

Abstract
Linkage to 7q has been the most robust genetic finding in familial autism. A previous scan of multiplex families with autism spectrum disorders found a linkage signal of genome-wide significance at D7S530 on 7q32. We searched a candidate imprinted region at this location for genetic variants in families with positive linkage scores. Using exon resequencing, we identified three rare potentially pathogenic variants in the TSGA14 gene, which encodes a centrosomal protein. Two variants were missense mutations (c.664C>G; p.P206A and c.766T>G; p.C240G) that changed conserved residues in the same protein domain; the third variant (c.192+5G>A) altered splicing, which resulted in a protein with an internal deletion of 16 residues and a G33D substitution. These rare TSGA14 variants are enriched in the affected subjects (6/348 patients versus 2/670 controls, Fisher's exact two tailed p= 0.022). This is the first report of a possible link of a gene with a centrosomal function with familial autism.
Keywords: autism spectrum disorders, chromosome 7q, TSGA14 gene, RNA splicing, centrosome
INTRODUCTION
Autism spectrum disorders (ASD) are a group of hereditary neurodevelopmental conditions characterized by impaired social interaction and communication, and accompanied by repetitive and stereotyped behavior. These life-long conditions have an early onset (at 2–3 years of age), high heritability (~90%) and a high male-to-female ratio (~4 to 1). ASD affects an estimated 1.5 million Americans and apparently has a polygenic etiology (Pickles and others 1995) though rare mutations and CNV in single genes cannot be excluded as being responsible for a subset of cases (Morrow and others 2008; Zhao and others 2007). The search for ASD susceptibility genes has included linkage analyses, candidate gene studies, analysis of copy-number variations (CNVs), and genome-wide association studies (GWAS). To date, both common and rare variants have been documented in ASD, but most of the evidence points toward the role of rare variants. Common ASD risk alleles have been identified from association studies of individual candidate genes with relevant neurobehavioral phenotypes (EN2 (Benayed and others 2005; Brune and others 2007) and OXTR (Jacob and others 2007; Liu and others; Wu and others 2005)) and of regions with linkage/chromosomal rearrangements (CNTNAP2 (Arking and others 2008)). Recently, new candidate locations have been identified in large-scale GWAS (Wang and others 2009a; Weiss and others 2009; Weiss and others 2008). Searches for rare mutations and CNV point to multiple contributing loci with very few recurrent variations. They are found in genes responsible for synaptic connectivity, such as NRXN1 (Feng and others 2006; Szatmari and others 2007), SHANK3 (Durand and others 2007; Moessner and others 2007), SHANK2 (Berkel and others 2010), X-linked synaptic genes (Jamain and others 2003; Lawson-Yuen and others 2008; Piton and others 2010), melatonin-related genes (Jonsson and others 2010) and genes involved in GTPase/Ras signaling pathways (Pinto and others 2010). The identified rare variants are either inherited with incomplete penetrance or occur de novo, and result in a broad spectrum of phenotypes including intellectual disability and various mental illnesses.
Linkage to chromosome 7q is among the most robust genetic findings in autism (Alarcon and others 2002; Ashley-Koch and others 1999; Hutcheson and others 2003; IMGSAC 1998; IMGSAC 2001; Lamb and others 2005; Schellenberg and others 2006). This region has had at least nominally significant scores in every linkage scan with a sample size of >50 affected sibling pairs (Cook 2001). Meta-analyses confirmed the 7q linkage and refined a genome-wide significant linkage to 7q22–32 under strict diagnosis of autism (Badner and Gershon 2002; Trikalinos and others 2006). We and others (Lamb and others 2005; Schellenberg and others 2006) obtained strong linkage signals at the identical location on 7q32 in non-overlapping samples. The ~20 cM common linkage peak area corresponds to 17 Mb of physical distance (242 genes; 535 transcripts).
Several linkage studies have detected parent-of-origin effects at 7q32, which corresponds to the D7S530–D7S640 interval (Ashley-Koch and others 1999; Lamb and others 2005), these findings suggest the involvement of an imprinted gene(s). A known cluster of imprinted genes at this location spans ~380 kb and encompasses at least 12 annotated genes/antisense transcripts (Supplement Table I). Among these, MEST, a paternally expressed gene, is of particular interest because of its documented role in the regulation of mammalian behavior (Lefebvre and others 1998). The gene is highly expressed in multiple areas of the developing brain. In addition, miR-335 in the second MEST intron represents a regulatory element that acts in early development on a network of neural targets and is vulnerable to environmental exposure (Sathyan and others 2007). In this study, we addressed the presence of common variants in the imprinted interval at 7q32 by family-based association testing, and we searched for rare deleterious mutations in the coding sequence of MEST and nearby genes by exon re-sequencing of subjects with autism from multiplex families positive for 7q32 linkage.
MATERIALS AND METHODS
Participants
Families with ASD (1640 individuals from 348 multiplex families with 2 or more affected children) were recruited for studies investigating the genetics of autism at the University of Washington. The set comprised 220 families analyzed in a previous linkage study (Schellenberg and others 2006) and an additional 128 families acquired more recently. Individuals were assessed using the Autism Diagnostic Interview-Revised (Lord and others 1994), the Autism Diagnostic Observation Schedule (Lord and others 2000), Diagnostic and Statistical Manual of Mental Disorders (DSM IV) (Association 1994), and a medical history review, as previously described (Schellenberg and others 2006). Strict criteria for Autism were met by 83% of affected children. All subjects with ASD were screened for the absence of Fragile × and for major chromosomal rearrangements. Using available information on 220 ASD families obtained in the whole genome linkage scan (Schellenberg and others 2006), 71 multiplex families with high scores (Z≥1) at 3 markers (D7S458, D7S530, D7S640) within 7q32 linkage peak were selected for association analysis. For the resequencing, 94 unrelated subjects with ASD (one per family) were used of which 47 were from families with high linkage scores (Z≥1) and 47 were from families with non-negative Z-scores (0<Z<1). For case-control association analysis of candidate variants, 348 unrelated subjects with ASD (one per family) were selected. For the analysis of variant transmission in families, all available members of 348 ASD families were genotyped.
We used two control groups which differed by sampling strategy: (1) volunteers from the University of Washington and Seattle Community Colleges (n = 170, no health information); (2) non-affected parents of dyslexic children (n =500). Only the second control group is known to contain no subjects with ASD. Ethnic structure of the ASD and control samples is provided in the Supplement Table II.
Resequencing of gene candidates
Exons and splice junctions of MEST, COPG2 and TSGA14 as well as the MEST intronic region harboring miR-335 were sequenced in both directions using fluorescent dye terminators (BigDye® Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems, Foster City, CA, USA) and an ABI Prism 3100 Genetic Analyzer (Applied Biosystems). Primers were selected using Primer3 software (Whitehead Institute, Cambridge, MA, USA). Variants were identified by sequence alignment using SEQUENCHER software (Gene Codes Corp, Ann Arbor, MI, USA).
SNP selection and genotyping assays
SNPs with a minor allele frequency over 0.2 in the Caucasian population were selected. One tag SNP was used per haploblock, the additional non-tag SNPs were included to allow for denser coverage of the region. SNPs were genotyped using TaqMan SNP genotyping assays (Applied Biosystems) and a 7900 Real-Time PCR System (Applied Biosystems). Genotyping was performed in 384-well plates with 5 ng of genomic DNA according to the manufacturer's protocol. Allele discrimination assays and/or custom “assay-by-design” TaqMan SNP assays (Applied Biosystems) were used to genotype TSGA14 variants. Primer sequences and PCR conditions are available upon request.
RNA isolation and cDNA synthesis
Postmortem brain tissues from cerebella of neurologically normal control subjects were obtained from the Neuropathology Core Brain Bank at the University of Washington. Average age of subjects was 70, and average postmortem interval was 4 hours. All tissue samples were obtained following informed consent, flash frozen at time of autopsy, and stored at −80 C. Total RNA from cultured cells or postmortem brain tissues was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) or RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNA synthesis with random primers was performed using the SuperScript III RT-PCR kit (Invitrogen).
Ex vivo RNA splicing assay
To create a minigene construct modeling the exon-intron architecture of TSGA14 (NCBI references: NM_018718.1; NP_061188.1), we performed 2-step cloning. First, exon 2 with 323 bp of the adjacent intron 2 sequence was amplified using primers integrating Hind III and Not I sites:
TSGAe2_Hind III_F: ATCAAGCTTATCTGATGAAAAGGATACCACAGAA
TSGAe2_NotI_R: GATGCGGCCGCCAAAATAGAGGCTGACCTCCTG
The double-digested PCR product was subcloned into a pRc/RSV vector (Invitrogen). Second, the 5.1 kb exon 3 – exon 4 region, which included 332 bp of adjacent intron 2 sequence was amplified using primers integrating Not I and Xba I sites and subcloned into the pRc/RSV-e2 construct:
TSGAe3-4_NotI_F : ATAGCGGCCGCAGGGGAGATAAAAGGAGGAG
TSGAe3-4_XbaI_R: CTCTAGATGCATCAGCTGGGCAAAAGTTGTAACT
c.192+5G>A mutation was introduced by site-directed mutagenesis using QuikChange XL site directed mutagenesis kit (Stratagene, La Jolla, CA, USA). Two independent clones with the c.192+5G>A mutation were used in the splicing assay.
Human embryonic kidney (HEK) 293T and rat pheochromocytoma (PC12) cell lines were maintained in DMEM medium supplemented with antibiotics (100 IU/ml penicillin, 100 μg/ml streptomycin) and 10% fetal calf serum (HEK 293T) or 5% fetal calf serum and 10% horse serum (PC12). Cells were seeded in triplicates in 35-mm dishes, grown to 50–70% confluence, and transfected with 2 μg of minigene constructs using Gene Porter (Genlantis, San Diego, CA, USA) or LipofectAMINE 2000 (Invitrogen) according to the manufacturer's protocol. Transfection efficiency was controlled by measuring the expression of 2 μg of pEGFP. Cells were harvested 16–20 h later, and total RNA was isolated. RT-PCR was performed using human-specific primers positioned in TSGA14 exon 2 (5'-CAGAACCCAAGATACCAGCA-3') and exon 4 (5'-ATCAGCTGGGCAAAAGTTGT-3'). PCR products were analyzed on 2% agarose gels, quantified and sequenced.
Transmission Disequilibrium Testing
To test for differential transmission of alleles, the FBAT program (v 1.7.1) was used (Laird and others 2000; Rabinowitz and Laird 2000). We evaluated the default additive model, which has reasonable power even when the underlying model is not additive and the “-e” empirical option which tests for association in the presence of linkage (Lake and others 2000).
Bioinformatic prediction of variant pathogenicity
The effect of identified variants on RNA splicing and protein structure or function was assessed using web-based prediction tools. Each variant was assessed by at least two predictions using independent algorithms. NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) (Brunak and others 1991), NNSplice (http://www.fruitfly.org/seq_tools/splice.html) (Reese and others 1997) and HSF (http://www.umd.be/HSF/) (Desmet and others 2009) were used to evaluate a variant's effect on splice sites. To estimate the effect of non-synonymous coding variants, we used Pmut (http://mmb2.pcb.ub.es:8080/PMut/) (Ferrer-Costa and others 2005), PolyPhen (http://genetics.bwh.harvard.edu/pph/) (Ramensky and others 2002), nnPredict (http://www.cmpharm.ucsf.edu/~nomi/nnpredict.html) (Kneller and others 1990), and Phyre (http://www.sbg.bio.ic.ac.uk/phyre/) (Kelley and Sternberg 2009).
RESULTS
Family-based association analysis
We assessed the presence of common risk variants in the imprinted region at 7q32 by performing family-based association analysis on a subset of 71 ASD families with the strongest linkage scores at 7q32 interval (see Materials and Methods). We focused on the ~220 kb region that is flanked by two recombination hotspots and contains TSGA14, MEST, COPG2, TSGA13, and KLF14 genes (Fig. 1). Fourteen SNPs were genotyped: 12 SNPs representing 3 haploblocks within the region (tag SNPs are rs12706933, rs1421140 and rs4067228) and 2 SNPs located outside (rs6467308 and rs290805 are tag SNPs for TSGA14 and KLF14 genic regions, respectively). While rs290805 is in a strong LD with rs4067228 and rs4067229 from the haploblock III, rs6467308 is not in LD with either haploblock (Supplement Table III). Hence, the 14 SNPs represent an effective number of 4 independent markers. Nominal association signals were observed in SNPs from two haploblocks: a small 9.3-kb block at the 5' end of TSGA14 (haploblock I) and a large 110-kb block that includes all of MEST and COPG2 and part of TSGA13 (haploblock II, Table I A). Upon confining analysis to strict diagnosis of autism, only rs2287371 signal from haploblock I was statistically significant. This signal remained significant upon analysis of Caucasians only (Table I B). There was no evidence of allelic fixation between haploblocks I and II in affected individuals, which suggests the presence of different founders in our linkage-positive ASD families (as tested by Hbat for all ethnicities, and for Caucasians only, data not shown). We then genotyped one SNP from haploblocks I and II each in the entire set of 348 multiplex families with ASD. Both SNPs showed trend of association (Table I), but did not reach significance after correction for multiple testing.
Fig. 1.
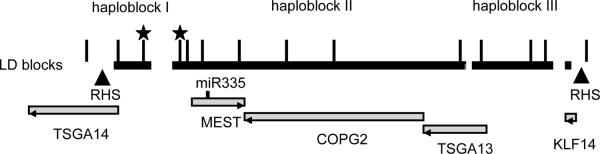
LD structure and genes in the 220 kb region at 7q32. LD blocks are indicated by black boxes. Triangles mark recombination hotspots. Long ticks mark approximate locations of 14 SNP from Table I on the physical map (not in scale). SNPs selected for genotyping of the entire set of ASD families are marked by stars. Grey shaded boxes denote protein-coding genes. miR-335 is marked by a short tick. Gene orientation is shown by arrowheads.
Table I.
Family-based association testing in multiplex families with ASD. (A) All ethnicities are analyzed. (B) Only Caucasians are analyzed. Settings: fbat; -e option, additive model. (ASD) all subjects with ASD count as affected, (AUT) only subjects with strict diagnosis of Autism count as affected, other diagnoses count as unknown. Underlined are P-values which reached significance after Bonferroni correction for multiple testing.
|
A
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Haploblock | Allele | Frequency | Linkage-positive families (N=71) | All multiplex families (N=348) | ||||||
|
| |||||||||||
| Z (ASD) | P (ASD) | Z (AUT) | P (AUT) | Z (ASD) | P (ASD) | Z (AUT) | P (AUT) | ||||
| rs6467308 | G | 0.42 | 0.65 | 0.519 | 0.75 | 0.451 | |||||
| rs2287371 | I | G | 0.50 | 2.42 | 0.016 | 2.81 | 0.005 | ||||
| rs12706933 | I | A | 0.48 | 2.48 | 0.013 | 2.68 | 0.007 | 2.39 | 0.017 | 2.05 | 0.040 |
| rs13245645 | II | G | 0.63 | 2.46 | 0.014 | 2.40 | 0.017 | 1.96 | 0.050 | 2.24 | 0.025 |
| rs1421140 | II | T | 0.67 | 2.43 | 0.015 | 2.40 | 0.016 | ||||
| rs2301335 | II | A | 0.61 | 2.18 | 0.029 | 1.95 | 0.051 | ||||
| rs2072573 | II | T | 0.61 | 1.98 | 0.047 | 1.83 | 0.067 | ||||
| rs2072575 | II | A | 0.58 | 2.18 | 0.030 | 2.02 | 0.043 | ||||
| rs2129905 | II | G | 0.59 | 2.32 | 0.020 | 2.09 | 0.036 | ||||
| rs1038638 | II | A | 0.59 | 2.14 | 0.032 | 1.91 | 0.056 | ||||
| rs4731699 | III | C | 0.74 | 1.77 | 0.077 | 1.81 | 0.070 | ||||
| rs4067228 | III | A | 0.62 | 1.87 | 0.061 | 1.46 | 0.146 | ||||
| rs4067229 | III | T | 0.49 | 1.88 | 0.060 | 1.52 | 0.129 | ||||
| rs290805 | III | C | 0.80 | 0.86 | 0.391 | 0.87 | 0.384 | ||||
|
B
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Haploblock | Allele | Frequency | Caucasian linkage-positive families (N=59) | All Caucasian multiplex families (N=260) | ||||||
|
| |||||||||||
| Z (ASD) | P (ASD) | Z (AUT) | P (AUT) | Z (ASD) | P (ASD) | Z (AUT) | P (AUT) | ||||
| rs6467308 | G | 0.40 | 0.16 | 0.875 | 0.33 | 0.741 | |||||
| rs2287371 | I | G | 0.50 | 2.54 | 0.011 | 3.05 | 0.002 | ||||
| rs12706933 | I | A | 0.49 | 2.46 | 0.014 | 2.71 | 0.007 | 2.16 | 0.031 | 2.03 | 0.043 |
| rs13245645 | II | G | 0.63 | 2.37 | 0.018 | 2.38 | 0.018 | 2.35 | 0.019 | 2.67 | 0.008 |
| rs1421140 | II | T | 0.67 | 2.47 | 0.013 | 2.50 | 0.013 | ||||
| rs2301335 | II | A | 0.61 | 2.14 | 0.033 | 1.95 | 0.051 | ||||
| rs2072573 | II | T | 0.61 | 1.93 | 0.054 | 1.82 | 0.069 | ||||
| rs2072575 | II | A | 0.59 | 2.13 | 0.033 | 2.03 | 0.043 | ||||
| rs2129905 | II | G | 0.60 | 2.29 | 0.022 | 2.10 | 0.035 | ||||
| rs1038638 | II | A | 0.60 | 2.09 | 0.036 | 1.91 | 0.057 | ||||
| rs4731699 | III | C | 0.75 | 1.83 | 0.067 | 1.89 | 0.058 | ||||
| rs4067228 | III | A | 0.63 | 1.65 | 0.100 | 1.27 | 0.205 | ||||
| rs4067229 | III | T | 0.49 | 1.70 | 0.089 | 1.39 | 0.165 | ||||
| rs290805 | III | C | 0.81 | 0.57 | 0.571 | 0.72 | 0.473 | ||||
Mutation search in the candidate genes
With the exception of TSGA13, the protein-coding genes and microRNA residing in the nominally associated region are expressed in the developing brain and/or have documented roles in CNS development (Supplement Table I). For each candidate gene, we sequenced the coding regions with adjacent splice sites in a discovery cohort of 94 unrelated subjects with ASD (see Materials and Methods).
The translated part of MEST contained only one synonymous SNP, rs61735155 (no co-transmission with disease, Supplement Table IV). We also examined intronic sequence variations surrounding miR-335. A known polymorphism situated 20 nt 3' of miR-335 (rs41272366) co-segregated with the strict diagnosis of autism in 7 of 8 families in our discovery cohort, but its frequency did not significantly differ between patient and control groups (8/94 patients with ASD versus 5/95 controls; Fisher's exact two tailed p = 0.40). Also, the rs41272366 minor allele was not associated with disease status in the whole ASD family sample (data not shown). In COPG2 we found two synonymous changes, one non-synonymous SNP and a 15 nt indel, which either were neutral and/or did not co-segregate with disease.
In TSGA14, we identified three variants that are not listed in SNP databases and that affected either mRNA splicing or conserved protein residues. All variants were all inherited from unaffected fathers and co-segregated with affected status in children (Fig. 2 A–D). The c.192+5G>A is predicted to abolish (NetGene2) or significantly weaken (NNSplice, SSF) the donor splice site. Two identified non-synonymous variants, P206A and C240G, are located in exons 8 and 9 which constitute a conserved Rhodanese Homology Domain (RHOD; NCBI ID: cd00158) of the TSGA14 protein. Pro-206 and Cys-240 are conserved across multiple species (Supplement Fig. 1). C240G has immediate pathogenic effect on the protein structure according to both Pmut and PolyPhen. P206A, which was found in two unrelated families, is classified as “probably damaging” by PolyPhen, “neutral” by Pmut, and likely to spread to the alpha-helical domain by nnPredict.
Fig. 2.
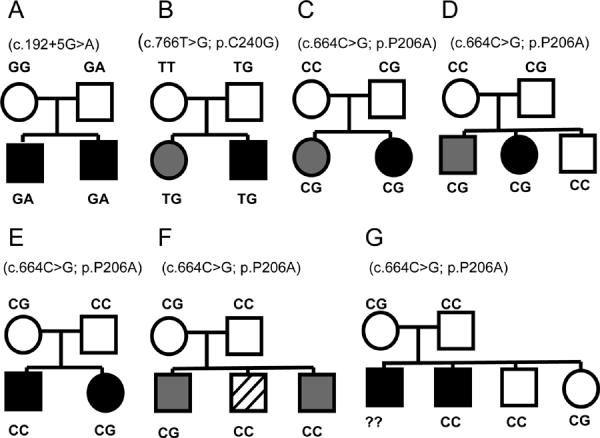
Transmission of TSGA14 variants in families with ASD. (A–D) Families with positive linkage scores, (E) zero linkage scores, (F–G) unknown linkage status. In (B) family, mother is Caucasian and father has more than 2 races, in all other families both parents are Caucasians. Circles indicates females, squares indicates males. White filling indicates unaffected, black filling indicates a diagnosis of autism, gray filling indicates a diagnosis of pervasive developmental disorder, and hatched filling indicates a learning disability. Corresponding TSGA14 variants are shown in parentheses.
Distribution of the TSGA14 variants in the entire ASD sample and control groups
We followed up on the detection of P206A, C240G and c.192+5G>A in the discovery cohort by genotyping our entire ASD sample and controls. 348 individuals with autism (1 subject from each family) were compared to 670 unrelated controls (Table II). When considered together, the TSGA14 variants were enriched in a linkage-positive fraction of ASD (4/94 versus 2/670 controls; Fisher's exact two-tailed P = 0.003), but when considered individually, they did not reach significance. Likewise, cumulative TSGA14 variants were enriched in total ASD cases (6/348 ASD versus 2/670 controls; Fisher's exact two tailed p = 0.022 for all ethnicities, and 5/260 ASD versus 1/624 controls; Fisher's exact two tailed p = 0.010 for Caucasians only).
Table II.
Distribution of TSGA14 variants in cases and controls. (A) All ethnicities are analyzed. (B) Caucasians only are analyzed. 348 unrelated affected subjects from 348 multiplex ASD families and 670 control subjects from 2 control groups (see Materials and Methods for the description of controls).
|
A
| |||||||
|---|---|---|---|---|---|---|---|
| Exon | Nt change | Aa change | Discovery cohort (N=94) | ASD sample (N=348) | Control 1 (N=170) | Control 2 (N=500) | Total control (N=670) |
| e3 + 5 nt | c.192+5 G> A | na | 1 | 1 | 0 | 0 | 0 |
| e9 | c.766T>G | C240G | 1 | 1 | 0 | 0 | 0 |
| e8 | C.664C> G | P206A | 2 | 4 | 1 | 1 | 2 |
|
| |||||||
| Cumulative TSGA14 variants | 4 | 6 | 1 | 1 | 2 | ||
|
B
| |||||||
|---|---|---|---|---|---|---|---|
| Exon | Nt change | Aa change | ASD sample (N=260) | Control 1 (N=134) | Control 2(N=490) | Total control (N=624) | |
| e3 + 5 nt | c.192+5 G> A | na | 1 | 0 | 0 | 0 | |
| e9 | c.766T>G | C240G | 0 | 0 | 0 | 0 | |
| e8 | C.664C> G | P206A | 4 | 0 | 1 | 1 | |
|
| |||||||
| Cumulative TSGA14 variants | 5 | 0 | 1 | 1 | |||
To trace transmission of TSGA14 variants within families we then genotyped all available members of the 348 ASD families (Fig. 2). Two variants, c.192+5G>A and C240G, were found only in ASD families positive for linkage at 7q32, and within these families these variants co-segregated with the autism phenotype in the children. P206A was present in five ASD families: two positive for linkage, one with zero scores and two with unknown linkage status. P206A of paternal origin was always co-transmitted with disease whereas maternal P206A behaved as a neutral variant.
Analysis of the TSGA14 allele-specific expression
Even though the TSGA14 is reported to have bi-allelic expression in the brain and other tissues, the proximity of imprinted genes could bias the expression in favor of one of the alleles (Hogart and others 2007). In such a case, underexpression of the maternal allele may increase the penetrance of otherwise recessive or hypomorphic variants in the paternal allele. To test this, we designed an allele specific expression (ASE) assay as previously described. (He and others 2005) (see Supplement Methods). Analysis of lymphoblastoid cell lines from an ASD family (Fig. 2C) in which P206A was transmitted from the father revealed that allelic differences in TSGA14 expression were not parent-of-origin specific (data not shown).
Functional analysis of the TSGA14 c.192+5G>A variant
We sought to confirm the occurrence and type of abnormally spliced TSGA14 transcripts bearing the c.192+5G>A variant by a cell-based splicing assay. TSGA14 splice forms include the ubiquitous long (L) form, which encodes a centrosome-binding protein, has 11 exons, and is abundant in the brain, and the short (S) form, which encodes a testis-specific peptide (Fig. 3A). Because the identified TSGA14 variants would only affect the L form we omitted from the minigene construct sequences instructing S form splicing. Minigene constructs carrying either G or A at position +5 of intron 3, were transiently transfected into rat PC12 and human HEK-293T cells. Bands corresponding to endogenous TSGA14 transcripts in brain tissues and non-transfected human cells were observed in human as well as in rat cell lines transfected with the wild-type minigene (Fig. 3B). Aberrant transcripts were detected only in the cells transfected with TSGA14 c.192+5G>A mutant construct. In both cell lines transfected with the mutant constructs aberrant transcripts constituted 58% of total expressed TSGA14. Sequencing of these RT-PCR products demonstrated the production of a normally spliced allele and an erroneously spliced mRNA in which the entire exon 3 was skipped. Thus, upon cell transfection, the c.192+5G>A variant produces a protein lacking 16 amino acids of its internal sequence and with an additional G33D substitution (Fig. 3C).
Fig. 3.
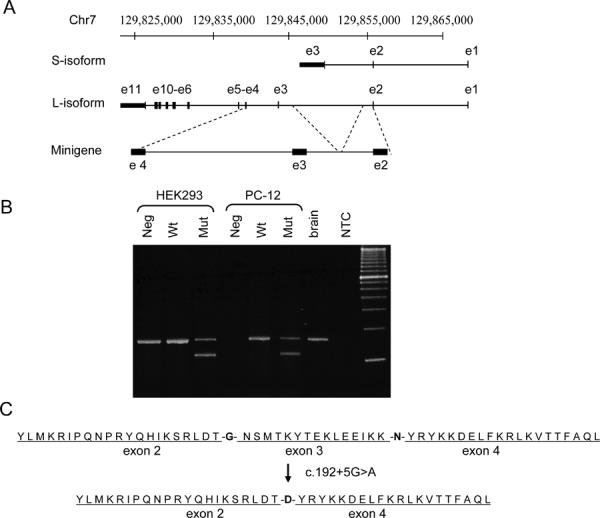
Abnormal TSGA14 splicing detected by ex vivo splicing assay. (A) Minigene construct. Scheme of the human TSGA14 genomic locus with the S isoform (SwissProt ID Q9BYV8-4), the L isoform (SwissProt IDr Q9BYV8-1), and a part of the TSGA14 gene cloned into expression vector pRcRSV. (B) RT-PCR products of spliced TSGA14 transcripts. Neg - untransfected cells, Wt - cells transfected with a minigene construct carrying wild-type allele (c.192+5G), Mut - cells transfected with a minigene construct carrying c.192+5A mutation, brain - RT-PCR product from control human brain mRNA, NTC - no template control PCR. (C) Exon 3 skipping, which results in G33D change and the deletion of 16 amino acids.
DISCUSSION
We hypothesized that both common and rare risk variants contribute to the ASD linkage signal at 7q32, and that they could be identified via combination of association analysis and mutation screening. Following the reported parent-of-origin biases at 7q32 we focused on the cluster of imprinted genes in the D7S530- D7S640 interval. Less than 2% of human genes are imprinted, yet they are heavily associated with neurodevelopmental pathology (Horsthemke and Buiting 2008; Keverne and Curley 2008). We analyzed the 220 kb region comprising imprinted MEST, COPG2, and KLF14 for association with ASD and detected nominal positive signals with two haploblocks. Only rs2287371 residing in the 5' non-translated region of TSGA14 passed a conservative Bonferroni correction for multiple testing (4 markers in 4 datasets) in 7q32-linked ASD families. Given that the trend of association in the whole sample is apparently driven by linkage-positive families, a common susceptibility variant(s) in this region seem unlikely. The observed association may be explained by the presence of less common or even rare causative variants which may create “synthetic associations” by occurring, stochastically, more often in association with one of the alleles at the common site versus the other allele (Dickson and others 2010). It is also plausible that low risk common variants cannot be detected in this study because of insufficient power of analysis (see Supplement Table V).
Candidate gene resequencing in linkage-positive ASD families revealed three putatively pathogenic mutations that affected mRNA splicing and conserved amino acids in the TSGA14 protein. P206A and C240G occur within a conserved Rhodanese domain and hence may confer similar defects to TSGA14 structure/function. A low allele frequency of an amino acid variant can, by itself, serve as a predictor of its functional significance (Kryukov and others 2007). c.192+5G>A has a disruptive effect on the donor splicing site in intron 3 confirmed by an ex vivo splicing assay. This mutation favors skipping of the entire exon 3 in both human and rat cell lines resulting in a protein with 16 missing amino acids of internal sequence and an additional G33D change that may confer a dominant negative phenotype. The effect observed in cell lines may differ from brain-specific regulation; however, exon 3 is a constitutive one, as defined by 363 GenBank accessions from 341 cDNA clones, including 59 from brain (AceView).
Cumulative frequency of TSGA14 variants in 7q32 positive ASD families significantly exceeded their frequency in the general population, although much larger case-control samples would be required to test each variant individually. For instance, when TSGA14 variants are counted cumulatively, the present study has a statistical power of 81% for Caucasians only, and of 74% for all ethnicities. Individual testing of the most abundant P206A variant at 80% power requires a minimum population size of 820 for both case and control groups. Accordingly, a minimum of 6820 cases/controls is required to test less frequent C240G and c.192+5g>A variants (assuming their frequency in a general population is 1 per 1000).
Interestingly, all three variants in the 7q32-linkage positive families were transmitted paternally. Transmission of P206A in the entire sample of ASD pedigrees may be consistent with origin-dependent penetrance, i.e., being benign or hypomorphic on the maternal allele, and being deleterious on the paternal allele. Such an effect could be due to differential expression of TSGA14 alleles or to a cis-interaction with another risk factor on the same allele (e.g., common variant in the regulatory element). A neighborhood effect on non-imprinted genes was observed for GABAA receptor genes located in the imprinted region at 15q11-13. Normally, these genes are biallelically expressed in the brain, but become subject to epigenetic dysregulation in ASD brains (Hogart and others 2007). Origin-specific expression bias was not found in lymphoblast cell lines of an ASD family carrying P206A in TSGA14, but we cannot rule out the possibility that origin-guided regulation of this gene occurs in the developing brain.
The TSGA14 mutations occur in the L isoform that encodes a centrosomal and microtubule-binding protein conserved in vertebrates (Andersen and others 2003; Gache and others 2010). Structural and functional integrity of the centrosome is critical for mammalian neurogenesis (Higginbotham and Gleeson 2007). In early neurodevelopment, asymmetric division of radial glia progenitors accounts for nearly all neurogenesis in the developing mammalian neocortex (Noctor and others 2004), and it is guided by asymmetry of centrosome partition (Wang and others 2009b). Mutations in a set of centrosomal proteins are responsible for microcephalies (Supplement Table VI), which are thought to result from depletion of progenitors essential for brain growth (Fish and others 2006; Griffith and others 2008; Rauch and others 2008).
Directed migrations of neurons along glial fibers are essential for the development of the laminar architecture of cortical regions of the mammalian brain and ultimately patterning of synaptic connectivity (Hatten 2002). In the migrating neuron, the centrosome is positioned ahead of the nucleus and guides the direction of migration. Mutations in another set of centrosomal and microtubule-associated proteins (Supplement Table VI), specifically affect nucleus–centrosome connection during neuronal migration (Kerjan and Gleeson 2007). This is exemplified in lissencephaly (literally “smooth brain”), in which the brains lack cortical furrowing.
Recent structural analyses (Andersen and others 2003) revealed that 75% of centrosomal proteins contain coiled-coil regions and only a few had any other motifs or domains. This raises the intriguing question of how these coiled-coil proteins cooperate to form a pericentriolar matrix. Secondary structure prediction (Supplement Fig. 2) indicates a significant decrease in apha-superhelix formation in the TSGA14 variant with deleted exon 3 suggesting altered folding and/or protein-protein interactions. Because TSGA14 protein is found in a salt-soluble fraction (Andersen and others 2003), the impaired coiled-domain formation of the mutant with deleted exon 3 may reduce TSGA14's loose association to the centrosomal scaffold.
In conclusion, we report an initial analysis of common and rare genetic variation in the imprinted region at 7q32 in families with ASD. Several rare substitutions of interest were identified in TSGA14, all inherited from unaffected parents. This is an expected finding in multiplex ASD families, in which risk alleles are likely to be inherited rather than occurring de novo. The variants identified in this study may represent functional risk factors in autism because (1) they are enriched in subjects with ASD; (2) they affect sequences conserved in multiple species; (3) changes in the protein are suggestive of malfunction in a dominant negative fashion; (4) amino acid substitutions located in a RHOD domain indicate a possible mutation hot spot; (5) the centrosomal and microtubule-binding function of TSGA14 makes it a plausible ASD candidate gene.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to the members of families with ASD who participated in our study. We thank Drs. Ulrike Schwarze, Brian Kraemer and Beate Peters for help with analysis of splicing and critical discussions, Dr. Chris Guthrie for help with cell cultures, Mrs. Elaine Loomis for her outstanding technical assistance, and Mr. Andrew David for his editorial assistance. This work was supported by grants from the National Institute of Child Health and Human Development (U19HD34565, P50HD066782, P50HD055782 and R01HD-55741) and the National Institute of Mental Health (U54MH066399).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- Alarcon M, Cantor RM, Liu J, Gilliam TC, Geschwind DH. Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. Am J Hum Genet. 2002;70(1):60–71. doi: 10.1086/338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426(6966):570–4. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy M, Lin S, Cook EH, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82(1):160–4. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley-Koch A, Wolpert CM, Menold MM, Zaeem L, Basu S, Donnelly SL, Ravan SA, Powell CM, Qumsiyeh MB, Aylsworth AS, et al. Genetic studies of autistic disorder and chromosome 7. Genomics. 1999;61(3):227–36. doi: 10.1006/geno.1999.5968. [DOI] [PubMed] [Google Scholar]
- Association AP. American Psychiatric Association; Arlington: 1994. Diagnostic and statistical manual of mental disorders; pp. 65–78. [Google Scholar]
- Badner JA, Gershon ES. Regional meta-analysis of published data supports linkage of autism with markers on chromosome 7. Mol Psychiatry. 2002;7(1):56–66. doi: 10.1038/sj.mp.4000922. [DOI] [PubMed] [Google Scholar]
- Benayed R, Gharani N, Rossman I, Mancuso V, Lazar G, Kamdar S, Bruse SE, Tischfield S, Smith BJ, Zimmerman RA, et al. Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. Am J Hum Genet. 2005;77(5):851–68. doi: 10.1086/497705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42(6):489–91. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220(1):49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- Brune CW, Korvatska E, Allen-Brady K, Cook EH, Jr., Dawson G, Devlin B, Estes A, Hennelly M, Hyman SL, McMahon WM, et al. Heterogeneous association between engrailed-2 and autism in the CPEA network. Am J Med Genet B Neuropsychiatr Genet. 2007 doi: 10.1002/ajmg.b.30585. [DOI] [PubMed] [Google Scholar]
- Cook EH., Jr. Genetics of autism. Child Adolesc Psychiatr Clin N Am. 2001;10(2):333–50. [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8(1):e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39(1):25–7. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, Cook EH, Jr., Skinner C, Schwartz CE, Sommer SS. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett. 2006;409(1):10–3. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21(14):3176–8. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci U S A. 2006;103(27):10438–43. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gache V, Waridel P, Winter C, Juhem A, Schroeder M, Shevchenko A, Popov AV. Xenopus meiotic microtubule-associated interactome. PLoS One. 2010;5(2):e9248. doi: 10.1371/journal.pone.0009248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, Vernay B, Al Sanna N, Saggar A, Hamel B, Earnshaw WC, et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet. 2008;40(2):232–6. doi: 10.1038/ng.2007.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME. New directions in neuronal migration. Science. 2002;297(5587):1660–3. doi: 10.1126/science.1074572. [DOI] [PubMed] [Google Scholar]
- He H, Olesnanik K, Nagy R, Liyanarachchi S, Prasad ML, Stratakis CA, Kloos RT, de la Chapelle A. Allelic variation in gene expression in thyroid tissue. Thyroid. 2005;15(7):660–7. doi: 10.1089/thy.2005.15.660. [DOI] [PubMed] [Google Scholar]
- Higginbotham HR, Gleeson JG. The centrosome in neuronal development. Trends Neurosci. 2007;30(6):276–83. doi: 10.1016/j.tins.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Hogart A, Nagarajan RP, Patzel KA, Yasui DH, Lasalle JM. 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum Mol Genet. 2007;16(6):691–703. doi: 10.1093/hmg/ddm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsthemke B, Buiting K. Genomic imprinting and imprinting defects in humans. Adv Genet. 2008;61:225–46. doi: 10.1016/S0065-2660(07)00008-9. [DOI] [PubMed] [Google Scholar]
- Hutcheson HB, Bradford Y, Folstein SE, Gardiner MB, Santangelo SL, Sutcliffe JS, Haines JL. Defining the autism minimum candidate gene region on chromosome 7. Am J Med Genet B Neuropsychiatr Genet. 2003;117(1):90–6. doi: 10.1002/ajmg.b.10033. [DOI] [PubMed] [Google Scholar]
- IMGSAC A full genome screen for autism with evidence for linkage to a region on chromosome 7q. International Molecular Genetic Study of Autism Consortium. Hum Mol Genet. 1998;7(3):571–8. doi: 10.1093/hmg/7.3.571. [DOI] [PubMed] [Google Scholar]
- IMGSAC Further characterization of the autism susceptibility locus AUTS1 on chromosome 7q. Hum Mol Genet. 2001;10(9):973–82. doi: 10.1093/hmg/10.9.973. [DOI] [PubMed] [Google Scholar]
- Jacob S, Brune CW, Carter CS, Leventhal BL, Lord C, Cook EH., Jr. Association of the oxytocin receptor gene (OXTR) in Caucasian children and adolescents with autism. Neurosci Lett. 2007;417(1):6–9. doi: 10.1016/j.neulet.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–9. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson L, Ljunggren E, Bremer A, Pedersen C, Landen M, Thuresson K, Giacobini M, Melke J. Mutation screening of melatonin-related genes in patients with autism spectrum disorders. BMC Med Genomics. 2010;3:10. doi: 10.1186/1755-8794-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4(3):363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kerjan G, Gleeson JG. Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly. Trends Genet. 2007;23(12):623–30. doi: 10.1016/j.tig.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Keverne EB, Curley JP. Epigenetics, brain evolution and behaviour. Front Neuroendocrinol. 2008;29(3):398–412. doi: 10.1016/j.yfrne.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Kneller DG, Cohen FE, Langridge R. Improvements in protein secondary structure prediction by an enhanced neural network. J Mol Biol. 1990;214(1):171–82. doi: 10.1016/0022-2836(90)90154-E. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80(4):727–39. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):S36–42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lake SL, Blacker D, Laird NM. Family-based tests of association in the presence of linkage. Am J Hum Genet. 2000;67(6):1515–25. doi: 10.1086/316895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb JA, Barnby G, Bonora E, Sykes N, Bacchelli E, Blasi F, Maestrini E, Broxholme J, Tzenova J, Weeks D, et al. Analysis of IMGSAC autism susceptibility loci: evidence for sex limited and parent of origin specific effects. J Med Genet. 2005;42(2):132–7. doi: 10.1136/jmg.2004.025668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson-Yuen A, Saldivar JS, Sommer S, Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet. 2008;16(5):614–8. doi: 10.1038/sj.ejhg.5202006. [DOI] [PubMed] [Google Scholar]
- Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet. 1998;20(2):163–9. doi: 10.1038/2464. [DOI] [PubMed] [Google Scholar]
- Liu X, Kawamura Y, Shimada T, Otowa T, Koishi S, Sugiyama T, Nishida H, Hashimoto O, Nakagami R, Tochigi M, et al. Association of the oxytocin receptor (OXTR) gene polymorphisms with autism spectrum disorder (ASD) in the Japanese population. J Hum Genet. 2010;55(3):137–41. doi: 10.1038/jhg.2009.140. [DOI] [PubMed] [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook EH, Jr., Leventhal BL, DiLavore PC, Pickles A, Rutter M. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30(3):205–23. [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24(5):659–85. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81(6):1289–97. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321(5886):218–23. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7(2):136–44. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Pickles A, Bolton P, Macdonald H, Bailey A, Le Couteur A, Sim CH, Rutter M. Latent-class analysis of recurrence risks for complex phenotypes with selection and measurement error: a twin and family history study of autism. Am J Hum Genet. 1995;57(3):717–26. [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton A, Gauthier J, Hamdan FF, Lafreniere RG, Yang Y, Henrion E, Laurent S, Noreau A, Thibodeau P, Karemera L, et al. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz D, Laird N. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered. 2000;50(4):211–23. doi: 10.1159/000022918. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319(5864):816–9. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311–23. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Sathyan P, Golden HB, Miranda RC. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J Neurosci. 2007;27(32):8546–57. doi: 10.1523/JNEUROSCI.1269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg GD, Dawson G, Sung YJ, Estes A, Munson J, Rosenthal E, Rothstein J, Flodman P, Smith M, Coon H, et al. Evidence for multiple loci from a genome scan of autism kindreds. Mol Psychiatry. 2006;11(11):1049–60. 979. doi: 10.1038/sj.mp.4001874. [DOI] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39(3):319–28. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikalinos TA, Karvouni A, Zintzaras E, Ylisaukko-oja T, Peltonen L, Jarvela I, Ioannidis JP. A heterogeneity-based genome search meta-analysis for autism-spectrum disorders. Mol Psychiatry. 2006;11(1):29–36. doi: 10.1038/sj.mp.4001750. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009a;459(7246):528–33. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tsai JW, Imai JH, Lian WN, Vallee RB, Shi SH. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature. 2009b;461(7266):947–55. doi: 10.1038/nature08435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461(7265):802–8. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Wu S, Jia M, Ruan Y, Liu J, Guo Y, Shuang M, Gong X, Zhang Y, Yang X, Zhang D. Positive association of the oxytocin receptor gene (OXTR) with autism in the Chinese Han population. Biol Psychiatry. 2005;58(1):74–7. doi: 10.1016/j.biopsych.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, Law K, Law P, Qiu S, Lord C, Sebat J, et al. A unified genetic theory for sporadic and inherited autism. Proc Natl Acad Sci U S A. 2007;104(31):12831–6. doi: 10.1073/pnas.0705803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.