

Abstract
Objective
The insulin-sensitizing agents referred to as thiazolidinediones (TZDs) possess anti-atherogenic and anti-inflammatory actions that contribute to protection against diabetic macrovascular complications. However, little is known about the effects of TZDs on retinal microvessel disorders. Here, we investigated whether TZDs modulate retinal vessel formation in a mouse model of oxygen- retinopathy.
Methods and Results
Neonatal mice were subjected to ischemia-induced retinopathy to produce pathological neovascular tuft formation. Pioglitazone (10 mg/kg/day), rosiglitazone (10 mg/kg/day) or vehicle was given by gavage once a day from postnatal day 7 (P7) to P17. Systemic treatment of wild-type (WT) mice with TZDs led to a significant decrease in pathological retinal neovascularization during ischemia compared with vehicle treatment, which was accompanied by increased plasma levels of the fat-derived hormone adiponectin. In contrast to WT mice, TZDs had no effects on ischemia-induced pathological retinal vessel formation in adiponectin-knockout (APN-KO) mice. Pioglitazone reduced tumor necrosis factor (TNF)-α expression in ischemic retina in WT mice but not in APN-KO mice. Furthermore, pioglitazone increased plasma adiponectin levels in TNF-α-KO mice but did not affect ischemia-induced pathological retinal neovascularization in this strain.
Conclusions
These data show that TZDs attenuate pathological retinal microvessel formation through adiponectin-mediated modulation of TNF-α production.
Keywords: pioglitazone, adiponectin, neovascularization, ischemia, angiogenesis
Introduction
Thiazolidinediones (TZDs), peroxisome proliferator-activated receptor (PPAR)-γ agonists (Pioglitazone and Rosiglitazone) have well-established anti-hyperglycemic effects in type 2 diabetes, which are mediated by a reduction of insulin resistance 1-3. Administration of TZDs inhibits the development of atherosclerotic lesion formation in animal models of atherosclerosis 4, 5 and suppresses neointimal thickening in response to injury in rabbits and rats 6, 7. TZDs are also reported to protect the heart from myocardial ischemia-reperfusion injury 8. These actions are believed to be mediated through the ability of TZDs to affect vascular function and modulate inflammation 1-3. While these data suggest that TZDs exert favorable effects on diabetic macrovascular complications, the epidemiological findings on TZD treatment and cardiovascular diseases are complex. The PROactive study and a meta-analysis reported that pioglitazone therapy has a beneficial impact on cardiovascular outcomes in diabetic populations 9, 10. A meta-analysis revealed that rosiglitazone is associated with adverse cardiovascular events in diabetic patients 11, but the more recent RECORD study did not find that rosiglitazone is linked to increased cardiovascular risk 12.
Ischemic retinopathies including retinopathy of prematurity and diabetic retinopathy are the main cause of blindness 13, 14. Retinal ischemia causes vascular injury and dysfunction, leading to a pathological blood vessel formation in the retina 14. Furthermore, hyperglycemia can contribute to the development of diabetic retinopathy by enhancing vascular permeability, inflammation and endothelial cell damage 13. These abnormal neovascular changes are associated with the overproduction of several growth factors and cytokines including vascular endothelial growth factor (VEGF) and tumor necrosis factor (TNF)-α 13, 14. Thus, inhibition of pathological retinal vessel formation is a logical strategy to prevent retinal injury and preserve visual acuity 15.
Adiponectin is an anti-inflammatory adipose-derived cytokine that is downregulated in patients with obesity-linked diseases including type 2 diabetes 16, 17. Increasing evidence indicates that TZD treatment increases plasma adiponectin levels in normal, obese, and type 2 diabetic subjects 18, 19 TZD treatment also increases adiponectin levels in diabetic db/db, ob/ob and wild-type mice 19-21. A number of experimental studies demonstrate that adiponectin plays a protective role in the development of insulin resistance and diabetic macrovascular complications including atherosclerosis and ischemic heart disease 22-25. Importantly, adiponectin-knockout (APN-KO) mice are refractory to the beneficial effects of TZDs on insulin resistance 20, 21. These findings suggest that the ability of TZDs to ameliorate insulin sensitivity is mediated, at least in part, by the ability of these drugs to upregulate adiponectin expression.
A recent clinical study showed that rosiglitazone treatment is associated with the delayed onset of proliferative diabetic retinopathy 26. In experimental models, it has been shown that intravitreal injection of TZDs suppresses pathological vessel formation in ischemic retina 27, and that TZDs suppress retinal leukostasis and vascular leakage 28. However, the molecular mechanisms of TZDs action in retinal vessel disease remain unclear.
Recently we have shown that adiponectin protects against pathological microvessel formation in retina in a mouse model of ischemic retinopathy 29. Thus, we hypothesized that TZDs protect against retinal vessel disorders through their ability to increase circulating adiponectin levels. Here we investigated the effects of TZDs on ischemia-induced retinal vessel formation in a mouse model of oxygen-induced retinopathy and assessed the participation of adiponectin and TNF-α in this process with loss-of-function genetic manipulations.
Methods
Mouse model of ischemia-induced retinopathy
Adiponectin-knockout (APN-KO) 23 and littermate wild-type (WT) mice in a C57BL/6 background, and TNF-α-KO and littermate WT mice in a C57BL/6/129 background (Jackson Laboratory) were used. Both genders were used. All animal studies were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the Institutional Animal Care and Use Committee in Boston University. To produce a clinically relevant ischemic retinopathy model, postnatal mice were subjected to oxygen-induced retinopathy 29, 30. Postnatal day 7 (P7) mice with their nursing mothers were exposed to 75±2% oxygen (hyperoxia) for 5 days (P7-12) to produce retinal vascular obliteration, and returned to room air (normoxia) at P12. When the mice are returned to room air at P12, the regions of vascular obliteration in retina are rendered hypoxic. Subsequently, hypoxic condition stimulates the development of pathological neovascular tufts within the vitreous body. P 17 is the time when the maximal neovascularization occurs in this model.
Pioglitazone treatment
Pioglitazone (10 mg/kg/day) 20, rosiglitazone (10 mg/kg/day) 21 or vehicles were placed in the stomach of pups by gavage once daily for 10 days from P7 to P14 or P17. Pioglitazone was obtained from Sigma, and rosiglitazone was purchased from Calbiochem. Mouse adiponectin levels were determined by ELISA kit (B-Bridge International Inc.). Glucose levels were measured with enzymatic kits (Wako Chemicals). TNF-α protein levels in ischemic retinas were measured by ELISA (R&D Systems).
Measurement of neovascularization and vascular obliteration
The degree of neovascularization and vascular obliteration were assessed by well-established methods 29, 31. Eyes were removed from mice at P17, and fixed in 4% paraformaldehyde. The retinas were dissected and stained with fluoresceinated Griffonia Bandereiraea Simplicifolia Isolectin B4 (Alexa Fluor 594, Molecular Probes) to detect vascular endothelial cells 29, 31. The retinas were radially cut from the edge of retina in the 4 quadrants, and mounted in mounting media (Vector Laboratories). Pictures of whole-mounts of retinas were taken at 5× magnification by fluorescence microscopy. Retinal segments were merged to generate a whole retinal image using Adobe Photoshop. Central non-perfused regions without capillaries and optic disc were defined as vascular obliteration. The extent of vascular obliteration and neovascular tuft formation was quantified by comparing the number of pixels in the affected areas with the total number of pixels in the retina. The physiological normal vascularization was calculated by subtracting the neovascular and vascular obliteration areas from total retinal areas 29. Investigators were masked to the mouse treatment.
Determination of mRNA levels
Total RNA was isolated from retinas of mice using a Qiagen kit, and cDNA was produced using ThermoScript RT-PCR Systems (Invitrogen). Quantitative Real-time PCR (QRT-PCR) was performed on iCycler iQ Real-Time PCR Detection System (BIO-RAD) using SYBR Green I as a double stranded DNA specific dye as described previously 22, 32. Primers for mouse TNF-α were purchased from Qiagen. Primers were: 5′-CTGTAACGATGAAGCCCTGGAG-3′ and 5′-TGGTGAGGTTTGATCCGCAT-3′ for mouse VEGF, 5′-ATTTCCCTGTTTGTGGCTGCTA-3′ and 5′-CAATCCCCTCCTGCAACTTC-3′ for mouse PPARα, 5′-TCCAGCCAATGCCTTTGC-3′ and 5′-TGGAGATTACTTTTCAGTGCAGAA-3′ for mouse CD36, 5′-TCACCTGGAAGACAGCTCCT-3′ and 5′-AATCCCCATTTACGCTGATG-3′ for mouse fatty acid binding protein 4, aP2, and 5′-AGAGGGAAATCGTGCGTGAC-3′ and 5′-CAATAGTGATGACCTGGCCGT-3′ for mouse β-actin.
Adenovirus-mediated gene transfer
Adenovirus vectors containing the gene for β-galactosidase (Ad-βgal) and full-length mouse adiponectin (Ad-APN) were prepared as described previously 23. The 4×107 plaque-forming units of Ad-APN or Ad-βgal were injected into the jugular vein of APN-KO mice at P10 29.
Western blot analysis
Retinal tissues were obtained at P14 and homogenized in RIPA buffer (Cell Signaling) with protease inhibitor cocktail (Roche). Tissue lysates were separated with denaturing SDS-PAGE. The membranes were immunoblotted with the indicated antibodies followed by the secondary antibody conjugated with horseradish peroxidase. ECL Western Blotting Detection kit (Amersham) was used for detection. Antibodies used in this study were phospho-AMPK (Thr172) antibody (Cell Signaling), pan-α-AMPK antibody (Cell Signaling) and β-actin antibody (Sigma). Quantitative analysis of relative phosphorylation levels was performed by using ImageJ program. Immunoblots were normalized to total loaded protein.
Statistical Analysis
All data are presented as means ± SEM. Differences were analyzed by Student's unpaired t test for 2 groups or ANOVA for multiple comparisons. A level of P<0.05 was accepted as statistically significant.
Results
Systemic delivery of pioglitazone inhibits ischemia-driven pathological retinal neovascularization in WT mice
A previous study analyzed the effects of TZDs on pathological retinal vascularization following intravitreal delivery 27. To test the impact of systemic pioglitazone administration, neonatal wild-type (WT) mice in a C57BL/6 background were exposed to hyperoxia (75% O2) for 5 days from postnatal day 7 (P7) to P12 and returned to room air at P12 to induce pathological neovascular tufts 29, 30. Pioglitazone or vehicle was given to mice by gavage from P7 to P17. Retinas were stained with fluoresceinated isolectin B4 to detect vascularization at P17. Figure 1A shows representative photographs of retinal whole mounts, stained with fluorescein-labeled isolectin B4. Quantitative analysis of neovascular tufts/total retinal area revealed that pioglitazone significantly reduced pathological retinal neovascular formation in this model (Figure 1B). Pioglitazone treatment also reduced vascular obliteration/total retinal area compared with vehicle treatment (Figure 1C). Pioglitazone increased areas of physiologically normal vascularization in ischemic retina (77.5±0.7%) compared with vehicle (71.2±0.8%). Thus, systemic administration of pioglitazone is protective in the context of ischemia-induced pathological retinal neovascularization. In contrast, under normoxic conditions, WT mice treated with pioglitazone or vehicle showed no central avascular area and no peripheral pathological vascular tuft formation (data not shown).
Figure 1.
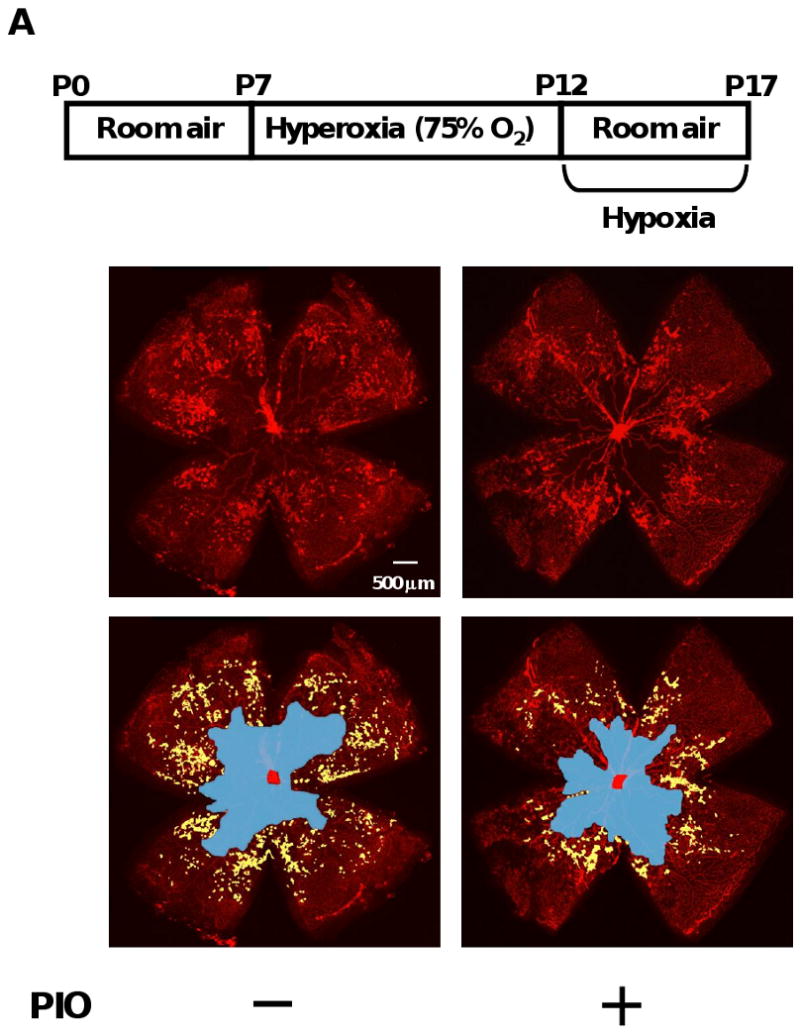

Pioglitazone suppresses pathological retinal neovascularization during ischemia in WT mice. A, Representative photographs of isolectin-stained retinas of WT mice treated with or without pioglitazone (PIO) at P17 are shown. A protocol for mouse model of oxygen-induced retinopathy is shown in an upper panel. Mice at P7 were reared in 75 % oxygen (hyperoxia) for 5 days followed by room air for 5 days. Pioglitazone or vehicle was administered to mice by gavage from P7 to P17. Retinas at P17 were stained with fluoresceinated isolectin B4. Vascular obliteration area is highlighted in blue. Neovascular tuft area is highlighted in yellow. Magnification, ×5. B and C, Quantification of retinal neovascularization (B) and vascular obliteration (C) in vehicle (n=15)- or pioglitazone (PIO, n=19)-treated WT mice at P17. Neovascular and vascular obliteration areas were quantified by comparing the number of pixels in the neovascular tuft and vascular obliteration areas, respectively with the total number of pixels in the retina. Data are presented as mean ± SEM.
Contribution of adiponectin to the suppressive action of pioglitazone on pathological neovascularization
Recently we have found that adiponectin is protective against the development of ischemic retinopathy 29. Because TZDs have been shown to increase adiponectin levels in mice and humans 18-21, we hypothesized that the beneficial actions of pioglitazone on pathological neovascularization in response to ischemia might be due to the induction of adiponectin. To test this hypothesis, we first measured plasma adiponectin levels during oxygen-induced retinopathy in the presence of pioglitazone or vehicle. Pioglitazone treatment significantly increased plasma adiponectin levels at P12, P14 and P17 compared with vehicle (Figure 2A). Plasma adiponectin levels were also slightly elevated at P14 and P17 in vehicle-treated WT mice (Figure 2A). Plasma glucose levels at P17 did not significantly differ between vehicle- and pioglitazone-treated WT mice (Glucose levels: 95 ± 6 mg/dl in vehicle-treated mice, 91 ± 8 mg/dl in pioglitazone-treated mice).
Figure 2.


Involvement of adiponectin induction in suppression of pathological vessel formation by pioglitazone. A, Upregulation of adiponectin by pioglitazone during ischemic retinopathy. Pioglitazone (PIO, n=10) or vehicle (n=8) was given to WT mice by gavage from P7 to P17. Plasma adiponectin levels were determined by ELISA. *, P<0.05; **, P<0.01 vs. vehicle-treated mice at P7. ***, P<0.01 vs. vehicle-treated mice. B, Administration of pioglitazone has no effects on pathological retinal neovascularization in APN-KO mice. APN-KO mice were subjected to ischemia-induced retinopathy. Pioglitazone (n=14) or vehicle (n=13) was given to APN-KO mice from P7 to P17. Adenoviral vectors expressing adiponectin (Ad-APN, n=9) or β-galactosidase (Ad-βgal, n=8) were delivered intravenously to APN-KO mice at P10. Neovascular areas were measured at P17. C, Effect of rosiglitazone on pathological neovascular formation in ischemic retina in WT and APN-KO mice. WT and APN-KO mice were treated with rosiglitazone (ROS) or vehicle from P7 to P17 (n=8 in each group). Results are presented as mean ± SEM. N.S., not statistically significant.
To test whether adiponectin mediates the protective actions of pioglitazone on ischemic retinopathy, we investigated the effect of pioglitazone on pathological vessel formation in adiponectin-knockout (APN-KO) mice in a C57BL/6 background. Compared with WT mice, APN-KO mice treated with vehicle exhibited increased pathological neovascular area (17.2 ± 0.6%), consistent with our previous observations 29. Pioglitazone treatment had no effect on retinal neovascular areas in APN-KO at P17 as compared with vehicle treatment (Figure 2B). In contrast, systemic delivery of adenoviral vectors expressing adiponectin (Ad-APN) attenuated retinal neovascular area in APN-KO at P17 compared with Ad-β-galactosidase (Ad-βgal) treatment (Figure 2B). While adiponectin levels were undetectable in Ad-βgal-treated APN-KO, Ad-APN-treated APN-KO had adiponectin levels of 11.4 ± 2.1 μg/ml at P17, which is similar to levels in WT mice. These data suggested that the inhibitory effect of pioglitazone on ischemia-induced neovascularization is dependent on its ability to upregulate adiponectin production.
We also evaluated the extent of pathological tuft formation in ischemic retina in WT and APN-KO mice after gavage administration of rosiglitazone, another TZD. Treatment of WT mice with rosiglitazone resulted in a significant increase in plasma adiponectin levels at P14 relative to vehicle treatment, which is comparable to the levels in pioglitazone-treated WT mice (Adiponectin levels: 13.7 ± 1.0 μg/ml in WT/vehicle and 28.1 ± 1.0 μg/ml in WT/rosiglitazone). Neovascular tuft formation in retinas of WT mice was attenuated by rosiglitazone treatment (Figure 2C). In contrast, rosiglitazone did not affect the areas of retinal neovascular formation in APN-KO mice (Figure 2C).
Pioglitazone reduces TNF-α expression in ischemic retina
To elucidate the mechanism by which pioglitazone protects against abnormal retinal neovascularization, we analyzed the expression of VEGF and TNF-α in ischemic retina in vehicle- and pioglitazone-treated WT mice at P14 by quantitative real-time PCR methods. VEGF mRNA levels did not significantly differ between vehicle- and pioglitazone-treated WT mice (Fold changes in transcripts in pioglitazone vs. vehicle: 0.92±0.10). In contrast, pioglitazone treatment significantly reduced retinal TNF-α mRNA levels in WT mice compared with vehicle treatment (Figure 3A). To test the involvement of adiponectin in suppression of retinal TNF-α expression by pioglitazone, we assessed retinal TNF-α mRNA expression at P14 in APN-KO mice in the presence of pioglitazone or vehicle. APN-KO mice treated with vehicle showed a significant increase in retinal TNF-α mRNA levels compared with vehicle-treated WT mice. In contrast to the inhibition of TNF-α levels in WT mice by pioglitazone, administration of pioglitazone to APN-KO mice did not lead to a significant reduction of TNF-α mRNA levels in ischemic retina (Figure 3A).
Figure 3.

Effect of pioglitazone on TNF-α expression in ischemic retina in WT and APN-KO mice. WT and APN-KO mice were treated with pioglitazone (PIO) or vehicle from P7 to P14. A, TNF-α mRNA levels in retina of WT (vehicle: n=8, PIO: n=8) and APN-KO (vehicle: n=9, PIO: n=9) mice at P14. TNF-α mRNA levels were determined by QRT-PCR analysis and expressed relative to β-actin mRNA levels. B, TNF-α protein levels in ischemic retina in WT (vehicle: n=6, PIO: n=6) and APN-KO (vehicle: n=6, PIO: n=6) mice at P14. Data are presented as mean ± SEM.
To corroborate the TNF-α transcript data, TNF-α protein levels were quantified by ELISA. Treatment of WT mice with pioglitazone significantly reduced TNF-α protein expression in ischemic retina (Figure 3B). In contrast, APN-KO mice exhibited increased levels of TNF-α production in ischemic retina, and pioglitazone treatment had little or no effect on these elevated levels of TNF-α protein (Figure 3B).
Role of TNF-α-deficiency in suppression of pathological neovascularization by pioglitazone
To investigate the role of TNF-α in retinal protection by pioglitazone, we assessed the impact of pioglitazone on abnormal retinal neovascularization under conditions of TNF-α-deficiency. TNF-α-KO and WT mice in a background of C57BL/6/129 were subjected to oxygen-induced retinopathy in the presence of pioglitazone or vehicle. As shown in Figure 4A, pioglitazone treatment increased plasma adiponectin levels at P14 by approximately 2-fold in both strains compared with vehicle treatment (Adiponectin levels: 11.2 ± 0.4 μg/ml in WT (C57BL/6/129)/vehicle, 19.7 ± 2.1 μg/ml in WT (C57BL/6/129)/pioglitazone, 7.9 ± 1.2 μg/ml in TNF-α-KO/vehicle, and 18.1 ± 1.2 μg/ml in TNF-α-KO/pioglitazone). Paradoxically, adiponectin levels were significantly lower in TNF-α-KO mice than in WT mice. TNF-α-KO mice treated with vehicle exhibited decreased retinal neovascular area compared with vehicle-treated WT mice, in agreement with previous reports 29, 31 (Figure 4B). Treatment of WT mice with pioglitazone resulted in a significant decrease in retinal neovascular areas at P17 (41%), but pioglitazone treatment had little or no effect on neovascular tuft formation in retina in TNF-α-KO mice (17% reduction).
Figure 4.

Role of TNF-α-deficiency in pioglitazone-mediated retinal protection. A, Plasma adiponectin levels of WT and TNF-α-KO mice after treatment with pioglitazone (PIO) or vehicle. WT and TNF-α-KO mice in a background of C57BL/6/129 (B6/129) were subjected to ischemia-induced retinopathy. Pioglitazone or vehicle was given to WT (vehicle: n=8, PIO: n=9) and TNF-α-KO (vehicle: n=14, PIO: n=10) mice from P7 to P14. Plasma adiponectin levels at P14 were determined by ELISA. B, Effect of pioglitazone on pathological retinal neovascularization in TNF-α-KO mice. Pioglitazone or vehicle was administered to WT (vehicle: n=14, PIO: n=13) and TNF-α-KO (vehicle: n=17, PIO: n=17) mice from P7 to P17. Data are presented as mean ± SEM (n=13-20).
Adiponectin mediates PPARα induction and AMPK activation caused by pioglitazone
Pioglitazone has been shown to stimulate PPARα 33 and AMPK activation in vivo 20, 21. Similarly, adiponectin is reported to increase PPARα expression and enhance AMPK activity in various tissues including liver and muscles 34. Therefore, we tested whether pioglitazone modulates PPARα expression and AMPK signaling during ischemic retinopathy in WT and APN-KO mice. QRT-PCR analysis revealed that PPARα mRNA levels were lower in retinas of APN-KO mice than in those of WT mice during vehicle treatment (Figure 5A). Treatment of WT mice with pioglitazone led to a significant increase in PPARα transcript levels in ischemic retinas, whereas no effects of pioglitazone were observed in APN-KO mice (Figure 5A). Western blot analysis demonstrated that pioglitazone enhanced phosphorylation of AMPK in ischemic retinas in WT mice, but not in APN-KO mice (Figure 5B). The levels of retinal AMPK phosphorylation tended to be lower in APN-KO mice than in WT mice after vehicle treatment, but the difference was not statistically significant. AMPK protein levels did not significantly change among four different groups. Finally we examined the effect of pioglitazone on mRNA levels of PPARγ-regulated genes, CD36 and fatty acid binding protein 4, aP2 in retinas of WT and APN-KO mice. No significant differences were observed in CD36 and aP2 mRNA levels between the two strains, and pioglitazone upregulated CD36 and aP2 transcript levels in ischemic retinas to similar extents in WT and APN-KO mice compared with vehicle (Figure 5C).
Figure 5.
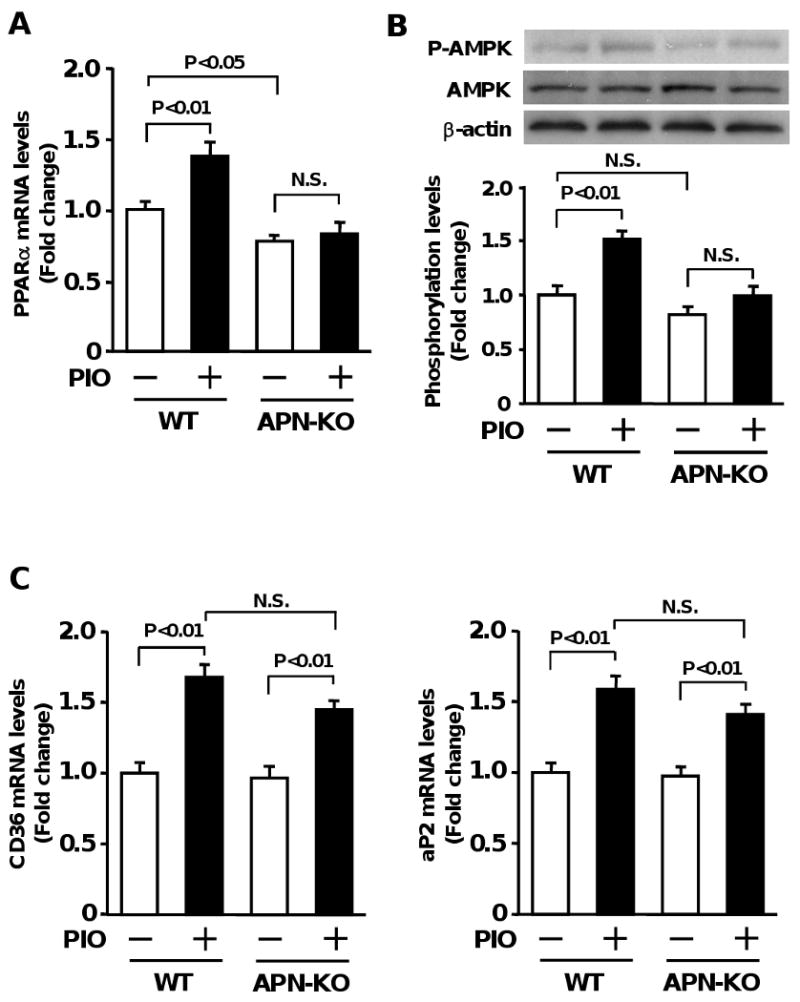
Contribution of adiponectin to pioglitazone-induced changes in PPARα expression and AMPK signaling during ischemic retinopathy. Retinas were harvested from eyes of WT or APN-KO mice after treatment with pioglitazone (PIO) or vehicle. A, Effect of pioglitazone on PPARα expression in ischemic retina in WT (vehicle: n=8, PIO: n=8) and APN-KO (vehicle: n=8, PIO: n=7) mice. PPARα mRNA levels at P14 were determined by QRT-PCR methods and expressed relative to β-actin levels. B, Effect of pioglitazone on AMPK signaling in ischemic retina in WT (vehicle: n=4, PIO: n=4) and APN-KO (vehicle: n=4, PIO: n=4) mice. Phosphorylation of AMPK (P-AMPK) in retinas at P14 was determined by western blot analysis. C, PPARγ-regulated gene expression in ischemic retina after treatment with pioglitazone. CD36 and aP2 mRNA levels in retina of WT (vehicle: n=8, PIO: n=8) and APN-KO (vehicle: n=8, PIO: n=7) mice at P14 were analyzed by QRT-PCR and expressed relative to β-actin levels. Data are presented as mean ± SEM.
Discussion
Clinical and experimental studies have shown that TZDs suppress pathological vessel formation in the retina 26-28. In the present study, we analyzed the effects of systemic TZD (pioglitazone and rosiglitazone) administration on pathological microvessel formation in retina in a mouse model of oxygen-induced retinopathy. The TZD-mediated suppression of pathological neovascularization was accompanied by elevations in plasma adiponectin levels, and mice lacking adiponectin were refractory to the therapeutic actions of TZDs in ischemic retina. Recently, we reported that the protective effects of adiponectin on pathological retinal vessel growth are mainly due to suppression of TNF-α production 29. Here we show that pioglitazone suppresses TNF-α expression in ischemic retinas of wild-type but not APN-KO mice. We also show that pioglitazone enhances plasma adiponectin levels in TNF-α-KO mice but pioglitazone was ineffective at suppressing the lower levels of pathological retinal neovascularization in this strain. Collectively, these data suggest that adiponectin upregulation mediates the beneficial actions of pioglitazone on ischemic retinopathy in vivo, and that the suppression of TNF-α by adiponectin is a mechanistic link in the action of this drug.
TNF-α is a major pro-inflammatory cytokine that promotes insulin resistance and diabetic vasculopathy 35-37. Accumulating evidence indicates that adiponectin negatively regulates TNF-α production in various disease states. APN-KO mice exhibit increased injury and TNF-α production in the heart following ischemia-reperfusion 22. Adiponectin-deficiency also contributes to exacerbation of diet-induced insulin resistance, which is associated with elevated TNF-α levels in adipose tissue 23. Conversely adiponectin supplementation reduces TNF-α expression in the vasculature of atherosclerotic mice and in the liver in a mouse model of steatosis 38, 39. Therefore, adiponectin-mediated inhibition of TNF-α represents an anti-inflammatory signaling axis in multiple target organs that leads to improvement of tissue injury.
TZDs have been shown to protect against pathological inflammation and tissue injury through suppression of pro-inflammatory genes, including TNF-α 36. It has been shown that inhibition of TNF-α largely contributes to the effects of TZD on inflammatory responses in adipose tissue by analysis of mice lacking TNF-α function 37. These findings are consistent with our observations showing that the retinal protection by pioglitazone is largely dependent on the suppression of TNF-α by adiponectin. Thus, it is tempting to speculate the therapeutic actions of TZDs in these different systems result in large part from the abilities of these drugs to influence the adiponectin-TNF-α regulatory axis.
Several in vitro studies show that adiponectin suppresses agonist-stimulated TNF-α production in multiple cell types. Adiponectin has been shown to block the increase in TNF-α expression in response to LPS in adipocytes and cardiac cells 22, 40. Adiponectin also reduces TNF-α production following LPS stimulation in macrophages through its ability to suppress nuclear factor κB activity 41, 42. Furthermore, adiponectin stimulates apoptotic body clearance by macrophages, leading to a reduction of TNF-α levels 43. Because TNF-α in ischemic retina has shown to be produced mainly by macrophage and macrophage-like microglia 31, the resolution of retinal vascular injury by adiponectin may be largely attributed to suppression of TNF-α in retinal macrophages.
In our study, treatment of neonatal WT mice with TZDs (10mg/kg/day) resulted in an increase in adiponectin level (∼2 fold increase), which is comparable with the adiponectin induction in adult obese mice following TZD treatment 20, 21. TZDs upregulate adiponectin expression in adipocytes through a number of mechanisms that are stimulated by PPARγ. TZDs stimulate adiponectin gene transcript in cultured adipocytes and in adipose tissues of obese mice in a PPARγ-dependent mechanism 19, 44, 45. TZDs are also reported to promote secretion of adiponectin from adipocytes 46. Recent studies have shown that TZDs promote the induction of ER chaperones, Ero1-Lα, ERp44 and DsbA-like protein, that are crucial for adiponectin secretion 47, 48.
It has been demonstrated that TZD treatment promotes AMPK activation in liver of obese mice in an adiponectin-dependent manner 20, 21. Consistent with these findings, we report that pioglitazone treatment leads to AMPK activation in ischemic retina of WT mice but not APN-KO mice. Likewise, we found that pioglitazone induced PPARα expression in WT but not APN-KO mice. Thus, adiponectin upregulation by adipocytes mediates the ability of pioglitazone to control a broad regulatory network in retina that can contribute to the resolution of vascular dysfunction and inflammation 33, 49. However, the causal relationships between TNF-α expression and activation of PPARα or AMPK have not been established, and the role of these signaling proteins in ischemic retinopathy is not known. Finally, in contrast to AMPK and PPARα, the genetic ablation of adiponectin did not affect the pioglitazone-induced expression of other PPARγ-responsive genes in retina, including CD36 and aP2. Collectively, these data indicate that TZDs exert both direct and indirect actions on retina. However, our study indicates that it is the indirect action of TZDs, via increased adiponectin expression by adipocytes, that mediates their beneficial actions on ischemic retina.
Previous experimental studies and our current data suggest that TZD therapy may be therapeutically beneficial for diseases of retinal inflammation and vascular dysfunction 27, 28. In this context, rosiglitazone therapy is shown to delay onset of diabetic proliferative retinopathy and reduce loss of visual acuity26. However, a recent study has shown that TZDs are associated with a moderately increased risk of diabetic macular edema 50. TZD treatment also leads to increases in circulating VEGF levels in diabetic subjects, which might exacerbate pathological retinal neovascularization 51. Thus, additional studies will be required to assess the safety and efficacy of TZDs for the treatment of diabetic retinal vessel disorders.
In conclusion, our study shows that systemic administration of pioglitazone protects against ischemic retinal injury by negatively regulating TNF-α expression. The ability of pioglitazone to suppress TNF-α expression and pathological neovascularization in ischemic retina in vivo is mediated through the upregulation of the anti-inflammatory adipokine adiponectin. Thus, pioglitazone treatment, or other approaches that stimulate adiponectin production, should be investigated further to assess their potential usefulness for the treatment of ischemia-induced retinal microvessel disease.
Acknowledgments
This study was funded by National Institutes of Health grants HL77774, HL86785, AG15052 and HL81587 to K. Walsh. N. Ouchi was supported by American Heart Association grant.
Footnotes
Conflict of Interest Disclosure: There is no conflict of interest that should be disclosed.
References
- 1.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 2.Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 3.Ceriello A. Thiazolidinediones as anti-inflammatory and anti-atherogenic agents. Diabetes Metab Res Rev. 2008;24:14–26. doi: 10.1002/dmrr.790. [DOI] [PubMed] [Google Scholar]
- 4.Collins AR, Meehan WP, Kintscher U, Jackson S, Wakino S, Noh G, Palinski W, Hsueh WA, Law RE. Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:365–371. doi: 10.1161/01.atv.21.3.365. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Ishibashi S, Perrey S, Osuga J, Gotoda T, Kitamine T, Tamura Y, Okazaki H, Yahagi N, Iizuka Y, Shionoiri F, Ohashi K, Harada K, Shimano H, Nagai R, Yamada N. Troglitazone inhibits atherosclerosis in apolipoprotein E-knockout mice: pleiotropic effects on CD36 expression and HDL. Arterioscler Thromb Vasc Biol. 2001;21:372–377. doi: 10.1161/01.atv.21.3.372. [DOI] [PubMed] [Google Scholar]
- 6.Shinohara E, Kihara S, Ouchi N, Funahashi T, Nakamura T, Yamashita S, Kameda Takemura K, Matsuzawa Y. Troglitazone suppresses intimal formation following balloon injury in insulin-resistant Zucker fatty rats. Atherosclerosis. 1998;136:275–279. doi: 10.1016/s0021-9150(97)00220-7. [DOI] [PubMed] [Google Scholar]
- 7.Joner M, Farb A, Cheng Q, Finn AV, Acampado E, Burke AP, Skorija K, Creighton W, Kolodgie FD, Gold HK, Virmani R. Pioglitazone inhibits in-stent restenosis in atherosclerotic rabbits by targeting transforming growth factor-beta and MCP-1. Arterioscler Thromb Vasc Biol. 2007;27:182–189. doi: 10.1161/01.ATV.0000251021.28725.e8. [DOI] [PubMed] [Google Scholar]
- 8.Yue Tl TL, Chen J, Bao W, Narayanan PK, Bril A, Jiang W, Lysko PG, Gu JL, Boyce R, Zimmerman DM, Hart TK, Buckingham RE, Ohlstein EH. In vivo myocardial protection from ischemia/reperfusion injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2001;104:2588–2594. doi: 10.1161/hc4601.099403. [DOI] [PubMed] [Google Scholar]
- 9.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. Jama. 2007;298:1180–1188. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- 10.Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Lefebvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Koranyi L, Laakso M, Mokan M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 11.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 12.Home PD, Pocock SJ, Beck-Nielsen H, Curtis PS, Gomis R, Hanefeld M, Jones NP, Komajda M, McMurray JJ. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 2009;373:2125–2135. doi: 10.1016/S0140-6736(09)60953-3. [DOI] [PubMed] [Google Scholar]
- 13.Sheetz MJ, King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. Jama. 2002;288:2579–2588. doi: 10.1001/jama.288.20.2579. [DOI] [PubMed] [Google Scholar]
- 14.Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438:960–966. doi: 10.1038/nature04482. [DOI] [PubMed] [Google Scholar]
- 15.Spaide RF, Fisher YL. Intravitreal bevacizumab (Avastin) treatment of proliferative diabetic retinopathy complicated by vitreous hemorrhage. Retina. 2006;26:275–278. doi: 10.1097/00006982-200603000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Walsh K. Obesity, adiponectin and vascular inflammatory disease. Curr Opin Lipidol. 2003;14:561–566. doi: 10.1097/00041433-200312000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 18.Yu JG, Javorschi S, Hevener AL, Kruszynska YT, Norman RA, Sinha M, Olefsky JM. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes. 2002;51:2968–2974. doi: 10.2337/diabetes.51.10.2968. [DOI] [PubMed] [Google Scholar]
- 19.Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I, Matsuzawa Y. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–2099. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- 20.Kubota N, Terauchi Y, Kubota T, Kumagai H, Itoh S, Satoh H, Yano W, Ogata H, Tokuyama K, Takamoto I, Mineyama T, Ishikawa M, Moroi M, Sugi K, Yamauchi T, Ueki K, Tobe K, Noda T, Nagai R, Kadowaki T. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281:8748–8755. doi: 10.1074/jbc.M505649200. [DOI] [PubMed] [Google Scholar]
- 21.Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L, Scherer PE. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- 22.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 24.Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T, Noda T. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277:25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura M, Izumiya Y, Higuchi A, Shibata R, Qiu J, Kudo C, Shin HK, Moskowitz MA, Ouchi N. Adiponectin prevents cerebral ischemic injury through endothelial nitric oxide synthase dependent mechanisms. Circulation. 2008;117:216–223. doi: 10.1161/CIRCULATIONAHA.107.725044. [DOI] [PubMed] [Google Scholar]
- 26.Shen LQ, Child A, Weber GM, Folkman J, Aiello LP. Rosiglitazone and delayed onset of proliferative diabetic retinopathy. Arch Ophthalmol. 2008;126:793–799. doi: 10.1001/archopht.126.6.793. [DOI] [PubMed] [Google Scholar]
- 27.Murata T, Hata Y, Ishibashi T, Kim S, Hsueh WA, Law RE, Hinton DR. Response of experimental retinal neovascularization to thiazolidinediones. Arch Ophthalmol. 2001;119:709–717. doi: 10.1001/archopht.119.5.709. [DOI] [PubMed] [Google Scholar]
- 28.Muranaka K, Yanagi Y, Tamaki Y, Usui T, Kubota N, Iriyama A, Terauchi Y, Kadowaki T, Araie M. Effects of peroxisome proliferator-activated receptor gamma and its ligand on blood-retinal barrier in a streptozotocin-induced diabetic model. Invest Ophthalmol Vis Sci. 2006;47:4547–4552. doi: 10.1167/iovs.05-1432. [DOI] [PubMed] [Google Scholar]
- 29.Higuchi A, Ohashi K, Kihara S, Walsh K, Ouchi N. Adiponectin suppresses pathological microvessel formation in retina through modulation of tumor necrosis factor-alpha expression. Circ Res. 2009;104:1058–1065. doi: 10.1161/CIRCRESAHA.109.194506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith LE, Wesolowski E, McLellan A, Kostyk SK, D'Amato R, Sullivan R, D'Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 31.Connor KM, SanGiovanni JP, Lofqvist C, Aderman CM, Chen J, Higuchi A, Hong S, Pravda EA, Majchrzak S, Carper D, Hellstrom A, Kang JX, Chew EY, Salem N, Jr, Serhan CN, Smith LE. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat Med. 2007;13:868–873. doi: 10.1038/nm1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouchi N, Shibata R, Walsh K. AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ Res. 2005;96:838–846. doi: 10.1161/01.RES.0000163633.10240.3b. [DOI] [PubMed] [Google Scholar]
- 33.Orasanu G, Ziouzenkova O, Devchand PR, Nehra V, Hamdy O, Horton ES, Plutzky J. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor-alpha-dependent manner in vitro and in vivo in mice. J Am Coll Cardiol. 2008;52:869–881. doi: 10.1016/j.jacc.2008.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 35.Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–949. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- 36.Abdelrahman M, Sivarajah A, Thiemermann C. Beneficial effects of PPAR-gamma ligands in ischemia-reperfusion injury, inflammation and shock. Cardiovasc Res. 2005;65:772–781. doi: 10.1016/j.cardiores.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Wellen KE, Uysal KT, Wiesbrock S, Yang Q, Chen H, Hotamisligil GS. Interaction of tumor necrosis factor-alpha- and thiazolidinedione-regulated pathways in obesity. Endocrinology. 2004;145:2214–2220. doi: 10.1210/en.2003-1580. [DOI] [PubMed] [Google Scholar]
- 38.Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, Ohashi K, Sakai N, Shimomura I, Kobayashi H, Terasaka N, Inaba T, Funahashi T, Matsuzawa Y. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106:2767–2770. doi: 10.1161/01.cir.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- 39.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ajuwon KM, Spurlock ME. Adiponectin inhibits LPS-induced NF-kappaB activation and IL-6 production and increases PPARgamma2 expression in adipocytes. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1220–1225. doi: 10.1152/ajpregu.00397.2004. [DOI] [PubMed] [Google Scholar]
- 41.Wulster Radcliffe MC, Ajuwon KM, Wang J, Christian JA, Spurlock ME. Adiponectin differentially regulates cytokines in porcine macrophages. Biochem Biophys Res Commun. 2004;316:924–929. doi: 10.1016/j.bbrc.2004.02.130. [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi N, Argueta JG, Masuhiro Y, Kagishita M, Nonaka K, Saito T, Hanazawa S, Yamashita Y. Adiponectin inhibits Toll-like receptor family-induced signaling. FEBS Lett. 2005;579:6821–6826. doi: 10.1016/j.febslet.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 43.Takemura Y, Ouchi N, Shibata R, Aprahamian T, Kirber MT, Summer RS, Kihara S, Walsh K. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest. 2007;117:375–386. doi: 10.1172/JCI29709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwaki M, Matsuda M, Maeda N, Funahashi T, Matsuzawa Y, Makishima M, Shimomura I. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes. 2003;52:1655–1663. doi: 10.2337/diabetes.52.7.1655. [DOI] [PubMed] [Google Scholar]
- 45.Combs TP, Wagner JA, Berger J, Doebber T, Wang WJ, Zhang BB, Tanen M, Berg AH, O'Rahilly S, Savage DB, Chatterjee K, Weiss S, Larson PJ, Gottesdiener KM, Gertz BJ, Charron MJ, Scherer PE, Moller DE. Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology. 2002;143:998–1007. doi: 10.1210/endo.143.3.8662. [DOI] [PubMed] [Google Scholar]
- 46.Bodles AM, Banga A, Rasouli N, Ono F, Kern PA, Owens RJ. Pioglitazone increases secretion of high-molecular-weight adiponectin from adipocytes. Am J Physiol Endocrinol Metab. 2006;291:E1100–1105. doi: 10.1152/ajpendo.00187.2006. [DOI] [PubMed] [Google Scholar]
- 47.Wang ZV, Schraw TD, Kim JY, Khan T, Rajala MW, Follenzi A, Scherer PE. Secretion of the adipocyte-specific secretory protein adiponectin critically depends on thiol-mediated protein retention. Mol Cell Biol. 2007;27:3716–3731. doi: 10.1128/MCB.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu M, Zhou L, Xu A, Lam KS, Wetzel MD, Xiang R, Zhang J, Xin X, Dong LQ, Liu F. A disulfide-bond A oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization. Proc Natl Acad Sci U S A. 2008;105:18302–18307. doi: 10.1073/pnas.0806341105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fisslthaler B, Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res. 2009;105:114–127. doi: 10.1161/CIRCRESAHA.109.201590. [DOI] [PubMed] [Google Scholar]
- 50.Fong DS, Contreras R. Glitazone use associated with diabetic macular edema. Am J Ophthalmol. 2009;147:583–586. e581. doi: 10.1016/j.ajo.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 51.Baba T, Shimada K, Neugebauer S, Yamada D, Hashimoto S, Watanabe T. The oral insulin sensitizer, thiazolidinedione, increases plasma vascular endothelial growth factor in type 2 diabetic patients. Diabetes Care. 2001;24:953–954. doi: 10.2337/diacare.24.5.953. [DOI] [PubMed] [Google Scholar]