a point-of-care assay, 31 showed that all but one subject receiving immediate release aspirin (Group 1) responded with inhibition of serum TxB2 formation by more than 95% (Table 3). 21 All subjects responded by suppression of the absolute post dose serum TxB2 concentration to below 10 ng/mL. 29 There was a similar increase in apparent non-response after enteric coated aspirin when measured 8 hours (Group 2; 24%) and 4 hours (Group 3; 59%) post dosing as observed with platelet aggregation using the relative inhibition of serum TxB2 by more than 95%.
Table 3.
“Non-response” to administration of 325 mg aspirin in study Phases 1 and 2 as assessed by serum TxB2 measurement. Regular or enteric coated aspirin was administered and the response assessed either 8 or 4 hours post dosing. “Non-responders” were arbitrarily defined as individuals who had less than 95% inhibition of serum TxB2, when comparing post and pre dosing assessments.
| % (N) | Group 1 | Group 2 | Group 3 |
|---|---|---|---|
| Plain aspirin, 8 hrs post drug | EC aspirin, 8 hrs post drug | EC aspirin, 4 hrs post drug | |
| N=40 | N=210 | N=150 | |
| Non-responders in Phase 1 | 3 | 24 | 59 |
| (1/40) | (35/210) | (88/150) | |
|
| |||
| Non-responders in Phase 1 AND Phase 2 | 0 | 7 | 26 |
| (0/40) | (14/199) | (39/146) | |
Phase 2 addressed the stability of the phenotype of “aspirin resistance”. Following a washout period, two cohorts identified in Phase 1 (108 “non-responders” and 149 “responders”) underwent repeat testing before and after a single dose of aspirin to assess the intraindividual variability of the drug response within each of the three Groups. While “non-response” defined either in terms of suppression of platelet aggregation or serum TxB2 was relatively common after enteric coated aspirin in Phase 1 of the study, the persistence of the phenotype in the same individuals in Phase 2 of the study was comparatively rare (Table 2). None of the volunteers who received plain aspirin (Group 1) was a “non-responder” in both phases of the study. Only 6% (Group 2) and 20% (Group 3) by arachidonic acid induced platelet aggregation and 7% (Group 2) and 26% (Group 3) by inhibition of serum TxB2 formation remained “nonresponders” in Phase 1 AND Phase 2 (Table 3).
Acute dose studies bias in favor of detection of “resistance”. Cumulative inhibition of platelet COX-1 during chronic drug administration would be expected to minimize the incidence attributable to insufficient drug exposure. Phase 3 determined whether resistance persisted during repeated daily administration of a reduced dose of enteric coated aspirin to mimic a typical, chronic low dose aspirin therapy. It also assessed whether the phenotype was specific for aspirin or extended to another platelet inhibitor, clopidogrel, which targets an adenosine diphosphate (ADP) receptor (the P2Y12). Twenty-seven individuals, who were “non-responders” and 25 who were “responders” based on arachidonic induced platelet aggregation in both Phase 1 AND Phase 2 received 81 mg aspirin for 7 days and 75 mg clopidogrel for 7 days, in a crossover design (Figure 1). Only one volunteer in the “non-responder” cohort failed to respond to the weeklong treatment with aspirin in Phase 3 as assessed by arachidonic acid induced platelet aggregation and serum TxB2 inhibition (Figure 2). However, platelets from this subject responded to aspirin addition ex vivo. Furthermore, this apparent “resistance” was not specific as this individual also failed to respond to clopidogrel (not shown). Interestingly, two individuals who entered Phase 3 as “controls” with demonstrated ability of their platelets to respond to aspirin administration in Phase 1 and 2 now failed to respond in Phase 3 (Figure 2). One of them also failed to respond to clopidogrel (not shown), but both responded to addition of aspirin ex vivo.
Figure 2.
Response to 7 days of low dose aspirin (81mg, enteric coated) in Phase 3. Assessment of platelet aggregation using arachidonic acid as the stimulus in the absence and presence of aspirin (100 μmol/L) added to isolated platelets ex vivo. (A) Individuals who qualified as “non-responders” in Phase 1 AND Phase 2 (n=27). One subject showed uninhibited platelet aggregation following one week of aspirin treatment, but responded to aspirin addition ex vivo. (B) Individuals who were classified as responders on two occasions of single dose aspirin administration in Phase 1 and Phase 2 (n= 25). Two subjects showed uninhibited platelet aggregation but responded to aspirin addition ex vivo.
Diagnosis of an individual's aspirin response status
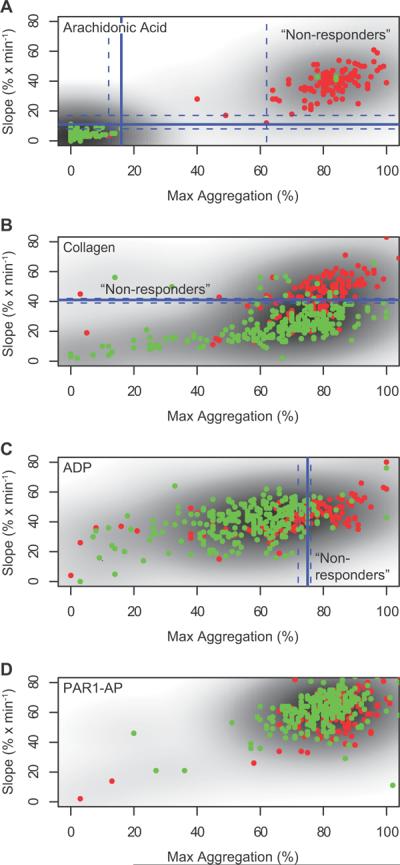
The study sought individuals who showed an inadequate mechanistic response to the administration of aspirin as assessed by platelet aggregation ex vivo, serum TxB2 formation – an index of the capacity of platelets to form COX-1 dependent TxA2 – and the urinary excretion of the thromboxane metabolite, 11-dehydro TxB2 (TxM) – an index of actual thromboxane biosynthesis. All three measurements detected a significant difference between the pre and post dose measurement in each treatment group (p<0·01, data not shown). Plotting their relative cumulative frequency distributions illustrates the utility of the drug response markers to determine response status at the individual level (Figure 3). The distribution curves for arachidonic acid induced platelet aggregation and serum TxB2 concentration, but not for urinary TxM excretion, allowed the data driven detection of a threshold value – a plateau in the distribution curve – which distinguishes between “responders” and “non-responders”. A threshold of 16% (95% confidence interval 12% to 62%) residual arachidonic acid induced platelet aggregation resulted in a test accuracy of >90% and an error rate of 10%. Indeed, the extended plateau in the frequency distribution renders arachidonic acid induced platelet aggregation essentially a dichotomous outcome measurement (see, for example, the post dose distribution of Group 3, the group with approximately 50% apparent “resistance”, in Figure 3C). Stimuli of platelet aggregation which activate platelet pathways in which COX-1 dependent events are downstream of the initial stimulus – collagen and ADP – were much less reliable diagnostic tools for the assessment of the response status (Figure 4). They resulted in accuracies of 80% and 75% and error rates of 21% and 36%. As expected, platelet activation with proteinase activated receptor-1 activating peptide (PAR-1AP), which ligates a thrombin receptor, did not associate with inhibition of serum Tx formation by aspirin.
Figure 3.

Relative cumulative frequency distributions of the arachidonic acid (325 mg) induced platelet aggregation response (A–C), serum thromboxane formation (D–F) and urinary thromboxane metabolite excretion (G–I) stratified by drug formulation and assessment time. The density of the data points is visualized by color coding – green high density/frequency, red – low density /frequency. Classification thresholds (“responders” vs. “non-responders”) as determined iteratively using random forest classifiers are shown as blue arrows, which also depict the fraction of responders in the study groups. Urinary TxM excretion failed to predict individual response status although pre- and postdose distributions segregated in each group (p<0.05).
Figure 4.

Consistency of the response to distinct stimuli of platelet aggregation. Four stimuli were used to assess the functional impact of aspirin treatment on platelet aggregation: arachidonic acid (A), collagen (B), adenosine diphosphate (ADP, C) and protease activated (thrombin) receptor 1 activating peptide (PAR-1 AP, D). Data are displayed as scattergrams of slope vs. maximum aggregation. Data points are color coded by the response status as determined by 95% inhibition of serum TxB2 concentration. Thus, “responders” are plotted in green and “non-responders” in red. Shades of gray represent densities of the two-dimensional distribution. Classification thresholds as determined iteratively using random forest classifiers and 95% confidence intervals are shown in blue. (A) The test sensitivity of arachidonic acid induced platelet aggregation was >90% with an error rate of 10% when volunteers with a residual platelet function following aspirin administration of 16% maximal aggregation (95% confidence interval 12% to 62%) or 11%/min slope (95% confidence interval 8%/min to 17%/min) were classified as “non-responders”. (B, C) Inducing platelet aggregation with collagen or ADP resulted in sensitivities of 80% and 75% and error rates of 25% and 36% at thresholds of 41%/min slope (95% confidence interval 39%/min to 42%/min) and 75% maximal aggregation (95% confidence interval 72% to 76%), respectively. Maximal aggregation was not a predictor of response status in collagen stimulated platelets; slope was not a predictor of response status in ADP stimulated platelets. (D) As expected, the PAR-1AP response did not associate with inhibition of serum Tx formation by aspirin.
Measurement of the absolute serum TxB2 concentration (rather than “serum TxB2 inhibition” based on the comparison of pre and post dose concentrations) is an assessment that could be performed in patients without stopping aspirin therapy. Analysis of the frequency distribution of the absolute serum TxB2 concentration also revealed a plateau that segregated the population into apparent responders and apparent non-responders (Figure 3). A threshold of 29 ng/mL (95% confidence interval 26 ng/mL to 78 ng/mL) serum TxB2 resulted in an accuracy of 98 % and an error rate of 2%.
The congruence between arachidonic acid induced aggregation and serum TxB2 formation for the diagnosis of “non-response” was high. One-hundred five of 108 (97%) subjects classified as “non-responsive” by arachidonic acid induced platelet aggregation in Phase 1 also showed incomplete suppression of serum TxB2 formation. However, measurement of serum TxB2 identified 32 of 400 (8 %) individuals as “non-responders” who had not been recognized by arachidonic induced platelet aggregation. Most of these had serum TxB2 levels between 10 and 100 ng/ml, which represents a partially suppressed range when compared to median pre and post dose concentrations of 4 ng/ml (IQR: 2 – 133 ng/ml) and 315 ng/ml (IQR: 193 – 450 ng/ml).
Analysis of the urinary TxM excretion data did not result in a threshold value that would allow for distinction of “responders” from “non-responders” at the individual level with any reasonable accuracy (Figure 3).
Discussion
Aspirin blocks just one pathway of platelet activation and treatment failures may result from the dominant role of other platelet agonists;11, 12, 32, 33 combination of aspirin with other antiplatelet drugs, such as antagonists of the P2Y12 receptor for ADP, sometimes confers a measurable incremental benefit.34 Other reasons for treatment failure include failure to consume the medication,10, 35 accelerated platelet turn over,36, 37 or interaction with reversible, competitive active site inhibitors of the COX-1 enzyme, such as ibuprofen13 or naproxen.14
The term “aspirin resistance” is often used to imply treatment failure although clinical outcomes have rarely been assessed – not unlike studies of the pharmacogenomics of clopidogrel.38 More commonly, a patient is classified as “resistant” to aspirin, if platelet aggregation, measured on a single occasion, is not depressed to an arbitrary degree. Estimates of the frequency of “aspirin resistance” have varied from 5% to 20% in most studies.16–20 Here we wished to determine the commonality of an internally consistent, specific phenotype of pharmacological resistance to aspirin that was stable over time, such as might result from gene variants in aspirin metabolizing enzymes or in the platelet COX-1-TxA2 synthesis/response network. This might be more readily discernible in a younger population without confounding disease variables and would be measurable by tests that reflected the mechanism of drug action.
The irreversible acetylation of platelet COX-1 is a sufficient mechanism to explain the cardioprotection afforded by aspirin, however direct assays39 have yet to be deployed to characterize resistance. The immediate consequence of drug action is suppression of formation of platelet TxA2, quantitatively reflected by the analysis of its inactive hydrolysis product, TxB2 in serum generated under controlled conditions.31 Measurement of serum TxB2 reflects dose dependent suppression by aspirin, cumulative inhibition during repeated daily dosing and delayed recovery of platelet thromboxane formation after cessation of aspirin, driven by platelet turnover time.40 Urinary Tx metabolites also reflect the pharmacodynamic impact of aspirin on platelets41 and, indeed, derive largely (~ 80%) from platelets under physiological conditions.42 However, other non-platelet sources of Tx biosynthesis are also reflected by urinary metabolites and their contribution may increase under conditions of disease or physiological perturbation.43 Here, we found that large variability in urinary TxM excretion renders it impracticable to assess even a healthy individual's aspirin response status although the reduction in mean urinary TxM by aspirin was clearly detectable in a group of treated individuals. Finally, platelet aggregation responses induced by the COX enzyme substrate, arachidonic acid, depend on its transformation to TxA2 and are suppressed by aspirin. Although other agonists aggregate platelets directly, secondary release of TxA2 may amplify the response and result in partial inhibition of the aggregation signal by aspirin.7 A complication is that the relationship between suppression of platelet thromboxane and aggregation is very nonlinear – the capacity of platelets to generate thromboxane has to be suppressed by >95% before functional inhibition of the aggregation response is attained.29 This explains why diagnostic approaches which tested the COX pathway directly had the most favorable test sensitivities and false classification rates for identification of “non-responders”.
We found that under acute dosing conditions, the frequency of “resistance” based solely on the failure to suppress the aggregation response to arachidonic acid was conditioned by the formulation of aspirin and the timing of the measurement. “Resistance” was measurable after enteric coated, but not immediate release formulations of aspirin and appeared more frequent after the former when measurements were performed 4 hours rather than 8 hours after dosing. This pattern of variance was reflected both by the aggregation response to arachidonic acid and by serum TxB2. By contrast, aspirin had minor effects on aggregation induced by ADP and collagen and no detectable effect on the activation of aggregation through the thrombin pathway by PAR-1AP. The incidence of apparent “resistance” to enteric coated aspirin based on a single measurement of aggregation was 49% at 4 hours (Group 3) dropping to 17% at 8 hours (Group 2) after dosing in Phase 1 of the study. Strikingly, there was marked discordance between the individuals who seemed “resistant” on the two occasions; only in 20% at 4 hours and 6% at 8 hours was “resistance” stable over time. Consistent with the suggestion that apparent “resistance” was largely reflective of variable drug exposure in the patients receiving enteric coated aspirin, we corrected the phenotype in 101 of 108 cases by adding aspirin ex vivo in Phase 1 or 2 and all of the remaining 7 cases responded to adding aspirin ex vivo in Phase 3.
In the Phase 3 of the study we compared cases who had remained “resistant” to inhibition of platelet aggregation by ingestion of aspirin in the first two phases of the study and matched them by gender and age with controls, then subjecting both groups to a weeklong administration of aspirin and clopidogrel in a cross over design. Because 15 of the original 400 subjects who fulfilled the inclusion criteria for Phase 3 were lost-to follow up, we may have underestimated the incidence of true biological resistance. However, of the 27 cases enrolled in this phase only one appeared now to be “resistant” to aspirin as assessed by arachidonic acid induced aggregation and this individual had responsiveness restored by addition of aspirin ex vivo. While this was consistent with “resistance” due to limited drug exposure in vivo, as might be caused by genetic variants in aspirin metabolism, the effect was not specific to aspirin: the volunteer failed also to exhibit clopidogrel induced inhibition of ADP induced platelet aggregation. The unreliability of basing a diagnosis of “aspirin resistance” on a single measurement of platelet aggregation was further emphasized by two individuals who appeared “resistant” amongst the controls in Phase 3, having been responsive to aspirin in the two earlier phases of the study. Here, again, responsiveness to aspirin was apparent by addition of the drug ex vivo and the subject was also unresponsive to clopidogrel. Thus, such “resistance” may have reflected a failure to ingest aspirin.
In summary, we performed studies in 400 healthy volunteers seeking to determine the commonality of a mechanistically consistent, temporally stable and specific phenotype of true aspirin resistance, such as might be attributable to genomic variation in aspirin metabolism or in the platelet COX-1 / TxA2 synthesis response pathway. We failed to find a single person who satisfied these criteria. By contrast, pseudoresistance, due to delayed and reduced drug absorption was common after ingestion of enteric coated aspirin.25, 26 These observations question the value of seeking to diagnose aspirin resistance with single point-of-care diagnostic approaches and support the finding of inconsistent platelet inhibition following enteric coated preparations of aspirin.44–46
Acknowledgements
G.A.F. is the Robert McNeill Professor of Translational Medicine and Therapeutics.
Funding Sources: This work was supported by the National Heart Lung and Blood Institute (HL 54500), the National Center for Research Resources (UL1-RR-024134), the American Heart Association (T.G.: Scientist Development Grant 0430148N), and Bayer Health Care (Morristown, NJ, Study No X05-215). The funding sources were not involved in the study design, or in the collection, analysis, and interpretation of data.
Footnotes
Conflict of Interest Disclosures: T.G. received consultancy fees from PLx Pharma. S.F. has no potential conflict of interest. J.A.L. has no potential conflict of interest. S.C.K has no potential conflict of interest. G.R.G has no potential conflict of interest. G.A.F. received research funding from Bayer Health Care to support partial funding of this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Collaborative overview of randomised trials of antiplatelet therapy-- iii: Reduction in venous thrombosis and pulmonary embolism by antiplatelet prophylaxis among surgical and medical patients. Antiplatelet trialists' collaboration. Bmj. 1994;308:235–246. [PMC free article] [PubMed] [Google Scholar]
- 2.Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. Bmj. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patrono C, Garcia Rodriguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353:2373–2383. doi: 10.1056/NEJMra052717. [DOI] [PubMed] [Google Scholar]
- 4.Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, Buring J, Hennekens C, Kearney P, Meade T, Patrono C, Roncaglioni MC, Zanchetti A. Aspirin in the primary and secondary prevention of vascular disease: Collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–1860. doi: 10.1016/S0140-6736(09)60503-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funk CD, Funk LB, Kennedy ME, Pong AS, FitzGerald GA. Human platelet/erythroleukemia cell prostaglandin g/h synthase: Cdna cloning, expression, and gene chromosomal assignment. Faseb J. 1991;5:2304–2312. [PubMed] [Google Scholar]
- 6.Hamberg M, Svensson J, Samuelsson B. Thromboxanes: A new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci U S A. 1975;72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FitzGerald GA. Mechanisms of platelet activation: Thromboxane A2 as an amplifying signal for other agonists. Am J Cardiol. 1991;68:11B–15B. doi: 10.1016/0002-9149(91)90379-y. [DOI] [PubMed] [Google Scholar]
- 8.Lewis HD, Jr., Davis JW, Archibald DG, Steinke WE, Smitherman TC, Doherty JE, 3rd, Schnaper HW, LeWinter MM, Linares E, Pouget JM, Sabharwal SC, Chesler E, DeMots H. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a veterans administration cooperative study. N Engl J Med. 1983;309:396–403. doi: 10.1056/NEJM198308183090703. [DOI] [PubMed] [Google Scholar]
- 9.RISCGroup Risk of myocardial infarction and death during treatment with low dose aspirin and intravenous heparin in men with unstable coronary artery disease. The risc group. Lancet. 1990;336:827–830. [PubMed] [Google Scholar]
- 10.Schwartz KA, Schwartz DE, Ghosheh K, Reeves MJ, Barber K, DeFranco A. Compliance as a critical consideration in patients who appear to be resistant to aspirin after healing of myocardial infarction. Am J Cardiol. 2005;95:973–975. doi: 10.1016/j.amjcard.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 11.Webster SE, Payne DA, Jones CI, Hayes PD, Bell PR, Goodall AH, Naylor AR. Anti-platelet effect of aspirin is substantially reduced after administration of heparin during carotid endarterectomy. J Vasc Surg. 2004;40:463–468. doi: 10.1016/j.jvs.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 12.Faraday N, Yanek LR, Mathias R, Herrera-Galeano JE, Vaidya D, Moy TF, Fallin MD, Wilson AF, Bray PF, Becker LC, Becker DM. Heritability of platelet responsiveness to aspirin in activation pathways directly and indirectly related to cyclooxygenase-1. Circulation. 2007;115:2490–2496. doi: 10.1161/CIRCULATIONAHA.106.667584. [DOI] [PubMed] [Google Scholar]
- 13.Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, Vyas SN, FitzGerald GA. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809–1817. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]
- 14.Capone ML, Sciulli MG, Tacconelli S, Grana M, Ricciotti E, Renda G, Di Gregorio P, Merciaro G, Patrignani P. Pharmacodynamic interaction of naproxen with low-dose aspirin in healthy subjects. J Am Coll Cardiol. 2005;45:1295–1301. doi: 10.1016/j.jacc.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 15.Hennekens CH, Schrör K, Weisman S, FitzGerald GA. Terms and conditions: Semantic complexity and aspirin resistance. Circulation. 2004;110:1706–1708. doi: 10.1161/01.CIR.0000142056.69970.DB. [DOI] [PubMed] [Google Scholar]
- 16.Gum PA, Kottke-Marchant K, Poggio ED, Gurm H, Welsh PA, Brooks L, Sapp SK, Topol EJ. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol. 2001;88:230–235. doi: 10.1016/s0002-9149(01)01631-9. [DOI] [PubMed] [Google Scholar]
- 17.Eikelboom JW, Hankey GJ. Aspirin resistance: A new independent predictor of vascular events? J Am Coll Cardiol. 2003;41:966–968. doi: 10.1016/s0735-1097(02)03013-9. [DOI] [PubMed] [Google Scholar]
- 18.Gum PA, Kottke-Marchant K, Welsh PA, White J, Topol EJ. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol. 2003;41:961–965. doi: 10.1016/s0735-1097(02)03014-0. [DOI] [PubMed] [Google Scholar]
- 19.DiChiara J, Bliden KP, Tantry US, Hamed MS, Antonino MJ, Suarez TA, Bailon O, Singla A, Gurbel PA. The effect of aspirin dosing on platelet function in diabetic and nondiabetic patients: An analysis from the aspirin-induced platelet effect (ASPECT) study. Diabetes. 2007;56:3014–3019. doi: 10.2337/db07-0707. [DOI] [PubMed] [Google Scholar]
- 20.Neubauer H, Kaiser AF, Endres HG, Kruger JC, Engelhardt A, Lask S, Pepinghege F, Kusber A, Mugge A. Tailored antiplatelet therapy can overcome clopidogrel and aspirin resistance - the bochum clopidogrel and aspirin plan (bocla-plan) to improve antiplatelet therapy. BMC Med. 2011;9:3. doi: 10.1186/1741-7015-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santilli F, Rocca B, De Cristofaro R, Lattanzio S, Pietrangelo L, Habib A, Pettinella C, Recchiuti A, Ferrante E, Ciabattoni G, Davi G, Patrono C. Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays: Implications for aspirin “resistance”. J Am Coll Cardiol. 2009;53:667–677. doi: 10.1016/j.jacc.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 22.Lawson JA, Patrono C, Ciabattoni G, FitzGerald GA. Long-lived enzymatic metabolites of thromboxane B2 in the human circulation. Anal Biochem. 1986;155:198–205. doi: 10.1016/0003-2697(86)90247-2. [DOI] [PubMed] [Google Scholar]
- 23.Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–1655. doi: 10.1161/01.cir.0000013777.21160.07. [DOI] [PubMed] [Google Scholar]
- 24.Eikelboom JW, Hankey GJ, Thom J, Bhatt DL, Steg PG, Montalescot G, Johnston SC, Steinhubl SR, Mak KH, Easton JD, Hamm C, Hu T, Fox KA, Topol EJ. Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: Determinants and effect on cardiovascular risk. Circulation. 2008;118:1705–1712. doi: 10.1161/CIRCULATIONAHA.108.768283. [DOI] [PubMed] [Google Scholar]
- 25.Muir N, Nichols JD, Clifford JM, Stillings MR, Hoare RC. The influence of dosage form on aspirin kinetics: Implications for acute cardiovascular use. Curr Med Res Opin. 1997;13:547–553. doi: 10.1185/03007999709113328. [DOI] [PubMed] [Google Scholar]
- 26.Bochner F, Somogyi AA, Wilson KM. Bioinequivalence of four 100 mg oral aspirin formulations in healthy volunteers. Clin Pharmacokinet. 1991;21:394–399. doi: 10.2165/00003088-199121050-00006. [DOI] [PubMed] [Google Scholar]
- 27.Patrignani P, Panara MR, Greco A, Fusco O, Natoli C, Iacobelli S, Cipollone F, Ganci A, Creminon C, Maclouf J, Patrono C. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J Pharmacol Exp Ther. 1994;271:1705–1712. [PubMed] [Google Scholar]
- 28.Fries S, Grosser T, Price TS, Lawson JA, Kapoor S, DeMarco S, Pletcher MT, Wiltshire T, FitzGerald GA. Marked interindividual variability in the response to selective inhibitors of cyclooxygenase-2. Gastroenterology. 2006;130:55–64. doi: 10.1053/j.gastro.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Reilly IA, FitzGerald GA. Inhibition of thromboxane formation in vivo and ex vivo: Implications for therapy with platelet inhibitory drugs. Blood. 1987;69:180–186. [PubMed] [Google Scholar]
- 30.Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 31.Patrono C, Ciabattoni G, Pinca E, Pugliese F, Castrucci G, De Salvo A, Satta MA, Peskar BA. Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb Res. 1980;17:317–327. doi: 10.1016/0049-3848(80)90066-3. [DOI] [PubMed] [Google Scholar]
- 32.Valles J, Santos MT, Aznar J, Osa A, Lago A, Cosin J, Sanchez E, Broekman MJ, Marcus AJ. Erythrocyte promotion of platelet reactivity decreases the effectiveness of aspirin as an antithrombotic therapeutic modality: The effect of low-dose aspirin is less than optimal in patients with vascular disease due to prothrombotic effects of erythrocytes on platelet reactivity. Circulation. 1998;97:350–355. doi: 10.1161/01.cir.97.4.350. [DOI] [PubMed] [Google Scholar]
- 33.Folts JD, Schafer AI, Loscalzo J, Willerson JT, Muller JE. A perspective on the potential problems with aspirin as an antithrombotic agent: A comparison of studies in an animal model with clinical trials. J Am Coll Cardiol. 1999;33:295–303. doi: 10.1016/s0735-1097(98)00601-9. [DOI] [PubMed] [Google Scholar]
- 34.Mehta SR, Yusuf S, Peters RJ, Bertrand ME, Lewis BS, Natarajan MK, Malmberg K, Rupprecht H, Zhao F, Chrolavicius S, Copland I, Fox KA. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The pci-cure study. Lancet. 2001;358:527–533. doi: 10.1016/s0140-6736(01)05701-4. [DOI] [PubMed] [Google Scholar]
- 35.Hennekens CH, Schneider WR, Hebert PR, Tantry US, Gurbel PA. Hypothesis formulation from subgroup analyses: Nonadherence or nonsteroidal anti-inflammatory drug use explains the lack of clinical benefit of aspirin on first myocardial infarction attributed to “aspirin resistance”. Am Heart J. 2010;159:744–748. doi: 10.1016/j.ahj.2009.11.033. [DOI] [PubMed] [Google Scholar]
- 36.Pascale S, Petrucci G, Dragani A, Habib A, Zaccardi F, Pagliaccia F, Pocaterra D, Ragazzoni E, Rolandi G, Rocca B, Patrono C. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood. 2012;119:3595–3603. doi: 10.1182/blood-2011-06-359224. [DOI] [PubMed] [Google Scholar]
- 37.Rocca B, Santilli F, Pitocco D, Mucci L, Petrucci G, Vitacolonna E, Lattanzio S, Mattoscio D, Zaccardi F, Liani R, Vazzana N, Del Ponte A, Ferrante E, Martini F, Cardillo C, Morosetti R, Mirabella M, Ghirlanda G, Davi G, Patrono C. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost. 2012;10:1220–1230. doi: 10.1111/j.1538-7836.2012.04723.x. [DOI] [PubMed] [Google Scholar]
- 38.Holmes MV, Perel P, Shah T, Hingorani AD, Casas JP. Cyp2C19 genotype, clopidogrel metabolism, platelet function, and cardiovascular events: A systematic review and meta-analysis. JAMA. 2011;306:2704–2714. doi: 10.1001/jama.2011.1880. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Fries S, Ricciotti E, Lawson JA, Blair AI, FitzGerald GA, Grosser T. Assessment of the pharmacological response to aspirin by quantitation of platelet cyclooxygenase acetylation. Circulation. 2010;122:A16834. [Google Scholar]
- 40.Pedersen AK, FitzGerald GA. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N Engl J Med. 1984;311:1206–1211. doi: 10.1056/NEJM198411083111902. [DOI] [PubMed] [Google Scholar]
- 41.Patrignani P, Filabozzi P, Patrono C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J Clin Invest. 1982;69:1366–1372. doi: 10.1172/JCI110576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.FitzGerald GA, Oates JA, Hawiger J, Maas RL, Roberts LJ, 2nd, Lawson JA, Brash AR. Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J Clin Invest. 1983;71:676–688. doi: 10.1172/JCI110814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Catella F, FitzGerald GA. Paired analysis of urinary thromboxane B2 metabolites in humans. Thromb Res. 1987;47:647–656. doi: 10.1016/0049-3848(87)90103-4. [DOI] [PubMed] [Google Scholar]
- 44.Maree AO, Curtin RJ, Dooley M, Conroy RM, Crean P, Cox D, Fitzgerald DJ. Platelet response to low-dose enteric-coated aspirin in patients with stable cardiovascular disease. J Am Coll Cardiol. 2005;46:1258–1263. doi: 10.1016/j.jacc.2005.06.058. [DOI] [PubMed] [Google Scholar]
- 45.Cox D, Maree AO, Dooley M, Conroy R, Byrne MF, Fitzgerald DJ. Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers. Stroke. 2006;37:2153–2158. doi: 10.1161/01.STR.0000231683.43347.ec. [DOI] [PubMed] [Google Scholar]
- 46.Peace A, McCall M, Tedesco T, Kenny D, Conroy RM, Foley D, Cox D. The role of weight and enteric coating on aspirin response in cardiovascular patients. J Thromb Haemost. 2010;8:2323–2325. doi: 10.1111/j.1538-7836.2010.03997.x. [DOI] [PubMed] [Google Scholar]