

Abstract
Several 7-peptide-substituted pterins were synthesized and tested as competitive active-site inhibitors of Ricin Toxin A (RTA). Focus began on dipeptide conjugates, and these results further guided the construction of several tripeptide conjugates. The binding of these compounds to RTA was studied via a luminescence-based kinetic assay, as well as through X-ray crystallography. Despite the relatively polar, solvent exposed active site, several hydrophobic interactions, most commonly π-interactions, not predicted by modeling programs, were identified in all of the best-performing inhibitors. Nearly all of these compounds provide IC50’s in the low μM range.
Keywords: Ricin inhibitors, Pterin, RTA, π-interactions
1) Introduction
The cytotoxin ricin, found in castor beans, is a prototypical type 2 ribosome inactivating protein.1 As such, it contains a catalytic A chain, Ricin Toxin A (RTA), which depurinates a specific adenosine in ribosomal RNA, as well as a lectin B chain which assists in cellular uptake.2 Ricin poisoning can occur through a variety of methods. As an example, when dispersed in aerosol form, it is toxic at doses below 1 μg/Kg body weight.3 Further adding to its threat, ricin can be easily extracted from castor beans, and the by-products of castor oil extraction. The low toxic dosage and ease of acquisition make ricin a potential weapons threat, and indeed ricin has been used as a weapon, most notably in the assassination of Georgi Markov.4 Furthermore, there were numerous cases of attempted use of ricin in the first decade of the 21st century, including contaminated letters sent to Washington.5
Countermeasures for ricin have included vaccines, inhibitors of cellular trafficking, and competitive active-site inhibitors. The last of these has been the method of choice in our work, as wide-spread vaccination carries many practical challenges, and the target for the cellular trafficking inhibitors is often not understood, complicating rational drug design.6 The structure of ricin, and RTA specifically, is known and has been refined to high resolution. The mechanism of action is well understood from structural and mutagenesis work.7 With this sound foundation, RTA is a reasonable candidate for rationally designed active-site inhibitors. There is one major concern with such a design project, however. The native substrate is the ribosome, and the RTA binding site is, not surprisingly, large and mostly polar. There are some hydrophobic areas, in particular the adenine binding “specificity” pocket, but in general specific interactions will require suitably polar inhibitors, inevitably leading to a penalty in desolvation upon binding. To date, successful small molecule RTA inhibitors are most commonly based upon pterins, guanines, pyrimidines, and stem-loop oligonucleotides.3, 8 Schramm and his colleagues have developed transition state analogs in an RNA framework that have low Kd values, but only bind at acidic pH, around 4.9
The use of pterins as RTA inhibitors began with pteroic acid (PTA), giving an IC50 of 600 μM, and a binding mode nearly identical to that predicted by virtual screening programs.8b Despite the modest potency, PTA remained the strongest RTA inhibitor in the pterin series for over ten years. This changed when a preference for 7-substituted pterins was discovered, with 7-carboxy pterin (7CP) showing an IC50 of 230 μM.8a Several amide-derivatives of 7CP were explored in an attempt to build off the initial lead structure. Optimization was slow due to the notorious insolubility associated with the pterin heterocycle, as traditional coupling techniques proved fruitless due to solvent restrictions.8a, 10 Structures of a previous key pterin-based RTA inhibitors are shown in Figure 1 below.
Figure 1.
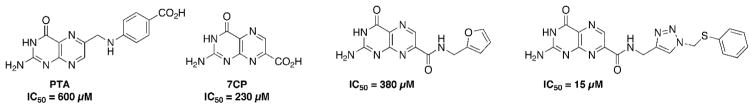
Key structures of previous RTA inhibitors
A great improvement in the complexity and rate of production of new pterin amides came through the use of 1,8-diazabicyclounde-7-cene (DBU), which we discovered converts 7-methoxycarbonyl-pterin (1) to the methanol-soluble DBU salt, and further activates the carbonyl in situ to catalyze amidation.11 No longer limited in the complexity of accessible pterin amides, we synthesized a variety of expanded furanyl and triazole linked pterins.11a While most derivatives exhibited IC50 values in the range of 150–400 μM, several derivatives showed IC50’s in the 15–70 μM range, and the improvements attributed to the triazole ring forming a stronger polar contact with the Tyr-80 residue. Many of the optimization strategies were aimed at accessing the so-called “secondary binding pocket” of RTA.6a, 9 In the ligand-bound form of ricin, the side-chain of Tyr-80 rotates from a “closed” conformation blocking access to the specificity pocket, to an open form. This allows access to the specificity pocket and divides the active-site into two pockets, with the secondary binding pocket accommodating an adjacent guanosine on RNA.9
In the practice of optimizing drug candidates, peptides are of particular interest due to their commercial availability and the diverse structural characteristics provided by individual amino acids.12 We postulated that the use of peptides would be a convenient method of mapping the active site of RTA for potential favorable interactions. We report herein the use of di- and tripeptide conjugated pterins, with the general design depicted in Scheme 1 and 2, with an emphasis on aromatic side-chains, as competitive binding inhibitors of RTA. Within the series, the best structure showed an IC50 of 6 μM.
Scheme 1.
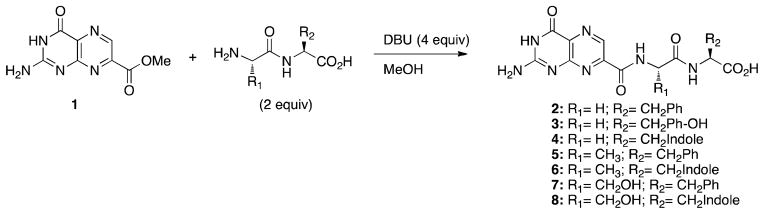
Synthesis of dipeptide conjugates 2-8. See Table 1 for full structures
Scheme 2.
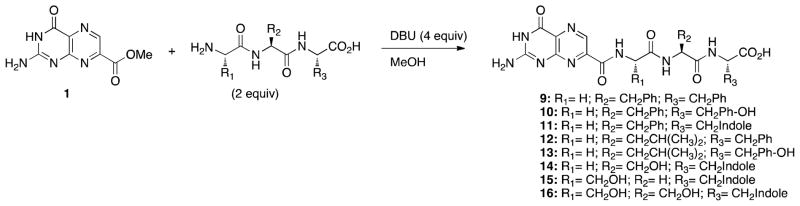
Synthesis of tripeptide conjugates 9-16. See Table 2 for full structures.
2) Chemistry
The synthesis of all compounds used in this study was carried out as shown in Schemes 1 and 2. Synthesis began with the construction of 7-methoxycarbonyl-pterin (1), which was prepared via acyl radical insertion.10 Compound 1 was suspended in a minimal amount of dry methanol, and solubilized through the addition of DBU, which also catalyzes the amidation reaction.11 Commercially available dipeptides (Scheme 1) or tripeptides (Scheme 2) were then added in a slight excess (2 equiv.). Through use of at least 4 equivalents of DBU the carboxylic acid of the peptides is deprotonated, while sufficient DBU remains for the dual role of solubility and nucleophilic catalysis. Yields for the reactions ranged from 29–98 %.
3) Results and Discussion
3.1. Dipeptide conjugated pterins
Previous results from our laboratory showed that bulky aromatic groups in the immediate proximity of the pterin head group were not well accommodated in the active site, while groups with extended linker length could better access favorable binding interactions.8a With those results in mind, we opted to begin exploring dipeptides rather than single amino acids attached to the pterin core. Strengthening this design plan, modeling by the virtual docking programs GOLD and ICM showed dipeptides were an appropriate length to move toward the secondary binding pocket. The first amino acid, which we shall refer to as P1, appeared by modeling to solely serve the role of a simple spacer; three representative P1 amino acids were chosen: glycine, alanine, and serine. Glycine represents a simple linker; alanine provides a more hydrophobic linker and introduces a source of conformational restrictions due to α-substitution; serine serves as a polar linker capable of hydrogen bonding. For the second amino acid, P2, emphasis was placed on aromatic side chains, which could, in principle, stack with the Tyr-80 residue that separates the two pockets.
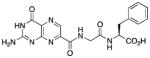
Containing a glycine-phenylalanine dipeptide, compound 2 represents the simplest of structures that meets the above criteria for our inhibitor design. Screening compound 2 against RTA in a luciferase-translational assay, we observed an IC50 of 20 μM (Figure 2) (See Experimental Section for details). This represents a ten-fold improvement in potency over the lead structure 7-carboxy-pterin (7CP), which had an IC50 of 200 μM.8a Intrigued as to the nature of the improved potency, a solution of 2 was soaked into pre-formed RTA crystals, and X-ray data collected. The binding mode of 2 is shown in Figure 3. As seen previously with pterin/RTA complexes, the pterin makes all the expected interactions with RTA. These include a tautomerization of the lactam NH, allowing for a hydrogen bond from the N1 position of the pterin to the carbonyl of Gly-121, and the N3 position accepting a hydrogen bond from Val-81. Furthermore, an amide NH from the peptide forms a hydrogen bond bridge with a water molecule to the side chain of Glu-177. In addition to these favorable polar interactions, an unexpected edge-to-face interaction between the aromatic side chain of the inhibitor’s P2 group with the invariant Trp-211 residue of RTA was observed. To accommodate the space filled by the bulky aromatic ring, the Tyr-80 residue has rotated by roughly 45° compared to that seen for previous 7-substituted pterins. This π-interaction was not predicted by the docking programs GOLD and ICM, highlighting the utility of empirically mapping the active site for favorable interactions.
Figure 2.
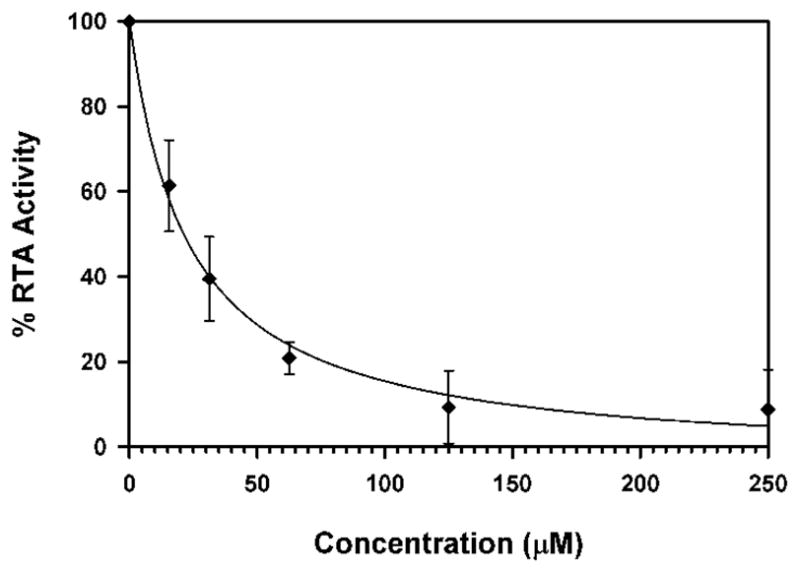
Dose-response curve for compound 2 with RTA. RTA activity was determined through normalization of luciferase counts in the presence and absence of 2. Percent activity is plotted as a function of inhibitor concentration.
Figure 3.
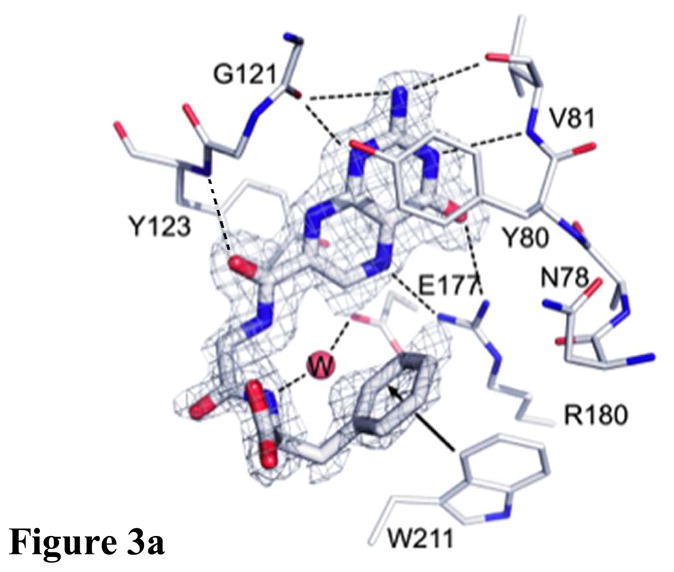

Figure 3a. Binding of 2 to RTA showing unexpected edge-to-face interaction
Figure 3b. Comparison of inhibitors with Gly-Gly and Gly-Phe pendants. The RTA active site is shown as the thinner bonds and the ligands with thicker bonds. Carbons of 2 are light, while those of the Gly-Gly ligand are dark. Waters are shown as spheres.
In trying to analyze the effects of chemical modification on ligand binding it is important to recall that a change in the free energy of binding of just 1.4 Kcal/mole changes binding affinity by a factor of 10. A three fold change in affinity is the result of a difference of just 0.6 Kcal/mole. The NG of binding reflects differences in enthalpy, typified by changes in hydrogen bond strength, and also reflects entropy. The latter is difficult to assess as it involves configurational entropy of the ligand as well as changes in solvation of both the ligand and the protein. In short, caution is required in interpreting how changes in this series of ligands affect the observed IC50 values. One valid assumption is that when all the atoms of a bound ligand can be seen in the X-ray structure it suggests that specific interactions are made that can compensate for the entropic cost of freezing out bond rotations within that ligand.
The IC50 for 7CP is about 200 μM, that for a pterin derivative bearing solely a Gly pendant is about 600 μM (data not shown), and that for the Gly-Phe derivative 2 is 20 μM. All three make the same strong interactions between the pterin and the RTA specificity pocket, however it appears that attaching only the P1 Gly linker is energetically unfavorable, likely for entropic reasons. However, adding the P2 Phe residue substantially improves binding. Presumably the favorable edge-to-face interaction can subsidize the cost of freezing the inhibitor and also contributes enough favorable interaction to drive the binding to a 30 fold increase over the monopeptide derivative. We can also compare compound 2, with its Gly-Phe pendant, to one with Gly-Gly. Such a compound has an IC50 of 300 μM; its binding is superimposed on 2 in Figure 3b. The pterin binding is identical and the pendant chains are similar. The C-terminal carboxylates occupy the same volume of space. The added phenyl side chain of 2 makes the edge-to-face interaction with the side chain of Trp211 as described above. Note that the side chain of Tyr 80 is displaced upward in 2 to make space for the ligand phenyl group. The phenyl group at P2 of compound 2 also displaces two waters, labeled W1 and W2, present in the Gly-Gly complex. It is plausible that the enthalpically favorable edge-to-face interaction combined with the entropically favorable release of water drives the binding of 2, which is about 2 Kcal/mol more favorable than that of the Gly-Gly ligand.
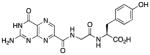
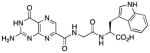
Encouraged by the above result, we synthesized compounds 3 and 4, which vary the π-donor ability of the aromatic ring.13 The tyrosine-terminated compound 3 performed quite well in the in vitro assay, showing an IC50 of 6 μM. X-ray analysis revealed compound 3 shared an identical binding mode as 2, displaying the same edge-to-face interaction. The indole ring of tryptophan has been identified as one of the best aromatic π-donors, and thus would be expected to further strengthen the edge-to-face interaction thereby strengthening inhibitor binding.13 However, the P2 tryptophan compound 4 showed slightly diminished potency compared to the previous two structures, with an IC50 around 40 μM. Examination of the crystal structure of the RTA/4 complex revealed a somewhat disordered binding, as the pterin ring was firmly locked in place and clearly visible, but the peptide showed very limited density, indicating non-specific interactions. Some evidence of the placement of the tryptophan side chain was observed through displaced water molecules previously seen in the active site. While 4 was an unexpectedly weaker inhibitor than 2 or 3, it is still superior to the vast majority of RTA inhibitors which function at physiological pH.3, 8a, b, 8d
Our next goal was to change the linkers for the phenylalanine and tryptophan based compounds as a means to explore side chain steric and hydrogen bonding effects. Compounds 5-8 were synthesized and tested for their activity against RTA. The use of an alanine as P1 produced stronger inhibitors than serine, as compounds 5 and 6 had IC50’s around 20 μM, while 7 and 8 had IC50’s closer to 100 μM (see Table 1). In this way, the slightly less potent P2 tryptophan compound 4 was improved through the use of alanine (eg; 6). Several attempts were made to synthesize an Ala-Tyr linked pterin, as it was an attractive target given the above results for alanine and the potency of 3. However, the alanine series proved more challenging synthetically. It appears the branched α-carbon severely diminishes reactivity, and compound 5 and 6 could only be synthesized through use of DMSO as a co-solvent at elevated temperatures. The serine series does not suffer such steric limitations, presumably due to the side-chain hydroxyl further activating the pterin carbonyl via hydrogen bonding. The use of numerous forcing conditions never proved successful in the synthesis of an Ala-Tyr derivative.14 While inhibition results from the serine-linked compounds (7-8) were less than satisfactory, some interesting structural features were observed when these compounds were soaked into RTA crystals and X-ray data collected. The hydroxyl group of P1 serine forms a hydrogen bond with the side chain of Asn-209, which appears to pull the di-peptide “out of place” from making the edge-to-face interaction just described for 2 and 3. This likely explains the decreased potency, and suggests that the gain of a P1 hydrogen bond contributes less to the binding affinity than the hydrophobic π-interaction. Interestingly, in the case of the serine-tryptophan conjugated pterin (8), the “out of place” positioning led to the ability to make a cation-π interaction with Arg-258 (Figure 4). However, this alternative π-interaction cannot compensate for the loss of the edge-to-face interaction. It is possible the glycine-tryptophan conjugated pterin (4) showed disordered binding due to the indole ring being partially involved in a cation-π, and partially in the edge-to-face interaction.
Table 1.
Summary of dipeptide conjugates
| Entry | Structure | IC50 (μM) |
|---|---|---|
| 2 |
|
20 |
| 3 |
|
6 |
| 4 |
|
40 |
| 5 |
|
21 |
| 6 |
|
19 |
| 7 |
|
90 |
| 8 |
|
115 |
Figure 4.

X-ray structure of the RTA/8 complex showing the cation-π interaction
To accommodate the anionic phosphate backbone in the natural RNA substrate, the linker space between the two binding pockets of RTA is lined with positive charge with three exposed arginine residues; in addition to the catalytic R180, these include arginines 134, 215, and 258.6a It is of note that compound 8 represents the first clear interaction with an arginine residue in the space between the two binding pockets and it is a cation-π interaction. To date, none of the terminal carboxylate of the di-peptides formed a salt bridge with a guanidinium of arginine. While the π-interactions observed in this study are rooted in electrostatics, one might not expect such a polar active site to show greater affinity for ligands seeking interactions more commonly associated with hydrophobic environments. When comparing the cation-π interaction of compound 8 to the edge-to-face interactions of 2 or 3, the diminished activity of 8 likely stems from the enthalpic penalty of desolvating a charged arginine; a penalty which was not compensated by the π-interaction. Desolvation of an aromatic side chain however, occurring for the edge-to-face interaction, should not carry a significant enthalpic penalty, leading to a net gain in binding strength for 2 and 3.
3.2. Tripeptide conjugated pterins
The results from the dipeptide series indicated that access to the second pocket would require a longer peptide pendant. With the terminal carboxylate of the dipeptides not involved in any strongly contributing interaction, further diversification to tripeptides could potentially give rise to increased binding affinity. Our efforts with the tripeptide derivatives can be divided into two main categories: those that build directly off results seen in the dipeptide series, and those suggested by virtual modeling of potential inhibitors. Compounds 9-13 were synthesized towards the first goal, while compounds 14-16 fit the latter direction (Scheme 2). Alanine was not explored as a linker in this series due to the synthetic challenges encountered in the dipeptide series. The results are summarized in Table 2.
Table 2.
Summary of tripeptide conjugates
| Entry | Structure | IC50 (μM) |
|---|---|---|
| 9 |
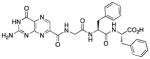
|
15 |
| 10 |
|
25 |
| 11 |
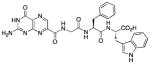
|
35 |
| 12 |
|
35 |
| 13 |
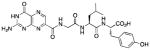
|
22 |
| 14 |
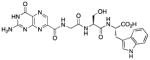
|
60 |
| 15 |
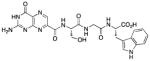
|
55 |
| 16 |
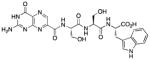
|
100 |
The Gly-Phe-Phe conjugated pterin 9 exhibited an IC50 of 15 μM, the same level of potency as the parent dipeptide 2. X-ray analysis for the RTA/9 complex revealed the P2 aromatic side chain was again making the energetically favorable edge-to-face interaction, while the terminal P3 aromatic side chain formed a slipped-stacked π-interaction with the P2 benzene ring (Figure 5). As the terminal end of the ligand is merely interacting with the ligand itself, and not the protein, it is not surprising that 9 should have similar potency as 2. In a relatively solvent-exposed area, the P3 ring is best able to interact with the inhibitor, not the protein target.
Figure 5.
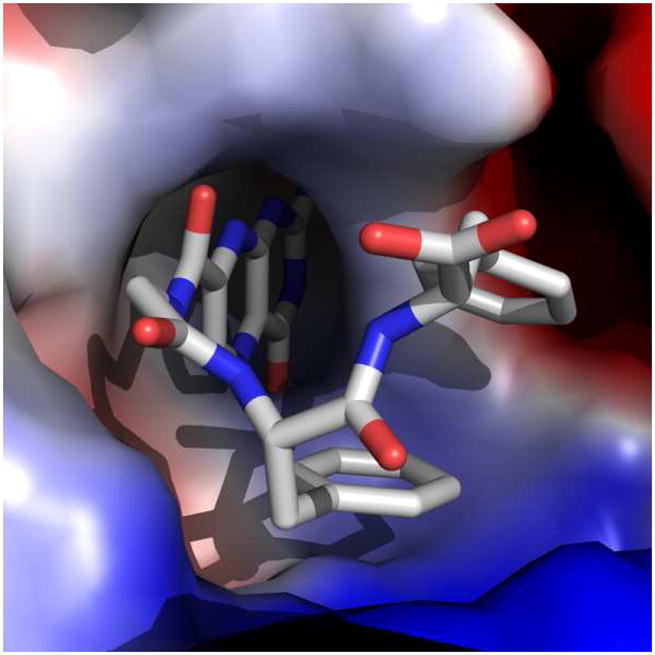
Surface representation of the RTA/9 complex, showing edge-to-face and slipped-stacked π-interactions. The RTA surface is shown as an electrostatic map.
Compounds 10 and 11 are structurally similar to 9, but vary the π-density at P3 and also have a potential for P3 to make an advantageous polar favorable polar interaction within the active site. The P3 tyrosine-terminated compound 10 showed activity on par with 2, and only slightly less potent than 9. The tryptophan-terminated pterin (11) had an IC50 of 35 μM, weaker still than the parent tripeptide conjugate 9. Efforts to obtain suitable crystal structures for both 10 and 11 have been unsuccessful to date.
In an effort to bypass the intramolecular slipped stack seen for 9, the tripeptide conjugate 12 was synthesized, containing a Gly-Leu-Phe pendent (Scheme 2). By replacing the P2 amino acid with leucine, we should replace the edge-to-face interaction with a generic hydrophobic interaction, reduce the P2-P3 stacking, and perhaps allow P3 to bind deeper in the RTA second pocket. Compound 12 displayed an IC50 of 35 μM; although slightly weaker than 9, 12 still represents a relatively strong interaction. The Gly-Leu-Tyr conjugate 13 was also synthesized; it had an IC50 around 20 μM. Crystallographic data were collected for the complexes of RTA with both of these modified tripeptide conjugates. In both cases the leucine side chain was clearly observed filling the space once occupied by the aromatic side chains of 2, 3, and 9, however neither structure displayed strong density for the terminal P3 amino acid. Without the driving force to slip-stack, the aromatic ring of P3 must rotate freely and may occupy numerous partially stabilizing geometries.
Modeling was undertaken to identify a tripeptide conjugate with the best potential of accessing the secondary binding pocket. Despite tryptophan being the least promising aromatic amino acid explored with the dipeptides, it was predicted to be the most promising as a third amino acid in accessing the second pocket by π-stacking with Tyr-80. In the choice of dipeptide linkers to a tryptophan, serine appeared best in the virtual docking studies. While serine-based compounds gave the poorest performance in the dipeptides, modeling suggested promise when serine was used for a tripeptide conjugate. Thus we chose to examine variations on glycine and serine as the first two amino acids of a family of tripeptides (eg; Gly-Ser for 14, Ser-Gly for 15, and Ser-Ser for 16). As summarized in Table 2, these structures showed inhibition in the range of 55–100 μM, worse than that seen for the dipeptide conjugates 2-6, and all previous tripeptides conjugates. Indeed these results were partly anticipated by earlier results with serine.
While it is easy to simply view the results from compounds 14-16 as an example of the inaccuracies of virtual drug modeling, it is more interesting when viewed in the context of the apparent preference towards hydrophobicity over hydrogen bonding seen previously. While there is some difference in the terminal aromatic group, the primary difference between compound 13 and the poorer performing compound 14 is replacement of the hydrophobic isopropyl group with a hydroxyl moiety, and yet the more hydrophobic structure is three times more potent. This further highlights a stark difference in the design of pterin inhibitors over previous reports which attributed improved potency to increased hydrogen bonding.8a, 11a
4) Conclusion
In recent years we have seen a two-order of magnitude improvement in the potency of pterin-based RTA inhibitors (600 μM for pteroic acid, 6 μM for 3). This optimization has been facilitated by the rapid generation of diverse structures through the use of di- and tripeptide conjugated pterins. The majority of compounds reported here had IC50’s at or below 50 μM. As such, these are some of the best competitive RTA inhibitors capable of functioning at physiological pH. Much of the success of these compounds over previous pterin inhibitors appears to stem from an unexpected preference towards hydrophobic and π-interactions, despite the primarily polar surface of the active site. These structures should guide future development of pterin-based RTA inhibitors, and can be transitioned to various forms of peptide isosteres for further optimization.
5) Experimental Details
All reagents used were of commercial quality and were obtained from Aldrich Chemical Co. and Fisher Scientific and were used as received. Peptides were purchased through GenScript and BAChem, and used as received. 1H and 13C NMR spectra were recorded in DMSO-d6 on a Varian spectrometer using the solvent as reference. Chemical shifts are given in parts per million (ppm). LC-MS data was recorded on an Agilent 6130 Quadrupole instrument. High resolution mass spectrometry was performed with a Varian 9.4T QFT-ESI ICR system. All solvents were removed by rotary evaporation under vacuum using a standard rotovap equipped with a dry ice condenser. All filtrations were performed with a vacuum. Purity of all final compounds was determined to be >95% on a Shimadzu HPLC with a 4.6 × 150 mm Phenomenex gemini 5 μm C18 column. The eluents were A, water + 0.1% TFA, and B, acetonitrile + 0.1% TFA. Gradient elution from 5% B to 50% B over 25 min with a final hold at 90% B for 5 min. Total run time was 30 min.
5.1. Synthesis
7-methoxycarbonyl-pterin (1) was synthesized by our previously reported acyl-radical insertion.10
General procedure for synthesis of peptide conjugated pterins
To a slurry of 7-methoxycarbonyl-pterin (1) in MeOH (0.5–1.0 mM) was added 4 eq. DBU to give a clear solution. Two equivalents of the corresponding peptide were then added to the mixture, which was stirred at room temperature for 4–24 h. Reaction completion was judged by LCMS analysis. After the reaction was complete, dilute aqueous HCl was added dropwise to afford a yellow precipitate. The solid was collected by filtration and washed several times with water and methanol to provide the product. In instances of remaining starting material, or the hydrolyzed byproduct 7-carboxy-pterin (7CP), the crude solid was redissolved in 0.5 mM aqueous NaOH and loaded onto an LH-20 gel filtration column and eluted with 0.1 M NaCl. The pure fractions were pooled and precipitated with HCl, followed by filtration to provide the pure final product.
N-{N-(pterin-7-yl)carbonylglycyl}-L-phenylalanine (2)
This compound was synthesized in 59% yield using the above method. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.80 (s, 1H), 11.59 (s, 1H), 8.86 (t, J = 5.9 Hz, 2H), 8.84 (s, 1H), 8.32 (d, J = 7.9 Hz, 1H), 7.28-7.12 (m, 5H), 7.01 (br. s, 2H), 4.44 (ddd, J = 8.5, 7.9, 5.1 Hz, 1H), 3.96 (dd, J = 16.7, 5.9 Hz, 1H), 3.88 (dd, J = 16.7, 5.9 Hz, 1H), 3.03 (dd, J = 13.8, 5.1 Hz, 1H), 2.87 (dd, J = 13.8, 8.5 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 172.7, 168.2, 162.8, 160.4, 156.1, 154.5, 147.2, 137.4, 136.5, 131.9, 129.2 (2C), 128.2 (2C), 126.5, 53.6, 42.1, 36.8; HR-ESIMS m/z [M−H]−: Calcd for (C18H16N7O5)− 410.12184; found 410.12152.
N-{N-(pterin-7-yl)carbonylglycyl}-L-tyrosine (3)
This compound was synthesized in 56% yield using the general method. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.76 (br. s, 1H), 11.77 (s, 1H), 9.20 (s, 1H), 8.90 (t, J = 5.9 Hz, 1H), 8.87 (s, 1H), 8.23 (d, J = 8.0 Hz), 7.12 (br. s, 2H), 6.99 (dt, J = 8.5, 2.0 Hz), 6.62 (dt, J = 8.5, 2.0 Hz), 4.37 (ddd, J = 8.4, 8.2, 5.1 Hz, 1H), 3.98 (dd, J = 16.8, 5.9 Hz, 1H), 3.90 (dd, J = 16.8, 5.9 Hz, 1H), 2.93 (dd, J = 13.8, 5.1 Hz, 1H), 2.78 (dd, J = 13.8, 8.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 172.9, 168.1, 162.8, 160.5, 155.9, 154.6, 147.2, 136.5, 131.9, 130.1 (2C), 127.4, 115.0 (2C), 109.6, 53.9, 42.1, 36.1; HR-ESIMS m/z [M−H]−: Calcd for (C18H16N7O6)− 426.11675; found 426.11625.
N-{N-(pterin-7-yl)carbonylglycyl}-L-tryptophan (4)
This compound was synthesized in 99% yield using the general method. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.72 (br. s, 1H), 11.67 (s, 1H), 10.86 (d, J = 2.0 Hz, 1H), 8.89 (t, J = 5.8 Hz, 1H), 8.87 (s, 1H), 8.30 (d, J = 7.8 Hz, 1H), 7.53 (dt, J = 7.9, 1.0 Hz, 1H), 7.32 (dt, J = 8.0, 1.0 Hz, 1H), 7.16 (d, J = 2.0 Hz, 1H), 7. 05 (br. s, 2H), 7.04 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.96 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H), 4.51 (td, J = 7.8, 5.2 Hz, 1H), 4.00 (dd, J = 16.7, 5.8 Hz, 1H), 3.93 (dd, J = 16.7, 5.8 Hz, 1H), 3.19 (dd, J = 14.6, 5.2 Hz, 1H), 3.05 (dd, J = 14.6, 7.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 173.1, 168.0, 162.6, 160.3, 155.9, 154.4, 147.1, 136.4, 135.9, 131.8, 127.1, 123.6, 120.7, 118.2, 118.0, 111.2, 109.4, 52.9, 42.0, 27.0; HR-ESIMS m/z [M−H]−: Calcd for (C20H17N8O5)− 449.13274; found 449.13204.
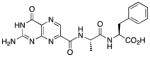
N-{N-(pterin-7-yl)carbonyl-L-alanyl}-L-phenylalanine (5)
This compound was synthesized in 55% yield using a modified version of the above method, whereby the reaction temperature was raised to 80 °C in a sealed vessel DMSO as a co-solvent. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.80 (br. s, 1H), 11.61 (s, 1H), 8.85 (s, 1H), 8.61 (d, J = 7.9 Hz, 1H), 8.47 (d, J = 8.0 Hz), 7.27-7.14 (m, 5H), 7.08 (br. s, 1H), 4.53 (dq, J = 7.9, 7.0 Hz, 1H), 4.46 (ddd, J = 9.2, 8.0, 5.0 Hz, 1H), 3.09 (dd, J = 13.8, 5.0 Hz, 1H), 2.91 (dd, J = 13.8, 9.2 Hz, 1H), 1.32 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 172.6, 171.4, 161.7, 160.3, 155.9, 154.5, 146.9, 137.4, 136.3, 132.0, 129.1 (2C), 128.1 (2C), 126.4, 53.5, 48.1, 36.6, 18.8; HR-ESIMS m/z [M−H]−: Calcd for (C19H18N7O5)− 424.13749; found 424.13692.
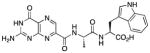
N-{N-(pterin-7-yl)carbonyl-L-alanyl}-L-tryptophan (6)
This compound was synthesized in 30% yield using the same modified procedure reported for 5. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.73 (br. s, 1H), 11.63 (s, 1H), 10.83 (d, J = 2.4 Hz, 1H), 8.85 (s, 1H), 8.64 (d, J = 7.8 Hz, 1H), 8.45 (d, J = 8.0 Hz, 1H), 7.53 (dt, J = 7.9, 1.0 Hz, 1H), 7.31 (dt, J = 8.0, 1.0 Hz, 1H), 7.16 (d, J = 2.4 Hz, 1H), 7.05 (br. s, 2H), 7.03 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.94 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H), 4.61-4.53 (m, 1H), 4.52 (ddd, J = 8.2, 8.0, 5.0, 1H), 3.20 (dd, J = 14.8, 5.0 Hz, 1H), 3.06 (dd, J = 14.8, 8.2 Hz, 1H), 1.34 (d, J = 7.0 Hz, 3H); 13C NMR (126 MHz, DMSO-d6) δ/ppm 172.9, 171.4, 161.6, 160.2, 155.9, 154.4, 146.9, 136.6, 135.9, 131.9, 127.1, 123.6, 120.8, 118.2, 118.0, 111.3, 109.5, 52.9, 40.0, 26.9, 18.7; HR-ESIMS m/z [M−H]−: Calcd for (C21H19N8O5)− 463.14839; found 463.14804.
N-{N-(pterin-7-yl)carbonyl-L-serinyl}-L-phenylalanine (7)
This compound was synthesized in 68% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.81 (br. s, 1H), 11.83 (s, 1H), 8.86 (s, 1H), 8.59 (d, J = 8.2 Hz, 1H), 8.37 (d, J = 8.3 Hz, 1H), 7.70-6.60 (m, 7H), 5.07 (br. s, 1H), 4.53 (dt, J = 8.2, 5.1 Hz, 1H), 4.46 (td, J = 8.3, 5.2 Hz, 1H), 3.71 (dd, J = 11.1, 5.1 Hz, 1H), 3.66 (dd, J = 11.1, 5.1 Hz, 1H), 3.07 (dd, J = 13.8, 5.2 Hz, 1H), 2.91 (dd, J = 13.8, 8.3 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 172.6, 169.2, 162.2, 160.2, 156.0, 154.8, 146.9, 137.4, 136.3, 132.0, 129.2 (2C), 128.2 (2C), 126.4, 61.7, 55.0, 53.6, 36.8; HR-ESIMS m/z [M−H]−: Calcd for (C19H18N7O6)− 440.13240; found 440.13191.
N-{N-(pterin-7-yl)carbonyl-L-serinyl}-L-tryptophan (8)
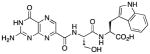
This compound was synthesized in 61% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.74 (br. s, 1H), 11.78 (br. s, 1H), 10.86 (d, J = 2.3 Hz, 1H), 8.88 (s, 1H), 8.61 (d, J = 8.1 Hz, 1H), 8.38 (d, J = 7.9 Hz, 1H), 7.52 (dd, J = 7.8, 1.0 Hz, 1H), 7.30 (dt, J = 7.8, 1.1 Hz, 1H), 7.21 (br. s, 2H), 7.16 (d, J = 2.3 Hz, 1H), 7.03 (ddd, J = 8.2, 7.8, 1.0 Hz, 1H), 6.94 (td, J = 8.2, 7.8, 1.1 Hz, 1H), 4.62-4.46 (m, 2H), 3.78-3.65 (m, 2H), 3.19 (dd, J = 14.7, 5.4 Hz, 1H), 3.07 (dd, J = 14.7, 7.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 173.0, 169.2, 162.1, 160.2, 155.1, 154.5, 146.9, 136.6, 136.0, 132.0, 127.3, 123.8, 120.9, 118.4, 118.2, 111.4, 109.5, 61.8, 54.9, 53.1, 27.1; HR-ESIMS m/z [M−H]−: Calcd for (C21H19N8O6)− 479.14330; found 479.14315.
N-[N-{N-(pterin-7-yl)carbonylglycyl}-L-phenylalanyl]-L-phenylalanine (9)
This compound was synthesized in 70% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.77 (br. s, 1H), 11.62 (s, 1H), 8.86 (s, 1H), 8.83 (t, J = 5.7 Hz, 1H), 8.40 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 8.5 Hz, 1H), 7.30-7.13 (m, 10H), 7.09 (br. s, 2H), 4.58 (ddd, J = 9.4, 8.5, 4.2 Hz, 1H), 4.44 (ddd, J = 8.8, 7.8, 5.3 Hz, 1H), 3.94 (dd, J = 16.7, 5.7 Hz, 1H), 3.82 (dd, J = 16.7, 5.7 Hz, 1H), 3.08 (dd, J = 13.9, 5.3 Hz, 1H), 3.00 (dd, J = 13.9, 4.2 Hz, 1H), 2.93 (dd, J = 13.9, 8.8 Hz, 1H), 2.73 (dd, J = 13.9, 9.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 172.7, 170.9, 167.8, 162.7, 160.4, 156.1, 154.5, 147.2, 137.6, 137.4, 136.5, 131.9, 129.3 (2C), 129.1 (2C), 128.2 (2C), 128.0 (2C), 126.5, 126.3, 53.6 (2 x CH2Ph), 42.2, 37.6, 36.7; HR-ESIMS m/z [M−H]−: Calcd for (C27H25N8O6)− 557.19025; found 557.19016.
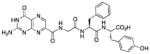
N-[N-{N-(pterin-7-yl)carbonylglycyl}-L-phenylalanyl]-L-tyrosine (10)
This compound was synthesized in 78% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.67 (s, 1H), 11.59 (s, 1H), 9.20 (s, 1H), 8.85 (s, 1H), 8.83 (t, J = 5.8 Hz, 1H), 8.31 (d, J = 7.8 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 7.22-7.12 (m, 5H), 7.10 (br. s, 2H), 7.02 (d, J = 8.5 Hz, 2H), 6.65 (d, J = 8.5 Hz, H), 4.58 (ddd, J = 4.2, 8.4, 9.4 Hz, 1H), 4.35 (ddd, J = 5.4, 7.8, 8.6 Hz, 1H), 3.93 (dd, J = 5.8, 16.6 Hz, 1H), 3.83 (dd, J = 5.8, 16.6 Hz, 1H), 3.00 (dd, J = 4.2, 13.9 Hz, 1H), 2.94 (dd, J = 5.4, 14.1 Hz, 1H), 2.81 (dd, J = 8.6, 14.1 Hz, 1H), 2.73 (dd, J = 9.4, 13.9 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 173.1, 171.2, 168.3, 163.1, 160.8, 156.1, 154.7, 147.4, 137.7, 136.9, 132.0, 130.3 (2C), 129.5 (2C), 128.3 (2C), 127.7 (2C), 126.6 (2C), 115.3, 54.2, 53.9, 42.5, 37.7, 36.2; HR-ESIMS m/z [M−H]−: Calcd for (C27H25N8O7)− 573.18517; found 573.18521.
N-[N-{N-(pterin-7-yl)carbonylglycyl}-L-phenylalanyl]-L-tryptophan (11)
This compound was synthesized in 78% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.67 (s, 1H), 11.59 (s, 1H), 10.87 (s, 1H), 8.86 (s, 1H), 8.83 (t, J = 5.9 Hz, 1H), 8.36 (d, J = 7.8 Hz, 1H), 8.21 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.33 (1H, d, J = 7.9 Hz, 1H), 7.24-7.13 (m, 8H), 7.06 (dd, J = 7.1, 7.9 Hz, 1H), 6.98 (dd, J = 7.1, 7.8 Hz, 1H), 4.61 (ddd, J = 4.1, 8.5, 9.2 Hz, 1H), 4.50 (ddd, J = 5.4, 7.8, 8.1Hz, 1H), 3.93 (dd, J = 5.7, 16.8 Hz, 1H), 3.84 (dd, J = 5.7, 16.8 Hz, 1H), 3.20 (dd, J = 5.4, 14.7 Hz, 1H), 3.07 (dd, J = 8.1, 14.7 Hz, 1H), 3.02 (dd, J = 4.1, 13.8 Hz, 1H), 2.74 (dd, J = 9.2, 13.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 173.1, 170.9, 167.9, 162.7, 160.3, 155.7, 154.4, 147.1, 137.6, 136.6, 136.0, 131.8, 129.3 (2C), 127.9 (2C), 127.2, 126.2, 123.7, 120.9, 118.4, 118.1, 111.3, 109.6, 53.6, 52.9, 42.2, 37.6, 27.0. HR-ESIMS m/z [M−H]−: Calcd for (C29H26N9O6)− 596.20115; found 596.20119.
N-[N-{N-(pterin-7-yl)carbonylglycyl}-L-leucinyl]-L-phenylalanine (12)
This compound was synthesized in 97% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ(ppm)= 8.80 (s, 1H), 7.20 – 7.09 (m, 5H), 4.35 – 4.27 (m, 2H), 3.87 (d, J = 5.7 Hz, 2H), 2.87 (m, 2H), 1.50 (m, 1H), 1.33 (m, 2H), 0.79 (dd, J = 12.6, 6.5 Hz, 6H); 13C NMR (125 MHz, DMSO-d6) δ(ppm)= 173.3, 172.2, 168.2, 163.2, 158.5, 155.1, 138.0, 137.7, 136.8, 132.3, 129.5 (2C), 128.5 (2C), 126.8, 109.9, 53.9, 51.2, 42.6, 41.4, 36.9, 24.5, 23.5, 22.1; HR-ESIMS m/z [M+H]+: Calcd for (C24H29N8O6)+ 525.22046; found 525.22028.
N-[N-{N-(pterin-7-yl)carbonylglycyl}-L-leucinyl]-L-tyrosine (13)
This compound was synthesized in 84% yield using the general method. 1H NMR (400 MHz, DMSO-d6) δ(ppm)= 9.18 (s, 1H), 8.88 (t, J = 5.5 Hz, 1H), 8.12 (dd, J = 13.6, 8.0 Hz, 2H), 6.99 (d, J = 8.5 Hz, 2H), 6.61 (d, J = 8.5 Hz, 2H), 4.41 – 4.24 (m, 2H), 3.95 (d, J = 7.6 Hz, 2H), 2.94 – 2.74 (m, 2H), 1.56 (m, 1H), 1.39 (m, 2H), 0.84 (dd, J = 12.1, 6.6 Hz, 6H); 13C NMR (150 MHz, DMSO-d6) δ(ppm)= 172.7, 171.6, 167.7, 162.7, 160.3, 155.8, 154.4, 147.1, 138.4, 131.5, 129.9 (2C), 127.4, 114.9 (2C), 53.7, 52.8, 50.7, 42.1, 35.8, 23.9, 23.0, 21.6; HR-ESIMS m/z [M+H]+: Calcd for (C24H29N8O7)+ 541.21537; found 541.21531.
N-[N-{N-(pterin-7-yl)carbonylglycyl}-L-serinyl]-L-tryptophan (14)
This compound was synthesized in 29% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.68 (s, 1H), 11.61 (s, 1H), 10.86 (d, J = 2.4 Hz, 1H), 8.93 (t, J = 5.8 Hz, 1H), 8.88 (s, 1H), 8.13 (t, J = 7.6 Hz, 2H), 7.52 (dd, J = 7.8, 1.1 Hz, 1H), 7.32 (dt, J = 7.6, 1.1 Hz, 1H), 7.17 (d, J = 2.4 Hz, 1H), 7.09-7.00 (m, 3H), 6.97 (ddd, J = 8.0, 7.6, 1.1 Hz, 1H), 4.87 (br. s, 1H), 4.48 (td, J = 7.6, 5.5 Hz, 1H), 4.41 (dt, J = 7.6, 5.8 Hz, 1H), 4.01 (d, J = 5.8 Hz, 2H), 3.59 (dd, J = 10.9, 5.8 Hz, 1H), 3.54 (dd, J = 10.9, 7.6 Hz, 1H), 3.18 (dd, J = 14.7, 5.5 Hz, 1H), 3.07 (dd, J = 14.7, 7.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 172.9, 169.6, 168.1, 162.7, 160.3, 155.9, 154.3, 147.1, 136.4, 135.9, 131.8, 127.1, 123.7, 120.7, 118.2, 118.0, 111.2, 109.4, 61.6, 54.8, 52.8, 42.2, 26.9; HR-ESIMS m/z [M−H]−: Calcd for (C23H22N9O7)− 536.16477; found 536.16459.
N-[N-{N-(pterin-7-yl)carbonyl-L-serinyl}glycyl]-L-tryptophan (15)
This compound was synthesized in 34% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.69 (br. s, 1H), 11.66 (s, 1H), 10.85 (d, J = 2.4 Hz, 1H), 8.88 (s, 1H), 8.69 (d, J = 7.8 Hz, 1H), 8.42 (t, J = 5.9 Hz, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.51 (dd, J = 8.0, 1.1 Hz), 7.35-7.28 (m, 1H), 7.14 (d, J = 2.4 Hz, 1H), 7.05 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 6.97 (ddd, J = 8.1, 7.0, 1.1 Hz, 1H), 5.24 (s, 1H), 4.52 (ddd, J = 7.8, 5.9, 5.7 Hz, 2H), 4.45 (ddd, J = 8.3, 8.0, 5.3 Hz, 1H), 3.81 (dd, J = 16.8, 5.9 Hz, 1H), 3.67 (dd, J = 16.8, 5.7 Hz, 1H), 3.17 (dd, J = 14.7, 5.3 Hz, 1H), 3.17 (d, J = 5.9 Hz, 2H), 3.01 (dd, J = 14.7, 8.3 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 173.2, 169.6, 168.6, 162.4, 160.4, 156.0, 154.6, 147.0, 136.4, 136.1, 132.0, 127.2, 123.8, 120.9, 118.4, 118.1, 111.4, 109.6, 61.7, 55.2, 53.1, 42.0, 27.1; HR-ESIMS m/z [M−H]−: Calcd for (C23H22N9O7)− 536.16477; found 536.16471.
N-[N-{N-(pterin-7-yl)carbonyl-L-serinyl}-L-serinyl]-L-tryptophan (16)
This compound was synthesized in 36% yield using the general procedure. 1H NMR (400 MHz, DMSO-d6) δ/ppm 12.66 (s, 1H), 11.66 (s, 1H), 10.85 (d, J = 2.4 Hz, 1H), 8.88 (s, 1H), 8.67 (d, J = 7.9 Hz, 1H), 8.29 (d, J = 7.8 Hz, 1H), 8.07 (d, J = 7.7 Hz, 1H), 7.51 (dd, J = 7.8 Hz, 1H), 7.32 (dd, J = 7.8, 1.0 Hz, 1H), 7.16 (d, J = 2.4 Hz, 1H), 7.07 (br. s, 2H), 7.05 (ddd, J = 8.2, 7.8, 1.0 Hz, 1H), 6.97 (ddd, J = 8.2, 7.8, 1.0 Hz, 1H), 5.30 (br. s, 1H), 4.89 (br. s, 1H), 4.64 (dt, J = 7.8, 5.2 Hz, 1H), 4.51-4.33 (m, 2H), 3.82 (dd, J = 11.0, 5.2 Hz, 1H), 3.70 (dd, J = 11.0, 5.2 Hz, 1H), 3.66-3.53 (m, 2H), 3.17 (dd, J = 14.6, 5.2 Hz, 1H), 3.04 (dd, J = 14.6, 7.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ/ppm 173.1, 169.7, 169.4, 162.2, 160.3, 156.0, 154.6, 146.9, 136.3, 136.0, 132.1, 127.2, 123.8, 120.9, 118.4, 118.1, 111.4, 109.5, 62.0, 61.5, 55.1, 54.8, 53.2, 27.0; HR-ESIMS m/z [M−H]−: Calcd for (C24H24N9O8)− 566.17533; found 566.17516.
5.2. In Vitro Luciferase Assay
The compounds to be tested were solubilized in 0.05 N KOH prior to their inclusion in the assay. After mixing RTA and various concentrations of compounds in the presence of BSA, translation reactions were initiated by mixing a portion of the RTA/compound mixture with reticulocyte lysate, and translation mix (amino acids, RNasin ribonuclease inhibitor, luciferase control mRNA). The reactions were incubated for 90 minutes at 30 °C, after which they were stopped by freezing them at −20 °C. After thawing at room temperature, 2.5 μl reaction mixture was mixed with 40 μl Luciferase Substrate Reagent (Promega), and their luminescence was measured on a Perkin Elmer Envision luminometer (Waltham MA).
For each concentration of inhibitor to be tested, reactions were run both in the presence and absence of RTA. To calculate the RTA activity, corrected for interference with the assay by the tested compounds, the percent differences in luminescence for the corresponding reaction pairs were divided by the percent difference for the controls with no inhibitor present. Values for IC50 were calculated by fitting the plot of RTA activity vs. inhibitor concentration to a hyperbolic decay function.
5.3. X-ray crystallography
Tetragonal RTA crystals were grown at 4°C using the sitting drop method from 7 mg/mL RTA and 1.3 M ammonium sulfate, 0.1 M sodium malonate, pH 5.0. For the purpose of determining the structure of RTA in complex with a compound, a crystal was transferred to a reservoir containing 1.5 mM compound in artificial mother liquor (1.3 M ammonium sulfate, 0.1 M sodium malonate, pH 5.0). Prior to data collection, a crystal was transferred briefly to a drop of 1.3 M ammonium sulfate, 1.3 M malonate, pH 6, for cryoprotection. The crystal mounted in a cryoloop (Hampton Research, Laguna Niguel, CA) was flash frozen in liquid nitrogen and mounted in the cold stream on the goniostat.
X-ray diffraction data were collected from a crystal soaked with 9 at 100 K on an R-AXIS IV++ image plate detector (Rigaku, The Woodlands, TX) with X-rays generated by a Rigaku MicroMax-007 HF rotating anode generator operated at 40 kV, 30 mA. Diffraction data from crystals soaked with 2, Pterin-Gly-Gly, and 8 were collected at 100 K at the Advanced Light Source beamline 5.0.2 at the Lawrence Berkeley National Laboratory. Diffraction images were processed and data reduced using HKL2000.15 The structures of the RTA-compound complexes were solved by molecular replacement with MOLREP16 using the structure of RTA (PDB accession code 1RTC)7c as the search model. Model building was carried out using Coot (Emsley et al, 2010).17 Refinement of models was done using Refmac 5.5010918 and PHENIX.19 There were several rounds of refinement followed by manual rebuilding of the model. To facilitate manual rebuilding, a difference map and a 2Fo−Fc map, σA-weighted to eliminate bias from the model20, were prepared. 5% of the diffraction data were set aside throughout refinement for cross-validation.21 MolProbity was used to determine areas of poor geometry and to make Ramachandran plots.22
Crystallographic data for the complexes are presented in Table 3. Coordinates of the refined model of RTA complexed with 2, Pterin-Gly-Gly, 8, and 9 have been deposited in the Protein Data Bank with accession codes 4HUO, 4HV7, 4HV3, and 4HUP, respectively.
Table 3.
Crystallographic Data
| Compound 2 | Pterin-Gly-Gly | Compound 8 | Compound 9 | |
|---|---|---|---|---|
| Space group | P41212 | P41212 | P41212 | P41212 |
| Cell constants | a=b=68.0, c=140.7 A˚́ | a=b=68.2, c=140.8 A˚́ | a=b=67.8, c=140.7 A˚́ | a=b=67.7, c=140.6 A˚́ |
| Resolution (Å) | 50.-1.52 (1.55-1.52) | 50.-1.89 (1.92-1.89) | 20.-1.54 (1.57-1.54) | 50.-1.70 (1.73-1.70) |
| Rmerge (%) (outer shell) | 0.098 (0.957) | 0.131 (0.630) | 0.056 (0.509) | 0.091 (0.863) |
| <I/σI> (outer shell) | 7.3 (2.1) | 6.3 (2.2) | 13.3 (4.4) | 10.4 (2.3) |
| Completeness (%)(outer shell) | 99.8 (100.0) | 100.0 (100.0) | 99.7 (98.6) | 91.5 (100.0) |
| Unique reflections | 51, 425 | 27,776 | 49, 375 | 33,847 |
| Redundancy | 13.8 | 13.8 | 13.1 | 12.3 |
| # of residues | 263 | 263 | 268 | 283 |
| # of protein atoms | 2083 | 2097 | 2122 | 2090 |
| # of ligand atoms | 30 | 23 | 35 | 41 |
| # of solvent atoms | 397 | 283 | 221 | 337 |
| Rworking | 0.201 | 0.202 | 0.201 | 0.209 |
| Rfree | 0.230 | 0.236 | 0.239 | 0.236 |
| Average B factor for protein atoms (Å2) | 21.1 | 29.6 | 20.4 | 24.7 |
| Average B factor for ligand atoms (Å2) | 20.5 | 34.6 | 17.0 | 34.0 |
| Average B factor for solvent atoms (Å2) | 34.5 | 38.4 | 27.8 | 34.6 |
| rms deviation from ideality | ||||
| bonds (Å) | 0.027 | 0.007 | 0.031 | 0.006 |
| angles (°) | 2.212 | 0.951 | 2.625 | 1.009 |
| % residues in most favored region | 98.1 | 98.9 | 97.7 | 98.5 |
| % residues in additional allowed region | 1.5 | 1.1 | 1.2 | 1.5 |
Values in parentheses correspond to highest resolution shell
Acknowledgments
This work was supported by National Institute of Health (NIH) grant AI 075509, by Robert A. Welch Foundation grant F1225, and by the College of Natural Sciences support to the Center for Structural Biology. Assistance for this work was provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, the Institute for Cellular and Molecular Biology, the University of Texas at Austin.
The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Abbreviations
- RTA
ricin toxin A
- PTA
pteroic acid
- 7CP
7-carboxy pterin
- DBU
1,8-diazabicyclounde-7-cene
- P1
first amino acid
- P2
second amino acid
- P3
third amino acid
References
- 1.Franz D. Textbook of Military Medicine: Aspects of Chemical and Biological Warfare. 1997. [Google Scholar]
- 2.(a) Lord JM, Roberts LM, Robertus JD. Ricin: structure, mode of action, and some current applications. Faseb J. 1994;8(2):201–208. [PubMed] [Google Scholar]; (b) Robertus J. The structure and action of ricin, a cytotoxic N-glycosidase. Semin Cell Biol. 1991;2(1):23–30. [PubMed] [Google Scholar]
- 3.Miller DJ, Ravikumar K, Shen H, Suh JK, Kerwin SM, Robertus JD. Structure-based design and characterization of novel platforms for ricin and shiga toxin inhibition. J Med Chem. 2002;45(1):90–98. doi: 10.1021/jm010186s. [DOI] [PubMed] [Google Scholar]
- 4.Loyd A, Fletcher M. Bin Ladin’s poison manual. London Times; London, England: Nov, 2001. p. 16. [Google Scholar]
- 5.Johnston D, Hulse C. The New York Times. Feb 4th, 2004. Ricin on Capital Hill: Finding Ricin in Office disrupts Senate. [Google Scholar]
- 6.(a) Jasheway K, Pruet J, Anslyn EV, Robertus JD. Structure-based design of ricin inhibitors. Toxins (Basel) 2011;3(10):1233–1248. doi: 10.3390/toxins3101233. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pincus SH, Smallshaw JE, Song K, Berry J, Vitetta ES. Passive and active vaccination strategies to prevent ricin poisoning. Toxins (Basel) 2011;3(9):1163–1184. doi: 10.3390/toxins3091163. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barbier J, Bouclier C, Johannes L, Gillet D. Inhibitors of the cellular trafficking of ricin. Toxins (Basel) 2012;4(1):15–27. doi: 10.3390/toxins4010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ready MP, Kim Y, Robertus JD. Site-directed mutagenesis of ricin A-chain and implications for the mechanism of action. Proteins. 1991;10(3):270–278. doi: 10.1002/prot.340100311. [DOI] [PubMed] [Google Scholar]; (b) Montfort W, Villafranca JE, Monzingo AF, Ernst SR, Katzin B, Rutenber E, Xuong NH, Hamlin R, Robertus JD. The 3-Dimensional Structure of Ricin at 2.8-A. J Biol Chem. 1987;262(11):5398–5403. [PubMed] [Google Scholar]; (c) Mlsna D, Monzingo AF, Katzin BJ, Ernst S, Robertus JD. Structure of recombinant ricin A chain at 2.3 A. Protein Sci. 1993;2(3):429–435. doi: 10.1002/pro.5560020315. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Monzingo AF, Robertus JD. X-ray analysis of substrate analogs in the ricin A-chain active site. J Mol Biol. 1992;227(4):1136–1145. doi: 10.1016/0022-2836(92)90526-p. [DOI] [PubMed] [Google Scholar]
- 8.(a) Pruet JM, Jasheway KR, Manzano LA, Bai Y, Anslyn EV, Robertus JD. 7-Substituted pterins provide a new direction for ricin A chain inhibitors. Eur J Med Chem. 2011;46:3608–3615. doi: 10.1016/j.ejmech.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yan X, Hollis T, Svinth M, Day P, Monzingo AF, Milne GWA, Robertus JD. Structure-based identification of a ricin inhibitor. Journal of Molecular Biology. 1997;266(5):1043–1049. doi: 10.1006/jmbi.1996.0865. [DOI] [PubMed] [Google Scholar]; (c) Chen XY, Link TM, Schramm VL. Ricin A-chain: kinetics, mechanism, and RNA stem-loop inhibitors. Biochemistry. 1998;37(33):11605–11613. doi: 10.1021/bi980990p. [DOI] [PubMed] [Google Scholar]; (d) Bai Y, Monzingo AF, Robertus JD. The X-ray structure of ricin A chain with a novel inhibitor. Arch Biochem Biophys. 2009;483:23–28. doi: 10.1016/j.abb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Ho MC, Sturm MB, Almo SC, Schramm VL. Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc Natl Acad Sci U S A. 2009;106(48):20276–20281. doi: 10.1073/pnas.0911606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pruet JM, Robertus JD, Anslyn EV. Acyl radical insertion for the direct formation of new 7-substituted pterin analogs. Tetrahedron Lett. 2010;51(18):2539–2540. doi: 10.1016/j.tetlet.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Pruet JM, Saito R, Manzano LA, Jasheway KR, Wiget PA, Kamat I, Anslyn EV, Robertus JD. Optimized 5-Membered Heterocycle-Linked Pterins for the Inhibition of Ricin Toxin A. ACS Medicinal Chemistry Letters. 2012;3(7):588–591. doi: 10.1021/ml300099t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Price KE, Larrivee-Aboussafy C, Lillie BM, McLaughlin RW, Mustakis J, Hettenbach KW, Hawkins JM, Vaidyanathan R. Mild and efficient DBU-catalyzed amidation of cyanoacetates. Org Lett. 2009;11(9):2003–2006. doi: 10.1021/ol900435t. [DOI] [PubMed] [Google Scholar]
- 12.(a) LeBeau AM, Banerjee SR, Pomper MG, Mease RC, Denmeade SR. Optimization of peptide-based inhibitors of prostate-specific antigen (PSA) as targeted imaging agents for prostate cancer. Bioorgan Med Chem. 2009;17(14):4888–4893. doi: 10.1016/j.bmc.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gujraty K, Sadacharan S, Frost M, Poon V, Kane RS, Mogridge J. Functional characterization of peptide-based anthrax toxin inhibitors. Mol Pharm. 2005;2(5):367–372. doi: 10.1021/mp050040f. [DOI] [PubMed] [Google Scholar]; (c) Powell LR, Dukes KD, Lammi RK. Probing the efficacy of peptide-based inhibitors against acid- and zinc-promoted oligomerization of amyloid-beta peptide via single-oligomer spectroscopy. Biophys Chem. 2012;160(1):12–19. doi: 10.1016/j.bpc.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen X, Wu J, Luo Y, Liang X, Supnet C, Kim MW, Lotz GP, Yang G, Muchowski PJ, Kodadek T, Bezprozvanny I. Expanded polyglutamine-binding peptoid as a novel therapeutic agent for treatment of Huntington’s disease. Chem Biol. 2011;18(9):1113–11125. doi: 10.1016/j.chembiol.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kumar A, Wang YH, Lin XF, Sun GQ, Parang K. Synthesis and evaluation of 3-Phenylpyrazolo[3,4-d]pyrimidine-peptide conjugates as Src kinase inhibitors. Chemmedchem. 2007;2(9):1346–1360. doi: 10.1002/cmdc.200700074. [DOI] [PubMed] [Google Scholar]; (f) Falciani C, Brunetti J, Pagliuca C, Menichetti S, Vitellozzi L, Lelli B, Pini A, Bracci L. Design and In vitro Evaluation of Branched Peptide Conjugates: Turning Nonspecific Cytotoxic Drugs into Tumor-Selective Agents. Chemmedchem. 2010;5(4):567–574. doi: 10.1002/cmdc.200900527. [DOI] [PubMed] [Google Scholar]; (g) Suhas R, Chandrashekar S, Gowda DC. Synthesis of elastin based peptides conjugated to benzisoxazole as a new class of potent antimicrobials - A novel approach to enhance biocompatibility. Eur J Med Chem. 2011;46(2):704–711. doi: 10.1016/j.ejmech.2010.12.005. [DOI] [PubMed] [Google Scholar]; (h) Bi W, Bi Y, Xue P, Zhang YR, Gao X, Wang ZB, Li M, Baudy-Floc’h M, Ngerebara N, Gibson KM, Bi LR. A new class of beta-carboline alkaloid-peptide conjugates with therapeutic efficacy in acute limb ischemia/reperfusion injury. Eur J Med Chem. 2011;46(5):1453–1462. doi: 10.1016/j.ejmech.2011.01.021. [DOI] [PubMed] [Google Scholar]; (i) Saidikoglou E, Magoulas G, Theodoropoulou C, Athanassopoulos CM, Giannopoulou E, Theodorakopoulou O, Drainas D, Papaioannou D, Papadimitriou E. Effect of conjugates of all-trans-retinoic acid and shorter polyene chain analogues with amino acids on prostate cancer cell growth. Eur J Med Chem. 2009;44(8):3175–3187. doi: 10.1016/j.ejmech.2009.03.029. [DOI] [PubMed] [Google Scholar]; (j) Krzysiak AJ, Scott SA, Hicks KA, Fierke CA, Gibbs RA. Evaluation of protein farnesyltransferase substrate specificity using synthetic peptide libraries. Bioorg Med Chem Lett. 2007;17(20):5548–5551. doi: 10.1016/j.bmcl.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma JC, Dougherty DA. The Cation-pi Interaction. Chem Rev. 1997;97(5):1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 14.The following conditions were explored in the attempted synthesis of an Ala-Tyr conjugated pterin: a) MeOH/DMSO, 80 oC, 36hrs. b) ethyleneglycol, 105 oC, 36hrs. c) DMF, 145 oC, 48hrs. d) DMSO, 140 oC, 6d. e) Ionic liquid (BMIM-Ms), rt, 7d.
- 15.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 17.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 19.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D. 2010;66(2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Read RJ. Improved Fourier Coefficients for Maps Using Phases from Partial Structures with Errors. Acta Crystallogr A. 1986;42:140–149. [Google Scholar]
- 21.Brunger AT. Assessment of Phase Accuracy by Cross Validation - the Free R-Value - Methods and Applications. Acta Crystallogr D. 1993;49:24–36. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- 22.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]