

Abstract
Neuroinflammation is a common pathological event observed in many different brain diseases, frequently associated with blood brain barrier (BBB) dysfunction and followed by cerebral edema. Neuroinflammation is characterized with microglia activation and astrogliosis, which is a hypertrophy of the astrocytes. Astrocytes express aquaporin 4, the water channel protein, involved in water homeostasis and edema formation. Aside from its function in water homeostasis, recent studies started to show possible interrelations between aquaporin 4 and neuroinflammation. In this review the roles of aquaporin 4 in neuroinflammation associated with BBB disruption and cerebral edema will be discussed with recent studies in the field.
Keywords: Aquaporin, Astrocyte, Blood brain barrier, Stroke, Traumatic brain injury, Neuroinflammation
The purpose of this review is not to discuss neuroinflammation mechanisms or an extensive review of aquaporin 4 (AQP4), for there are numerous reviews covering these two topics independently. Rather, we address the question whether AQP4 is a common player between edema and neuroinflammation by reviewing the recent literature in the field. In recent years, AQP4 has been associated with neuroinflammation in chronic and acute brain diseases [1-12]. Since AQP4 is mostly expressed on the astrocytes, and because neuroinflammation is characterized by both phenotypic changes of resting astrocytes to astrogliosis and microglial activation, we believe that tackling this question can lead to a better understanding of many brain diseases.
General introduction
Aquaporin 4 - overview
Aquaporin (AQP) is a family of water channel protein ubiquitously expressed in various cell types and organisms [13]. The aquaporin family exhibits a common structure with six membrane spanning alpha helical domains, a consensus motif composed of Asparagine-Proline-Alanine (NPA) constituting part of the pore, and an approximate molecular weight of 30 kDa [14]. AQP4 is the most abundant AQP found in the primate and rodent brains, mainly in the perivascular astrocyte endfeet [13]. AQP4 is assembled in homo-tetramers where each individual aquaporin represents a water channel (Figure 1A) [15]. The assemblage of four molecules of AQP4 forms a central pore, through which water, cations, and gases such as CO2 flow [16]. Interestingly, AQP4 proteins are major constituents of a higher structural arrangement within astrocyte endfoot named the orthogonal arrays of particles (OAPs) observed using electron microscopy after cryo-fracture preparation [17] (Figure 1B). The size of the OAPs is determined by the ratio between the two main isoforms of AQP4: the long (AQP4-m1) and short (AQP4-m23) splice variants (Figure 1B) [17]. The AQP4-m23 isoform stabilizes the OAP structure [17,18]. The exact functional roles of the OAPs remain unknown both in normal and pathological brains. An increase in the AQP4 m1 variant disrupts the structure of the OAPs [17,18], which is observed in stroke [19,20] and parallels blood–brain barrier (BBB) disruption. The role of AQP4 within the perivascular space and in BBB structure is still a matter of discussion and unresolved [21,22]. Interestingly, OAPs were proposed to play a role in potassium buffering [23,24]. Strengthening this hypothesis, AQP4 found in the astrocyte endfeet facing cerebral blood vessels co-localized with the potassium channel, Kir4.1 [25,26]. AQP4 knockout mice (AQP4−/−) showed a delay in potassium reuptake suggesting that AQP4 has a role in potassium homeostasis in facilitating water diffusion along the potassium gradient during brain activity [27]. Not only involved in water movement, AQP4 may also contribute to cell adhesion [28] and migration [29]. These data underline the diversity and complexity in AQP4 functions, and the subsequent sections will show another possible function of AQP4 in the process of neuroinflammation.
Figure 1.

Structural organization of AQP4 in the astrocyte membrane. (A) Schematic drawing of the AQP4 homo-tetramer assembly within the lipid membrane from a lateral view resulting in a central pore permeable to cations and gases (green arrows) [16]. Each individual aquaporin facilitates bi-directional water movement that is dependent on the osmotic gradient (blue arrows) (modified from Badaut et al. [57]). (B) In normal brain, association between AQP4-m1 (red circles) and AQP4-m23 (blue circles) isoforms contribute to form orthogonal array of particles (OAPs). Higher expression of AQP4-m23 contributes to the formation of large OAPs, and should facilitate the gas, ion (green arrows), and water diffusion (water arrows) through the astrocyte membrane. (C) In brain injury, increase of AQP4-m1 [19] should contribute to disruption of OAPs (modified from Badaut et al. [57]). Changes in OAP size may decrease the number of central pore and possibly affect not only water movement but also the ion and gas movements.
Microglia, astrocytes, and AQP4 in the context of neuroinflammation
Neuroinflammation is largely described in the acute phase after brain injury, along with edema, as well as in chronic brain diseases like multiple sclerosis. The term, ‘neuroinflammation’, encompasses several molecular and cellular modifications without a clear, unified definition amongst the various brain diseases and injuries. However, it is important to realize that neuroinflammation is distinct from peripheral inflammation [30-32], even if they share some of the same molecular players, particularly due to microglia and astrocytes, which are cells specific to the CNS. Since the inflammatory process may differ from organ to organ [31], Graeber and colleagues recently drew the attention to the potential danger of using the two terminologies ‘neuroinflammation’ and ‘inflammation’ interchangeably.
Although microglia is the cell type considered to be primarily responsible for the innate immune response in the CNS [33,34], it is premature to conclude that a decrease in microglial activation is the only evidence needed for the treatment of neuroinflammation with anti-inflammatory drugs [30]. Activation status for both astrocytes and microglia, along with secretion of cytokines and chemokines, should be considered for neuroinflammation. Microglial activation is characterized by morphological changes in which the usually ramified microglia becomes round with no ramifications [33]. There seems to be a dual role for microglial activation in which acute activation is beneficial [35] but a chronic one is detrimental [33,36]. In fact, activated microglia is responsible for producing pro-inflammatory cytokines such as IL-1β, IL-6, TNFα and also anti-inflammatory cytokines such as IL-4, IL-10 and TFGβ [37].
Activated astrocytes also play a key role in neuroinflammation with their involvement in astrogliosis, although whether astrogliosis is beneficial or detrimental seem to depend on the situation [38], much like microglial activation. The process of astrogliosis includes the hyptertrophy of astrocytes with different morphological fates depending on the severity of the injury [39]. The absence of AQP4 in astrocytic endfeet may lead to decreased hypertrophy of astrocytes due to decreased water entry and migration toward the site of the injury [29,40]. Like microglia, activated astrocytes contribute to the secretion of chemokines and cytokines (see examples above), possibly involved in BBB disruption and vasogenic edema. Interestingly, AQP4 is upregulated in the vasogenic edema resolution phase visualized by normalization of magnetic resonance (MR) signals in several disease models [2,41]. Furthermore, AQP4 has also been reported to be present in reactive microglia after intranigral injection of lipopolysaccharide (LPS) in rats, although the functional significance of this de novo microglial AQP4 expression is unknown [42]. These changes in AQP4 during the inflammatory process suggest changes in water movement related to neuroinflammation.
AQP4 and neuroinflammation in autoimmune diseases
Experimental autoimmune encephalomyelitis (EAE) and AQP4
Recent data in a model of experimental autoimmune encephalomyelitis (EAE) in which homogenized guinea-pig whole spinal cord was injected into rats showed upregulation of AQP4 starting at 10 days until the onset and peak of cerebellar enlargement. At these timepoints, significantly positive correlation was observed between AQP4 and BBB disruption in the cerebellum, associated with a decrease of tight junction proteins such as occludin [7]. This detrimental role of AQP4 in EAE is supported by a less severe clinical and tissue inflammation score after EAE and LPS-injection in AQP4−/− mice than WT animals [1]. This is most likely the cause of reduced production of the pro-inflammatory cytokines, TNFα and IL-6, observed in AQP4−/− mice astrocyte cultures [1].
AQP4−/− mice studies have also suggested that AQP4 could be contributing to the production of CD4+ and CD25+ T regulator cells; and lack of AQP4 may be disrupting the immunosuppressive regulators in Parkinson’s disease, leading to increased microglial activation and a worse outcome due to more dopaminergic neuronal loss after induction of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [5]. Interestingly, AQP4 expression is present in the spleen, lymph nodes, and thymus, hinting towards a more direct role of AQP4 in systemic immune responses, and perhaps not just confined to neuroinflammation [5].
Neuromyelitis optica (NMO) and AQP4
The possible link between neuroinflammation and AQP4 was advertised with neuromyelitis optica (NMO), a demyelinating disease. NMO is a pathological condition characterized by abnormal signals most often observed in the spinal cord and optic nerve, and in the form of blindness and paralysis. Interestingly, AQP4 has been identified as the target for NMO-IgG, a unique feature of the disease which differentiates it from multiple sclerosis [43-45], making it a very useful differential diagnostic tool in the clinics. More specifically, there is plausible evidence that NMO-IgG specifically targets AQP4 within the OAP structures, rather than free AQP4 isoforms [6,46,47]. Whether the presence of an autoantibody against AQP4 is the cause of the disease or a collateral consequence of some secondary pathological mechanisms still lacks an unanimous answer, but studies performed where immunoglobulins taken from AQP4 antibody positive NMO patients were administered to rats with EAE showed NMO pathology seen in the clinics [48,49], suggesting that the presence of AQP4 autoantibody in patients already suffering from neuroimmune disease worsens the condition and leads to the NMO pathology observed. Interestingly, several clinical observations have been reported in which patients with myasthenia gravis (MG) also suffer from auto-AQP4-antibody positive NMO simultaneously [50-56]. Thus pointing out the possibility of a common autoimmune origin for both diseases, or the aforementioned worsening effect of the AQP4 autoantibody in patients with pre-existing immune diseases; previously unrecognized because of the lack of knowledge about the NMO IgG auto-AQP4 antibody as a diagnostic tool for NMO. This link could point to the involvement of AQP4 in the peripheral immune system as well.
In summary, these recent data from NMO and AQP4−/− mice models are encouraging to propose that AQP4 is a player in inflammation and neuroinflammation. But considering AQP4 properties as a water channel, its function in these processes are still unclear.
Neuroinflammation and edema in brain injury: astrocyte AQP4
BBB breakdown and vasogenic edema
AQP4 is one of the key players in edema formation and resolution [57,58] and increase in its expression is observed in reactive astrocytes after injury. Edema is frequently observed in brain injuries and is associated with BBB disruption [57,59]. Compromised BBB integrity leads to plasma protein leakage and extravascular fluid accumulation [57]. The breakdown of the BBB is a complex process partially caused by the activation of matrix metalloproteinases (MMPs), which is part of the neuroinflammatory response [60-62]. Pro-inflammatory cytokines such as IL-1β and TNFα has been shown to produce MMP-9 and MMP-3 in cultured astrocytes and microglia (reviewed in [62]). MMP-9 aggravates vasogenic edema development by degrading the basal lamina located between the astrocytic endfeet and endothelia [62]. Of particular interest is the link of MMP with AQP4; MMP-2 and MMP-9 are known to degrade agrin and MMP-3 degrades dystroglycan [63], two proteins that have a critical role in the maintenance of the OAP [64-67]. So, when MMP are upregulated after a neuroinflammatory response, more AQP4-OAPs will be disorganized, leading to a possible disruption of the BBB and edema. Vasogenic edema development can further damage the endothelia by increased water volume and therefore increased hydrostatic pressure. Thus, if there is decreased BBB disruption, there will be less pro-inflammatory cytokines, MMPs, and edema (Figure 2).
Figure 2.
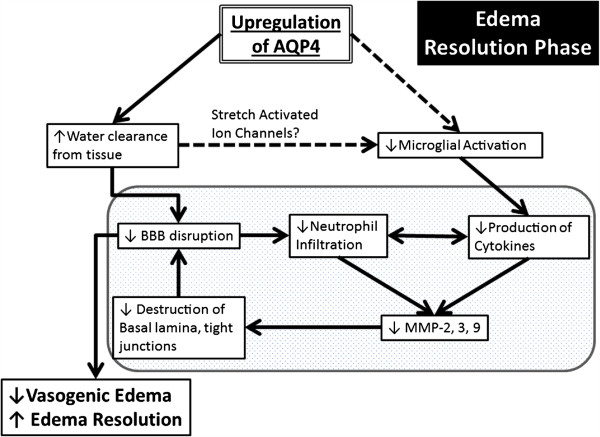
Schematic summary of a beneficial role AQP4 upregulation plays during the edema resolution phase. The upregulation of AQP4 causes increased water clearance from the tissue, which in turn causes decreased BBB disruption because of decreased pressure, and there is less neutrophil infiltration and decreased pro-inflammatory cytokines. This cause decreased MMP production [62], which possibly results in less destruction of the basal lamina and tight junctions, causes an even greater decrease of the BBB. In another pathway (dotted lines), the increased water clearance from the tissue and extracellular space causes changes in the osmotic pressure, changing the activation state of the stretch activated ion channels expressed in microglia [86-88], causing less microglial activation, thus causing decreased pro-inflammatory cytokine. The resulting decrease in BBB disruption/permeability leads to decreased vasogenic edema or better edema resolution. Finally, this scheme outlines the potential link between AQP4, edema and neuroinflammation.
Magnetic resonance imaging and AQP4
One useful modality in assessing injury severity and outcome in cerebral pathological conditions both in clinics and research is magnetic resonance imaging (MRI). Because MRI detects changes observed via the excitation of water molecules, the presence of the water channel protein, AQP4 in astrocytes suggests a possible involvement in MRI. Diffusion-weighted magnetic resonance imaging (DWI) is widely used as a diagnostic tool in clinical and research settings to assess edematous damage after various brain pathologies from ischemic stroke to various neuroinflammatory diseases [57,68-71]. The apparent diffusion coefficient (ADC) is obtained from DWI and is used to evaluate cerebral changes in clinical and experimental models. A decrease in the ADC is classically associated with a decrease in the extracellular space during cell swelling after brain injury [70]. More recently, ADC changes have been hypothesized to be linked with the level of expression of AQP4. Several experiments have shown increases in AQP4 expression and increased ADC [71] and decreased AQP4 expression with decreased ADC [58,72]. Of note, Tourdias et al. [2] have recently shown in a model of focal inflammation that AQP4 upregulation was associated with early edema formation via increased ADC, peak BBB disruption, and increased pro-inflammatory cytokine secretion. Diffusion tensor imaging (DTI) takes into account the non-uniform directionality of water flow (anisotropy) in the brain. This anisotropy has mainly been attributed to myelinated neuronal axons in the white matter tract, but recent evidence has hinted towards the possible role of astrocytes and glial scars in DTI signal changes after traumatic brain injury [73]. In fact, increased anisotropy was correlated with reactive astrocytes and not with axonal changes in the perilesional cortex [73]. This idea is supported by a study showing a correlation with changes in DTI signals associated with hypertrophic astrocytes and increase of AQP4 [74]. As AQP4 expression changes after brain injury in astrocytes and microglia, it is rational to think that MRI may be a useful tool to evaluate the evolution of the neuroinflammatory process, especially in conjunction with AQP4 and edema.
Edema resolution in inflammatory conditions and AQP4
In focal brain inflammation, AQP4 expression is upregulated during the edema resolution phase at 2 to 14 days post injury [2]. However the exact role of this increase in AQP4 is still a matter of discussion. In brain injection of l-α-lysophosphatidylcholine, a significant increase in AQP4 expression was observed at the edema resolution phase (7, 14, and 20 days post injection) compared to the edema build-up phase (1 and 3 days post injection). In this model, the edema resolution phase was defined as a return to baseline for ADC values and a lower IL-1β mRNA level, compared to the edema build-up phase [2]. Interestingly, a similar observation was made in a model of juvenile traumatic brain injury with upregulation of AQP4 observed during the edema resolution period when ADC is returning to normal [41]. These data suggest that the presence of AQP4 plays a positive role during edema resolution by facilitating water extravasations from the brain parenchyma to liquid compartments including CSF and blood vessels (Figure 2).
In stroke pathophysiology, animals with a pre-existing inflammatory condition had aggravated stroke outcomes as seen by more edema and BBB damage at 24 h after injury compared to groups with no pre-existing inflammation in the periphery [75]. Pre-existing systemic inflammation induced a surge in the levels of IL-1 in the ischemic cerebral cortex [75]. Interestingly, increase in IL-1α expression bordering dilated blood vessels in the ipsilateral cortex was observed, signifying a possible direct role of pro-inflammatory cytokines on edema formation [75]. Because IL-1β has been shown to induce AQP4 in astrocytes [76,77], and blocking either AQP4 through gene deletion [78] or IL-1β through anti-IL-1β antibody [79] was seen to decrease edema, AQP4 may be a possible target for systemic inflammation leading to increased edema. In a mouse model of atherosclerosis (APOE−/− mice under high fat diet), development of chronic inflammation due to adhesion of a large number of T cells and macrophages in the vasculature [80], as well as microglial activation in the brain is observed [81]. These mice upon aging showed BBB leakage and higher astrogliosis associated with increased AQP4 [4]. These changes may contribute to a worse outcome in aged atherosclerotic patients who suffer an ischemic stroke because of higher risk of edema.
AQP4 and microglial activation after injury
There are recent interesting data concerning the relationship between AQP4 and microglial activation. A link between neuroinflammation and AQP4 was described using the AQP4−/− mice that are more susceptible to seizures (decreased seizure latency and increased seizure severity) compared to WT 1 month after TBI and associated with a decrease in neuroinflammatory processes [82]. This difference is related to the neuroinflammatory response showing less astrogliosis and increased microglial activation in AQP4−/− compared to WT mice. Minocycline injection in AQP4−/− inhibited the increase in microglia and also mitigated the severity of the post-traumatic seizure [82]. Similar observations were reported in a model of cryoinjury with increased microglia and reduced astrogliosis in AQP4−/− mice compared to WT at 7 and 14 days post injury. In this model the authors reported a decrease in the lesion volume and lower neuronal loss in AQP4−/− mice compared to WT at 1 day after injury, and the opposite result at 7 and 14 days [83]. Similarly, in adult rats, intravenous minocycline administration after TBI [84] and subarachnoid hemorrhage [85] resulted in less BBB disruption associated with decreased MMP9 and AQP4 at 1 day post injury. In our lab, we have also observed that treatment with small interference RNA (siRNA) targeted against AQP4 (siAQP4) after juvenile TBI showed a decrease in AQP4 associated with less BBB disruption, edema, more NeuN positive cells, and better behavior outcomes compared to the control group at 3 days post injury (unpublished data). As shown in the adult model, we have also noticed a significant increase of activated microglia cells and decreased astrogliosis around the lesion at 3 days post injury in siAQP4-treated rats compared to controls (unpublished data). All together, these data underline that changes in AQP4 expression are associated with changes in astrogliosis and microglia activation in acute brain injury (Figure 3). Astrogliosis may require the presence of AQP4 to facilitate the water movement necessary for the migration [29,40] and hypertrophy. However, the mechanism behind the decrease of the AQP4 and activation of microglia is less obvious and still unknown. One possible mechanism behind the changes observed in post-traumatic or ischemic microglia activation and cytokine release in response to AQP4 downregulation or inhibition may be partly due to the presence of stretch-activated Cl- channels expressed in microglia [86,87]. Stretch-activated/swelling-activated Cl- channels are activated by osmotic stress [88]. It has been observed that the activation of these channels contributes the maintenance of the non-activated (ramified) phenotype of microglia [86]. Because AQP4 is responsible for water transport, inhibition of AQP4 either through genetic deletion or siRNA will alter the osmotic stress within the extracellular space surrounding the microglia, changing the activation status of the swelling activated chloride channels, resulting in microglial activation (Figures 2 and 3). Another possibility lies in the cross-talk that occurs between astrogliosis and microglial activation [34]. It is possible that the decreased extent of injury-induced reactive astrogliosis as a result of knocking down AQP4 caused increased microglial activity.
Figure 3.
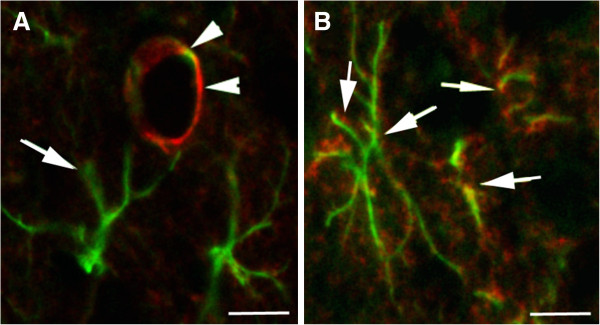
AQP4 distribution in the astrocyte in normal cortex and after brain injury. (A) Confocal picture of AQP4 immunostaining (red, arrow heads) in normal brain shows the presence of the water channel protein on the astrocyte endfoot (GFAP staining, green, arrow) in contact to the blood vessels in the cortex. (B) Confocal pictures of the AQP4 immunostaining (red) on reactive astrocytes revealed with GFAP immunolabelling (green) in the cortex after traumatic brain injury. The presence of the AQP4 staining is not only localized on the endfeet in contact to the blood vessels but also distributed in all astrocyte processes (arrows). Scale bar 10 μm.
In summary, the presence of AQP4 seems to play a detrimental role acutely, but at a later phase starting from around 7 days post injury for at least 1 month, AQP4 may play a beneficial role that seems to be involved with inhibiting activation of microglia and promoting edema resolution.
Conclusion
As reviewed, AQP4 has a key role in the edema process, which may be followed by ADC changes in different brain pathologies. However, whether the presence of AQP4 is beneficial or detrimental seem to depend on the timepoint and injury models. In vasogenic edema, where the BBB is compromised, AQP4 seems to play a beneficial role in eliminating accumulating water from the extracellular space of the CNS (Figure 2). Edema is frequently associated with neuroinflammation with activation of the microglia and astrogliosis. The upregulation of AQP4 previously associated with edema recovery may also contribute to the neuroinflammatory process in astrogliosis and microglia inactivation. In fact, absence or decrease of AQP4 is associated with decrease of astrogliosis and increase of microglia activation. However, the molecular mechanisms of the water channel, AQP4, with the inflammation process are still unknown. But these recent data are encouraging to hypothesize that AQP4 could be a common denominator between edema and neuroinflammation, and underscores the importance of independent investigation to understand how AQP4 is contributing to the neuroinflammatory and edematous process.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
AF and JB wrote and edited the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Andrew M Fukuda, Email: afukuda@llu.edu.
Jerome Badaut, Email: jbadaut@llu.edu.
Acknowledgements
This work was supported by the National Institute of Health NINDS R01HD061946. We would like to thank Dr. Melanie Price (Neurology Department, Lausanne University Hospital, Switzerland) for critical reading and constructive suggestions.
References
- Li L, Zhang H, Varrin-Doyer M, Zamvil SS, Verkman AS. Proinflammatory role of aquaporin-4 in autoimmune neuroinflammation. FASEB J. 2011;25:1556–1566. doi: 10.1096/fj.10-177279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourdias T, Mori N, Dragonu I, Cassagno N, Boiziau C, Aussudre J, Brochet B, Moonen C, Petry KG, Dousset V. Differential aquaporin 4 expression during edema build-up and resolution phases of brain inflammation. J Neuroinflammation. 2011;8:143. doi: 10.1186/1742-2094-8-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS. Aquaporins in clinical medicine. Annu Rev Med. 2012;63:303–316. doi: 10.1146/annurev-med-043010-193843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Copin JC, Fukuda AM, Gasche Y, Schaller K, da Silva RF. Increase of arginase activity in old apolipoprotein E deficient mice under Western diet associated with changes in neurovascular unit. J Neuroinflammation. 2012;9:132. doi: 10.1186/1742-2094-9-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y, Fan Y, He L, Liu W, Wen X, Zhou S, Wang X, Zhang C, Kong H, Sonoda L, Tripathi P, Li CJ, Yu MS, Su C, Hu G. Novel role of aquaporin-4 in CD4+ CD25+ T regulatory cell development and severity of Parkinson’s disease. Aging Cell. 2011;10:368–382. doi: 10.1111/j.1474-9726.2011.00677.x. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, Fallier-Becker P, Noell S, Lennon VA. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci USA. 2012;109:1245–1250. doi: 10.1073/pnas.1109980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XN, Wang WZ, Fu J, Wang HB. The relationship between aquaporin-4 expression and blood–brain and spinal cord barrier permeability following experimental autoimmune encephalomyelitis in the rat. Anat Rec (Hoboken) 2011;294:46–54. doi: 10.1002/ar.21286. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, Kumanogoh A, Kajiyama K, Yoshikawa H, Sakoda S. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen-specific T cells. Biochem Biophys Res Commun. 2010;394:205–210. doi: 10.1016/j.bbrc.2010.02.157. [DOI] [PubMed] [Google Scholar]
- Villaran RF, de Pablos RM, Arguelles S, Espinosa-Oliva AM, Tomas-Camardiel M, Herrera AJ, Cano J, Machado A. The intranigral injection of tissue plasminogen activator induced blood–brain barrier disruption, inflammatory process and degeneration of the dopaminergic system of the rat. Neurotoxicology. 2009;30:403–413. doi: 10.1016/j.neuro.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Pham H, Doerrbecker J, Ramp AA, D’Souza CS, Gorasia DG, Purcell AW, Ayers MM, Orian JM. Experimental autoimmune encephalomyelitis (EAE) IN C57Bl/6 mice is not associated with astrogliosis. J Neuroimmunol. 2011;232:51–62. doi: 10.1016/j.jneuroim.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Zador Z, Stiver S, Wang V, Manley GT. Role of aquaporin-4 in cerebral edema and stroke. Handb Exp Pharmacol. 2009;190:159–170. doi: 10.1007/978-3-540-79885-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang H, Verkman AS. Greatly attenuated experimental autoimmune encephalomyelitis in aquaporin-4 knockout mice. BMC Neurosci. 2009;10:94. doi: 10.1186/1471-2202-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Lasbennes F, Magistretti PJ, Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab. 2002;22:367–378. doi: 10.1097/00004647-200204000-00001. [DOI] [PubMed] [Google Scholar]
- Gonen T, Walz T. The structure of aquaporins. Q Rev Biophys. 2006;39:361–396. doi: 10.1017/S0033583506004458. [DOI] [PubMed] [Google Scholar]
- Yu J, Yool AJ, Schulten K, Tajkhorshid E. Mechanism of gating and ion conductivity of a possible tetrameric pore in aquaporin-1. Structure. 2006;14:1411–1423. doi: 10.1016/j.str.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Musa-Aziz R, Chen LM, Pelletier MF, Boron WF. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc Natl Acad Sci USA. 2009;106:5406–5411. doi: 10.1073/pnas.0813231106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Davidson KGV, Yasumura T, Furman CS. Freeze-fracture and immunogold analysis of aquaporin-4 (AQP4) square arrays, with models of AQP4 lattice assembly. Neuroscience. 2004;129:915–934. doi: 10.1016/j.neuroscience.2004.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman CS, Gorelick-Feldman DA, Davidson KG, Yasumura T, Neely JD, Agre P, Rash JE. Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci USA. 2003;100:13609–13614. doi: 10.1073/pnas.2235843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt L, Ternon B, Price M, Mastour N, Brunet JF, Badaut J. Protective role of early aquaporin 4 induction against postischemic edema formation. J Cereb Blood Flow Metab. 2009;29:423–433. doi: 10.1038/jcbfm.2008.133. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Iwasaki Y, Yamamoto T, Konno H, Yoshimoto T, Suzuki J. Disintegration of orthogonal arrays in perivascular astrocytic processes as an early event in acute global ischemia. Brain Res. 1984;300:141–145. doi: 10.1016/0006-8993(84)91348-9. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Tait MJ, Reza A, Davies DC, Bell BA, Verkman AS, Papadopoulos MC. AQP4 gene deletion in mice does not alter blood–brain barrier integrity or brain morphology. Neuroscience. 2009;161:764–772. doi: 10.1016/j.neuroscience.2009.03.069. [DOI] [PubMed] [Google Scholar]
- Zhou J, Kong H, Hua X, Xiao M, Ding J, Hu G. Altered blood–brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport. 2008;19:1–5. doi: 10.1097/WNR.0b013e3282f2b4eb. [DOI] [PubMed] [Google Scholar]
- Grange-Messent V, Bouchaud C. Effects of soman on cerebral astrocyte plasma membranes: a freeze-fracture study. Neurosci Lett. 1994;178:77–80. doi: 10.1016/0304-3940(94)90294-1. [DOI] [PubMed] [Google Scholar]
- Grange-Messent V, Raison D, Bouchaud C. Compared effects of extracellular K + ions and soman, a neurotoxic, on cerebral astrocyte morphology. An in vitro study. J Submicrosc Cytol Pathol. 1996;28:151–159. [PubMed] [Google Scholar]
- Badaut J, Nehlig A, Verbavatz J, Stoeckel M, Freund-Mercier MJ, Lasbennes F. Hypervascularization in the magnocellular nuclei of the rat hypothalamus: relationship with the distribution of aquaporin-4 and markers of energy metabolism. J Neuroendocrinol. 2000;12:960–969. doi: 10.1046/j.1365-2826.2000.00539.x. [DOI] [PubMed] [Google Scholar]
- Badaut J, Verbavatz JM, Freund-Mercier MJ, Lasbennes F. Presence of aquaporin-4 and muscarinic receptors in astrocytes and ependymal cells in rat brain: a clue to a common function? Neurosci Lett. 2000;292:75–78. doi: 10.1016/S0304-3940(00)01364-1. [DOI] [PubMed] [Google Scholar]
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K. Implications of the Aquaporin-4 structure on array formation and cell adhesion. J Mol Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. 2005;118:5691–5698. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Li W, Rodriguez ML. Role of microglia in CNS inflammation. FEBS Lett. 2011;585:3798–3805. doi: 10.1016/j.febslet.2011.08.033. [DOI] [PubMed] [Google Scholar]
- Kelley KW, Dantzer R. Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav Immun. 2011;Suppl 1:S13–S20. doi: 10.1016/j.bbi.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Liu W, Tang Y, Feng J. Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci. 2011;89:141–146. doi: 10.1016/j.lfs.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, Vexler ZS. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. 2011;31:12992–13001. doi: 10.1523/JNEUROSCI.2102-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman ML, Puig KL, Combs CK, Rosenberger TA. Acetate reduces microglia inflammatory signaling in vitro. J Neurochem. 2012;123:555–567. doi: 10.1111/j.1471-4159.2012.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, Verkman AS. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 2007;21:108–116. doi: 10.1096/fj.06-6848com. [DOI] [PubMed] [Google Scholar]
- Fukuda AM, Pop V, Spagnoli D, Ashwal S, Obenaus A, Badaut J. Delayed increase of astrocytic aquaporin 4 after juvenile traumatic brain injury: Possible role in edema resolution? Neuroscience. 2012;222:366–378. doi: 10.1016/j.neuroscience.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas-Camardiel M, Venero JL, de Pablos RM, Rite I, Machado A, Cano J. In vivo expression of aquaporin-4 by reactive microglia. J Neurochem. 2004;91:891–899. doi: 10.1111/j.1471-4159.2004.02759.x. [DOI] [PubMed] [Google Scholar]
- Marignier R, Giraudon P, Vukusic S, Confavreux C, Honnorat J. Anti-aquaporin-4 antibodies in Devic's neuromyelitis optica: therapeutic implications. Ther Adv Neurol Disord. 2010;3:311–321. doi: 10.1177/1756285610382478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- Nicchia GP, Mastrototaro M, Rossi A, Pisani F, Tortorella C, Ruggieri M, Lia A, Trojano M, Frigeri A, Svelto M. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia. 2009;57:1363–1373. doi: 10.1002/glia.20855. [DOI] [PubMed] [Google Scholar]
- Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J Biol Chem. 2011;286:16516–16524. doi: 10.1074/jbc.M111.227298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, Gilden D, Nessler S, Stadelamann C, Hemmer B. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66:617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, Adzemovic M, Bauer J, Berger T, Fujihara K, Itoyama Y, Lassmann H. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66:630–643. doi: 10.1002/ana.21837. [DOI] [PubMed] [Google Scholar]
- Uzawa A, Mori M, Iwai Y, Kobayashi M, Hayakawa S, Kawaguchi N, Kuwabara S. Association of anti-aquaporin-4 antibody-positive neuromyelitis optica with myasthenia gravis. J Neurol Sci. 2009;287:105–107. doi: 10.1016/j.jns.2009.08.040. [DOI] [PubMed] [Google Scholar]
- Ogaki K, Hirayama T, Chijiiwa K, Fukae J, Furuya T, Noda K, Fujishima K, Hattori N, Takahashi T, Okuma Y. Anti-aquaporin-4 antibody-positive definite neuromyelitis optica in a patient with thymectomy for myasthenia gravis. Neurologist. 2012;18:76–79. doi: 10.1097/NRL.0b013e318247bc91. [DOI] [PubMed] [Google Scholar]
- Jarius S, Paul F, Franciotta D, de Seze J, Munch C, Salvetti M, Ruprecht K, Liebetrau M, Wandinger KP, Akman-Demir G, Melms A, Kristoferitsch W, Wildemann B. Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler. 2012;18:1135–1143. doi: 10.1177/1352458511431728. [DOI] [PubMed] [Google Scholar]
- Leite MI, Coutinho E, Lana-Peixoto M, Apostolos S, Waters P, Sato D, Melamud L, Marta M, Graham A, Spillane J, Villa AM, Callegaro D, Santos E, da Silva AM, Jarius S, Howard R, Nakashima I, Giovanni G, Buckley C, Hilton-Jones D, Vincent A, Palace J. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology. 2012;78:1601–1607. doi: 10.1212/WNL.0b013e31825644ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemadifar M, Abtahi SH, Dehghani A, Abtahi MA, Akbari M, Tabrizi N, Goodarzi T. Myasthenia gravis during the dourse of neuromyelitis optica. Case Rep Neurol. 2011;3:268–273. doi: 10.1159/000334128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaknin-Dembinsky A, Abramsky O, Petrou P, Ben-Hur T, Gotkine M, Brill L, Brenner T, Argov Z, Karussis D. Myasthenia gravis-associated neuromyelitis optica-like disease: an immunological link between the central nervous system and muscle? Arch Neurol. 2011;68:1557–1561. doi: 10.1001/archneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- Kay CS, Scola RH, Lorenzoni PJ, Jarius S, Arruda WO, Werneck LC. NMO-IgG positive neuromyelitis optica in a patient with myasthenia gravis but no thymectomy. J Neurol Sci. 2008;275:148–150. doi: 10.1016/j.jns.2008.06.038. [DOI] [PubMed] [Google Scholar]
- Badaut J, Ashwal S, Obenaus A. Aquaporins in cerebrovascular disease: a target for treatment of brain edema? Cerebrovasc Dis. 2011;31:521–531. doi: 10.1159/000324328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Ashwal S, Adami A, Tone B, Recker R, Spagnoli D, Ternon B, Obenaus A. Brain water mobility decreases after astrocytic aquaporin-4 inhibition using RNA interference. J Cereb Blood Flow Metab. 2011;31:819–831. doi: 10.1038/jcbfm.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezowski V, Fukuda AM, Cecchelli R, Badaut J. Endothelial cells and astrocytes: a concerto en duo in ischemic pathophysiology. Int J Cell Biol. 2012;2012:176287. doi: 10.1155/2012/176287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell A, Ortega-Aznar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina CA, Lo EH, Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood–brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.STR.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983–994. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg-Buchholz K, Mack AF, Steiner E, Pfeiffer F, Engelhardt B, Wolburg H. Loss of astrocyte polarity marks blood–brain barrier impairment during experimental autoimmune encephalomyelitis. Acta Neuropathol. 2009;118:219–233. doi: 10.1007/s00401-009-0558-4. [DOI] [PubMed] [Google Scholar]
- Fallier-Becker P, Sperveslage J, Wolburg H, Noell S. The impact of agrin on the formation of orthogonal arrays of particles in cultured astrocytes from wild-type and agrin-null mice. Brain Res. 2011;1367:2–12. doi: 10.1016/j.brainres.2010.09.092. [DOI] [PubMed] [Google Scholar]
- Noell S, Wolburg-Buchholz K, Mack AF, Beedle AM, Satz JS, Campbell KP, Wolburg H, Fallier-Becker P. Evidence for a role of dystroglycan regulating the membrane architecture of astroglial endfeet. Eur J Neurosci. 2011;33:2179–2186. doi: 10.1111/j.1460-9568.2011.07688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noell S, Fallier-Becker P, Deutsch U, Mack AF, Wolburg H. Agrin defines polarized distribution of orthogonal arrays of particles in astrocytes. Cell Tissue Res. 2009;337:185–195. doi: 10.1007/s00441-009-0812-z. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Wolburg-Buchholz K, Mack A, Fallier-Becker P. Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood–brain barrier. Neuroscientist. 2009;15:180–193. doi: 10.1177/1073858408329509. [DOI] [PubMed] [Google Scholar]
- Tourdias T, Hiba B, Raffard G, Biran M, Nishiguchi T, Aussudre J, Franconi JM, Brochet B, Petry KG, Dousset V. Adapted focal experimental autoimmune encephalomyelitis to allow MRI exploration of multiple sclerosis features. Exp Neurol. 2011;230:248–257. doi: 10.1016/j.expneurol.2011.04.023. [DOI] [PubMed] [Google Scholar]
- Chastain CA, Oyoyo UE, Zipperman M, Joo E, Ashwal S, Shutter LA, Tong KA. Predicting outcomes of traumatic brain injury by imaging modality and injury distribution. J Neurotrauma. 2009;26:1183–1196. doi: 10.1089/neu.2008.0650. [DOI] [PubMed] [Google Scholar]
- Obenaus A, Ashwal S. Magnetic resonance imaging in cerebral ischemia: focus on neonates. Neuropharmacology. 2008;55:271–280. doi: 10.1016/j.neuropharm.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Tourdias T, Dragonu I, Fushimi Y, Deloire MS, Boiziau C, Brochet B, Moonen C, Petry KG, Dousset V. Aquaporin 4 correlates with apparent diffusion coefficient and hydrocephalus severity in the rat brain: a combined MRI-histological study. Neuroimage. 2009;47:659–666. doi: 10.1016/j.neuroimage.2009.04.070. [DOI] [PubMed] [Google Scholar]
- Meng S, Qiao M, Lin L, Del Bigio MR, Tomanek B, Tuor UI. Correspondence of AQP4 expression and hypoxic-ischaemic brain oedema monitored by magnetic resonance imaging in the immature and juvenile rat. Eur J Neurosci. 2004;19:2261–2269. doi: 10.1111/j.0953-816X.2004.03315.x. [DOI] [PubMed] [Google Scholar]
- Budde MD, Janes L, Gold E, Turtzo LC, Frank JA. The contribution of gliosis to diffusion tensor anisotropy and tractography following traumatic brain injury: validation in the rat using Fourier analysis of stained tissue sections. Brain. 2011;134:2248–2260. doi: 10.1093/brain/awr161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsan LA, Poulet P, Guignard B, Parizel N, Skoff RP, Ghandour MS. Astrocytic hypertrophy in dysmyelination influences the diffusion anisotropy of white matter. J Neurosci Res. 2007;85:935–944. doi: 10.1002/jnr.21201. [DOI] [PubMed] [Google Scholar]
- Denes A, Ferenczi S, Kovacs KJ. Systemic inflammatory challenges compromise survival after experimental stroke via augmenting brain inflammation, blood–brain barrier damage and brain oedema independently of infarct size. J Neuroinflammation. 2011;8:164. doi: 10.1186/1742-2094-8-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Yamamoto N, Arima H, Hirate H, Morishima T, Umenishi F, Tada T, Asai K, Katsuya H, Sobue K. Interleukin-1beta induces the expression of aquaporin-4 through a nuclear factor-kappaB pathway in rat astrocytes. J Neurochem. 2006;99:107–118. doi: 10.1111/j.1471-4159.2006.04036.x. [DOI] [PubMed] [Google Scholar]
- Laird MD, Sukumari-Ramesh S, Swift AE, Meiler SE, Vender JR, Dhandapani KM. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? J Neurochem. 2010;113:637–648. doi: 10.1111/j.1471-4159.2010.06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Clausen F, Hanell A, Israelsson C, Hedin J, Ebendal T, Mir AK, Gram H, Marklund N. Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2011;34:110–123. doi: 10.1111/j.1460-9568.2011.07723.x. [DOI] [PubMed] [Google Scholar]
- Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke. 2006;37:1923–1932. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- Crisby M, Rahman SM, Sylven C, Winblad B, Schultzberg M. Effects of high cholesterol diet on gliosis in apolipoprotein E knockout mice. Implications for Alzheimer’s disease and stroke. Neurosci Lett. 2004;369:87–92. doi: 10.1016/j.neulet.2004.05.057. [DOI] [PubMed] [Google Scholar]
- Lu DC, Zador Z, Yao J, Fazlollahi F, Manley GT. Aquaporin-4 reduces post-traumatic seizure susceptibility by promoting astrocytic glial scar formation in mice. J Neurotrauma. 2011. Sep 22 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Shi WZ, Zhao CZ, Zhao B, Zheng XL, Fang SH, Lu YB, Zhang WP, Chen Z, Wei EQ. Aquaporin-4 deficiency attenuates acute lesions but aggravates delayed lesions and microgliosis after cryoinjury to mouse brain. Neurosci Bull. 2012;28:61–68. doi: 10.1007/s12264-012-1063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, Ding JY, Dornbos D, Li X, Guthikonda M, Rossi NF, Ding Y. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood–brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114:92–101. doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- Wang Z, Meng CJ, Shen XM, Shu Z, Ma C, Zhu GQ, Liu HX, He WC, Sun XB, Huo L, Zhang J, Chen G. Potential contribution of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 to blood–brain barrier disruption and brain edema after experimental subarachnoid hemorrhage. J Mol Neurosci. 2012;48:273–280. doi: 10.1007/s12031-012-9769-6. [DOI] [PubMed] [Google Scholar]
- Eder C, Klee R, Heinemann U. Involvement of stretch-activated Cl- channels in ramification of murine microglia. J Neurosci. 1998;18:7127–7137. doi: 10.1523/JNEUROSCI.18-18-07127.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichter LC, Mertens T, Liu B. Swelling activated Cl- channels in microglia: Biophysics, pharmacology and role in glutamate release. Channels (Austin) 2011;5:128–137. doi: 10.4161/chan.5.2.14310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS, Ross PE, Cahalan MD. Chloride channels activated by osmotic stress in T lymphocytes. J Gen Physiol. 1993;101:801–826. doi: 10.1085/jgp.101.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]