

Abstract
The Atg1/ULK1 complex plays a central role in starvation-induced autophagy, integrating signals from upstream sensors such as MTOR and AMPK and transducing them to the downstream autophagy pathway. Much progress has been made in the last few years in understanding the mechanisms by which the complex is regulated through protein-protein interactions and post-translational modifications, providing insights into how the cell modulates autophagy, particularly in response to nutrient status. However, how the ULK1 complex transduces upstream signals to the downstream central autophagy pathway is still unclear. Although the protein kinase activity of ULK1 is required for its autophagic function, its protein substrate(s) responsible for autophagy activation has not been identified. Furthermore, examples of potential ULK1-independent autophagy have emerged, indicating that under certain specific contexts, the ULK1 complex might be dispensable for autophagy activation. This raises the question of how the autophagic machinery is activated independent of the ULK1 complex and what are the biological functions of such noncanonical autophagy pathways.
Keywords: AMPK, Atg1, kinase, MTOR, regulation
Introduction
Autophagy is a conserved catabolic process that utilizes lysosomal activity to turn over cellular proteins or organelles. Mammalian cells can undergo three primary types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy. These modes of autophagy differ in the type of cargo to be degraded and the methods of delivering them to lysosomes. Chaperone-mediated autophagy makes use of a protein translocation pathway that selectively feeds individual substrate proteins directly into the lysosome. Microautophagy occurs through direct sequestration of cargo involving the formation of tubular invaginations at the lysosomal membrane. Macroautophagy delivers cargo to the lysosomes through the formation and transport of specific intracellular membrane vesicles termed autophagosomes.1-3
Of the three types of autophagy, macroautophagy is thought to be the predominant form and is the most studied.2 The process of macroautophagy, herein referred to as autophagy, starts with the formation of a cup-shaped membrane structure, termed the phagophore. The phagophore elongates, sequesters cytosolic components (cargos), and eventually seals to form a double-membrane vesicle called the autophagosome. Cargo loading into autophagosomes may be selective for specific proteins, organelles or pathogens through the use of receptors such as Atg32 (yeast) and SQSTM1/p62 (mammals) or non-specifically through bulk loading of cytoplasmic contents.4 Autophagosomes fuse with lysosomes, resulting in cargo degradation and concomitant release of the metabolic byproducts such as amino acids and other molecules through permeases in the lysosomal membrane. As a membrane-trafficking pathway, autophagy shares several points of convergence with endocytosis, although these converging points, such as lysosomal fusion, can be differentially regulated in autophagy and endocytosis.5 The turnover of long-lived, aggregated or damaged proteins and organelles by autophagy is essential for maintaining cellular homeostasis. While autophagy occurs at basal levels under normal conditions, it is activated in response to cellular stresses such as nutrient starvation, hypoxia, growth factor withdrawal, endoplasmic reticulum (ER) stress, and pathogen infection. Upon these stresses, the cell employs autophagy either to redeploy its resources to tide over the period of stress or to degrade harmful components (such as damaged mitochondria or invading pathogens) via lysosomal degradation.6 Dysregulation of autophagy has been implicated in a range of diseases including neurodegenerative disorders most typically involving the accumulation of pathogenic proteins, inflammatory disorders such as Crohn disease, and cancer.7,8
The study of autophagy has accelerated in the last decade because of the discovery of autophagy-related (ATG) genes by yeast genetics9-11 and subsequently, their mammalian homologs.4,12 More than 36 ATG genes have been identified through genetic screens, and the ATG genes making up the core machinery of autophagy can be classified into several functional units including: the Atg1/ULK1 complex, commonly considered as an initiator in the autophagic cascade; the Vps34/PIK3C3 phosphatidylinositol 3-kinase (PtdIns3K) complex; two ubiquitin-like conjugation systems (Atg12–Atg5, Atg8/LC3–PE); the PtdIns3P effector Atg2-Atg18 complex; and the transmembrane protein Atg9. In this review we focus on the Atg1/ULK1 initiator complex, because of its important role as a bridge between nutrient sensing and induction of an autophagic response both in yeast and mammalian cells.
The yeast Atg1 Complex vs. the Mammalian ULK1 Complex
Atg1 and ULK1
In yeast, ATG1 was originally identified from genetic screens performed by the Ohsumi lab,9 Thumm lab,11 and Klionsky lab.10 Through these studies, multiple complementation groups affecting both autophagosome accumulation9,11 and cytoplasm-to-vacuole targeting (Cvt)10 were found. atg1 mutants fail to accumulate autophagic bodies in the presence of the protease inhibitor PMSF and die at a faster rate during nutrient starvation, linking autophagy and Atg1 with cellular metabolism.9 Atg1 is a serine-threonine kinase and the only protein kinase among the 36 yeast ATG genes described so far.
As with many other Atg proteins, Atg1 also has its mammalian ortholog. But the identification and validation of the mammalian orthologs took a few twists and turns. Atg1 shares strong homology with C. elegans uncoordinated-51 (UNC-51),13 which has two mammalian homologs known as Unc-51 like kinase-1 (ULK1) and ULK2 (Fig. 1). Therefore, ULK proteins had long been suspected to be mammalian counterparts of Atg1.14 The direct experimental evidence came from an RNAi-based screen by Tooze and colleagues to identify protein kinases involved in mammalian autophagy.15 In this screen they identified ULK1, but not ULK2, as an essential component for amino acid starvation-induced autophagy in HEK293 cells. Detailed mechanistic analysis confirms that ULK1 is indeed the functional equivalent of yeast Atg1.15,16 Because the sequence homology between ULK1 and ULK2 is significant (55% identical sequence), it is somewhat a surprise that RNAi knockdown of ULK1 was sufficient to block starvation-induced autophagy in HEK293 cells, whereas RNAi of ULK2 had no effect.15 Later studies indicate that ULK1 and ULK2 possess redundant roles in autophagy,17,18 and indeed, it takes a double knockout of Ulk1 and Ulk2 to completely block amino acid starvation-induced autophagy in mouse embryonic fibroblasts (MEFs).19 Mechanistically, both ULK1 and ULK2 are recruited to phagophores upon autophagy induction, both are able to bind to the regulatory proteins ATG13 and RB1CC1/FIP200, and their kinase-dead mutants have dominant negative properties (can block autophagy) when overexpressed in cells.20-22 Therefore, both ULK1 and ULK2 are the functional homologs of yeast Atg1, and the predominant autophagic role of ULK1 observed in HEK293 cells might simply be due to the much higher expression of ULK1 protein in these cells compared with that of ULK2. Additionally, it is highly likely that the existence of two Atg1 homologs provides additional mechanisms for differential regulation of autophagy in mammals.
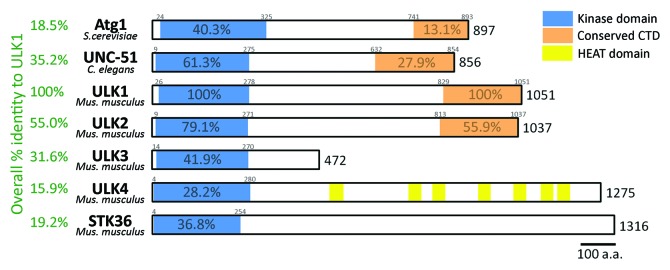
Figure 1. Atg1 and its homologous proteins in different organisms. The figure is a schematic representation of Atg1 (yeast), UNC-51 (C. elegans) and ULKs (mice). These proteins share high sequence identity and/or similarity mainly in the kinase domains. Significant similarity is also seen in the tail of Atg1, UNC-51, ULK1, and ULK2, defining a C-terminal domain of 150–250 amino acids. Overall sequence identity to mouse ULK1 as calculated by ClustalW is shown on the left-hand side.
ULK1 and ULK2 share significant homology in their C-terminal regions in addition to the N-terminal kinase domains. The role of the C-terminal regions of ULK1 and ULK2 in autophagy is intriguing. Domain studies of ULK1 have shown that its C-terminal domain (CTD) is required for both interaction with ATG13 and RB1CC1 (see below for more detail) and translocation of ULK1 to phagophores.15,20,21 Mammals have three additional protein kinases that are homologous to the kinase domains of ULK1 and ULK2. They are ULK3, ULK4 and STK36 (the latter is also known as “fused”) (Fig. 1). These additional ULKs do not have the conserved C-terminal sequence and are not thought to be involved in starvation-induced autophagy. It is, however, possible that they may play a role in other forms of autophagy. In support of this, ULK3 has been recently linked to autophagy induction during senescence.23
The Atg1 complex and the ULK1 complex
In yeast, Atg1 has been reported to interact with up to eight other Atg proteins, some of which are specific to autophagy (Atg13, Atg17, Atg29, Atg31) while others are required primarily for the Cvt pathway (Atg11, Atg20, Atg24 and Vac8).24 Both pathways are morphologically distinct, although similar, and at the molecular level they share many of the same Atg proteins. It is thought that Atg1 may function as a switch between autophagy and the Cvt pathway through changes in post-translational modifications or interacting partners. Atg17, Atg29 and Atg31 form a stable complex regardless of nutritional status, whereas recruitment of Atg13 to Atg1 may be triggered by nutrient starvation (Fig. 2).25-27 As will be discussed in more detail below, under nutrient-rich conditions, Atg13 is hyperphosphorylated in part by the nutrient-sensing kinase TOR, which is proposed to prevent its interaction with Atg1. According to this model, upon nutrient starvation, TOR is inactivated, leading to dephosphorylation of Atg13 and subsequent Atg1-Atg13 interaction and autophagy activation. Therefore, the regulated interaction between Atg13 and Atg1 dictates the autophagic function of Atg1, and the Atg1 complex. Contrary to this model, a recent paper by Kraft et al. suggests that Atg1 is constitutively bound to Atg13.28 In such a scenario, the phosphorylation of Atg13 may modulate Atg1 kinase activity through a change in conformation rather than regulated interaction. In addition, Atg1 kinase activity is reported to increase during starvation and requires Atg13 and Atg17 for maximal activation.25,29

Figure 2. Regulation of the Atg1 complex in yeast and the ULK1 complex in mammalian cells. In yeast, one model suggests that TORC1 dictates autophagy activation through regulating Atg1 complex formation: TORC1 phosphorylates Atg13 under nutrient-rich conditions, preventing complex formation (for simplicity, only Atg1, Atg13, and Atg17 are shown here); upon starvation, TORC1 is inactivated, thus the inhibitory phosphorylation on Atg13 is removed, triggering complex formation with Atg1 and Atg17. However, recent reports indicate that the Atg1-Atg13-Atg17 complex is constitutively formed in yeast. In mammals, the ULK1 complex is stable even under nutrient-rich conditions. Inhibitory phosphorylation by MTOR on ULK1 and ATG13 prevents complex activation, likely through a specific conformational change of the ULK1 complex. A red “P” indicates inhibitory phosphorylation, whereas blue “P” indicates potential activating phosphorylation.
In mammalian cells, there appears to be no equivalent of the yeast Cvt pathway. The main autophagic ULK1 complex consists of ULK1, ATG13, and RB1CC1 to mediate the signal of the nutrient-sensing kinase MTOR, as discovered by the laboratories of Mizushima,21 Jiang,30 and Kim.22 Mammalian ATG13 possesses mild similarity with C. elegans ATG-13, but its similarity with yeast Atg13 is rather limited;20,30,31 this is perhaps the reason why mammalian ATG13 was a relatively late discovery. RB1CC1 does not have an obvious sequence homolog in yeast, but functional analysis suggests it is the counterpart of yeast Atg17. Complex formation with ATG13 and ULK1 appears to promote the stability of ULK1, as a decrease in ULK1 protein levels is detected in atg13−/− cells and cells with ATG13 or RB1CC1 knockdown.21,30,32 In yeast, Atg13 is thought to mediate the interaction between Atg1 and Atg17, as Atg1 can interact with Atg13 in atg17∆ cells, but not with Atg17 in atg13∆ cells.25,33 In mammalian cells, ATG13 has been proposed to enhance the interaction between ULK1 and RB1CC1 as well.32 However, RB1CC1-ULK1 interaction, as detected by co-immunoprecipitation, is not affected in cells where ATG13 has been knocked down compared with control-depleted cells. Furthermore, ULK1 can interact with RB1CC1 or ATG13 in an in vitro binding assay, even in the absence of the other, demonstrating that ULK1 can interact directly with RB1CC1 in a manner independent of ATG13, and vice versa.30 Additionally, similar to the yeast scenario, both ATG13 and RB1CC1 can enhance the kinase activity of ULK1.22,30
Albeit functioning in a highly similar fashion, regulation of the yeast Atg1 complex and that of the mammalian ULK1 complex also have obvious dissimilarity (Fig. 2). Perhaps the most striking difference is that under both nutrient-rich and nutrient-starved conditions, the majority of ULK1 exists as part of a macromolecular complex containing ATG13 and RB1CC1.30,32 While starvation-induced Atg13 dephosphorylation and subsequent Atg1-Atg13 interaction may provide a simple mechanism for controlling the autophagy function of Atg1 in yeast, how does dephosphorylation of mammalian ATG13 activate ULK1, since ULK1-ATG13-RB1CC1 complex formation is constitutive? Can the phosphorylation status of ATG13 influence the conformation of the ULK1 complex, although not affect complex formation? Is the ULK1 complex subjected to more complex regulation through other post-translational modifications or interaction with other proteins? Do ATG13 and RB1CC1 have other functions beyond stabilizing ULK1 and enhancing its kinase activity, such as acting as scaffolds for recruitment of other proteins including the ULK1 substrate(s) whose phosphorylation by ULK1 is required for autophagy? Indeed, several other interacting partners of ULK1, some of which do not have putative homologs in baker’s yeast, have been reported, such as C12orf44/ATG101,34,35 AMBRA1,36 HSP90-CDC37,37 SYNGAP1,38 GABARAP,39 and FEZ1.40 It is yet to be firmly demonstrated whether and/or how these interactions are relevant to the role of ULK1 in autophagy.
Sensing the Upstream Signal
Previous work in both yeast and mammalian systems suggests that the ULK1 complex senses upstream signals, particularly the nutrient and energy signals, mainly via change of its phosphorylation status. However, this does not appear to be the complete story. Recent studies demonstrate the significance of other regulatory mechanisms, which provide additional versatility for cells to control the intensity and timing of ULK1-mediated autophagy (Figs. 3 and 4). In this section, we will review the current knowledge concerning how the ULK1 complex responds upon upstream stimulation. It should be emphasized that in order to monitor the effect of an upstream signal to the ULK1 complex, one should not only measure the eventual autophagy, but also the direct impact on the ULK1 complex per se, such as stimulation of ULK1 kinase activity and translocation of the ULK1 complex to phagophores.30

Figure 3. Domain structure and posttranslational modifications on ULK1. (A) ULK1 contains an N-terminal kinase domain, a C-terminal domain (CTD) that is conserved with the yeast and C. elegans counterparts, and a serine/proline-rich region in between that is the site for many post-translational modification events. ATG13 and RB1CC1/FIP200 interact with ULK1 through its CTD. The mapped interaction sites with members of the LC3 family and AMPK are indicated. The site of interaction with RPTOR is unresolved, as its interaction with either the kinase domain or the serine/proline-rich domain has been reported. (B) Post-translationally modified residues on ULK1. As many as 30 phosphorylation events have been reported for ULK1. For brevity, only events that have been experimentally verified are listed in the table. Similarly, only the two experimentally verified acetylation sites are listed in the table.

Figure 4. The ULK1 complex integrates upstream nutrient and energy signals to coordinate the induction of autophagy. The nutrient-sensing pathways for growth factors, amino acids and energy converge on ULK1 through unique post-translational modifications, which regulate the activity of this autophagy-induction complex. MTOR inhibits the induction of autophagy through regulation of the ULK1 complex, which undergoes global dephosphorylation upon starvation. The acetyltransferase TIP60, which is regulated by the growth factor-sensitive kinase GSK3B, catalyzes acetylation of ULK1 to bolster its kinase activity. The energy-sensing kinase AMPK can promote autophagy through inhibition of MTOR and may fine-tune the autophagic response through regulation of the ULK1 complex as well. Dark blue arrows indicate events that promote autophagy, while light blue arrows indicate events that are inhibitory to autophagy. The gray arrow between AMPK and ULK1 indicates the possibility that AMPK phosphorylation of ULK1 exerts multiple regulatory effects on ULK1.
Phosphorylation
In addition to ATG13, ULK1 is also hyperphosphorylated in nutrient-rich conditions and undergoes global dephosphorylation upon starvation. At least 30 phosphorylation sites have been identified on ULK1, although the majority of the responsible kinases and the functions of these phosphorylation events remain to be identified.41 Regardless, this suggests that phosphorylation is an important mode of ULK1 regulation.
As a major nutrient/energy sensor, MTOR has been reported to directly phosphorylate ULK1 and ATG13. The MTOR complex 1 (MTORC1), consisting of MTOR, RPTOR and MLST8, is responsive to growth factors and amino acid levels and phosphorylates both ULK1 and ATG13.22,30,32 The MTORC1 complex binds to ULK1 directly through RPTOR under nutrient-rich conditions, and dissociates from the ULK1 complex upon starvation.32,42 MTOR-driven ULK1 phosphorylation correlates with autophagy inhibition, and with weaker ULK1 kinase activity.22,30,32 It is unclear if MTOR association occurs in the cytosol or on certain autophagosome precursor membrane structures. MTOR phosphorylation events may influence the localization of the ULK1 complex and exert its inhibitory effects by sequestering or physically separating the ULK1 complex from its enzymatic substrates. In yeast, Atg13 phosphorylation by TOR complex 1 (TORC1) under nutrient-rich conditions may inhibit the assembly of the Atg1 complex and recruitment to the phagophore assembly site (PAS).29 Inhibition of TORC1 by rapamycin also stimulates dimerization/oligomerization of Atg1 in an Atg13-dependent manner, which likely leads to subsequent activation of Atg1 kinase activity.43 Importantly, Ohsumi and colleagues showed that an Atg13 mutant defective in phosphorylation by TORC1 is sufficient to trigger autophagy in yeast in under nutrient-rich conditions.44 In contrast, the functional significance of MTOR phosphorylation sites on mammalian ATG13 and ULK1 are less clear, as complex formation is not dependent upon MTOR inactivation, and the phosphorylation sites of mammalian ATG13 by MTOR have not been completely mapped, making mutational analysis similar to that performed in yeast impossible at this stage. Furthermore, as ULK1 is also under regulation by MTOR, it is possible that preventing ATG13 phosphorylation by MTOR will not be sufficient for autophagy activation.
In yeast, protein kinase A (PKA) also phosphorylates Atg1 and Atg13 in vitro.45,46 PKA phosphorylation has no apparent effect on Atg1 kinase activity, but appears to regulate its association with the PAS, because phospho-mutants of PKA sites are constitutively located at the PAS.45 Inhibition of PKA is sufficient to induce autophagy, indicating that TORC1 and PKA regulate autophagy through distinct pathways.46 It remains to be seen whether mammalian PKA regulates ULK1. In Arabidopsis thaliana, nutrient starvation induces dephosphorylation and protein turnover of both ATG1 and ATG13. It is likely that this regulation might function as a negative feedback mechanism to tightly connect autophagy with nutritional status in the cell.47
It has been known for some time that another important cellular energy sensor, AMPK, can play a role in autophagy induction by phosphorylating TSC2 and RPTOR to inactivate MTOR, hence indirectly activating the ULK1 complex. Recently, AMPK has also been shown to directly interact with and phosphorylate ULK1 in a nutrient-dependent manner.41,42,48-50 Phosphorylation sites on ULK1 have been mapped by several groups and in some cases attributed to either MTOR or AMPK, but with very few overlapping sites among the groups.48-51 Of the potential phosphorylation sites, Ser555, Ser637 and Ser757 were reported by three or more independent groups (Fig. 3). AMPK phosphorylation site Ser555 is thought to recruit phospho-binding protein 14–3-3 to the ULK1 complex,42,50 whereas MTOR site Ser757 is required for AMPK binding, as its mutation disrupts ULK1-AMPK interaction.48,49 The effect of AMPK phosphorylation on the ULK1 complex and autophagy are not well established, with conflicting reports on whether it leads to stimulation or inhibition of autophagy. The study by Guan and colleagues48 suggests that phosphorylation of AMPK sites on ULK1 is stimulated by glucose starvation, contributing to ULK1 activation. In this case, ULK1 kinase activity (measured by ULK1 autophosphorylation) increases upon glucose starvation, and is correlated with AMPK activation. However, the study by Shang et al.49 identified AMPK sites that are dephosphorylated upon amino acid starvation; phospho-mutants defective in AMPK binding exhibit faster protein degradation upon autophagy stimulation, leading them to the conclusion that AMPK phospho-sites may be inhibitory to autophagy induction. The differences in their observations might reflect the distinct autophagy role of AMPK when sensing different triggers (i.e., glucose starvation vs. amino acid starvation); they could also be due to the monitoring of different phosphorylation sites in these two studies. A recent attempt to elucidate the role of AMPK in autophagy suggests that the kinase may regulate ATG9 localization together with ULK1.41 In yet another study by Shaw and colleagues, AMPK regulation of ULK1 was linked to mitophagy.50 This study identified four ULK1 sites that are phosphorylated upon treatment with the AMPK activators metformin and phenformin. AMPK-deficient primary mouse hepatocytes accumulate aberrant mitochondria, suggesting a mitophagy defect. ULK1 with compound phospho-deficient mutations at these AMPK sites cannot reconstitute starvation-induced SQSTM1/p62 degradation in ulk1−/− MEFs, suggesting that the AMPK phosphorylation on ULK1 is required for ULK1 function in autophagy. Given the number of groups that have independently validated AMPK interaction and phosphorylation of ULK1, it is likely that AMPK has a regulatory role on the ULK1 complex. The fact that ampk−/− MEFs can still undergo autophagy, rules out an essential autophagic role for this kinase (at least in response to amino acid starvation).41,48 It is, however, possible that AMPK fine-tunes ULK1 activity and the subsequent autophagy outcome in response to various energy requirements, or is involved in autophagy regulation in a more subtle way such as regulation/regeneration of autophagic components during prolonged autophagy.
Intriguingly, there have also been reports on the reverse, i.e., ULK1 regulation of MTOR and AMPK. ULK1 was reported to phosphorylate RPTOR in vitro to negatively regulate MTOR activity. In cells, knockdown of ULK1 results in increased MTORC1 signaling as assessed by the phosphorylation of MTORC1 substrates.52,53 This could constitute a feed-forward mechanism that ensures rapid shutdown of MTOR signaling during autophagy induction and maintenance of MTOR inhibition during nutrient-limiting conditions. Interestingly, RB1CC1 is also reported to regulate MTOR through its interaction with TSC1.54,55 Overexpression of RB1CC1 alone has no effect on tsc1−/− MEFs, but in wild-type MEFs it causes an increase of RPS6 kinase phosphorylation, which correlates with a mild (5%) increase in average cell size. It is unclear if ULK1 is involved in the RB1CC1-dependent regulation of MTOR or vice versa. However, ULK1 and RB1CC1 appear to have opposite regulatory effects on MTOR, and are likely to represent distinct mechanisms, as ULK1 inhibition of MTOR activity can still be observed in tsc2−/− MEFs.53 In terms of AMPK, ULK1 has been reported to be able to phosphorylate all three AMPK subunits.56 ULK1 phosphorylation was found to be inhibitory to AMPK activity and was proposed to be a form of negative feedback to dampen autophagy-induction signals.
Dephosphorylation of ULK1 complex components upon autophagy activation requires more than just suppressing their protein kinases such as MTOR, yet the protein phosphatase(s) involved in ULK1 or ATG13 dephosphorylation is currently unknown, let alone whether such a phosphatase(s) is regulated in a coordinated manner with other components. The phosphatase inhibitor okadaic acid has long been described as an inhibitor of autophagy,57-59 though its mechanism of action is not known. It may function through inhibiting dephosphorylation of inhibitory phospho-sites, such as the MTOR sites in the ULK1 complex.49 Contrary to the reported effects of okadaic acid in eukaryotic cells, a yeast study reported Tap42-PP2A, a target of okadaic acid, as a negative regulator of autophagy, although it did not identify the substrate of the phosphatase.60 Furthermore, a recent screen of PP2A subunit mutant fly strains identified two PP2A complexes required for starvation-induced autophagy in Drosophila larval fat body cells and proposed the Atg1 complex to be a target of PP2A regulation.59
Acetylation
Aside from phosphorylation, acetylation is another post-translational modification that has recently been found to play a role in autophagy.61,62 Acetylation involves the transfer of acetyl groups from acetyl-CoA to the lysine residue of the target protein. The modification regulates diverse processes and has been shown to have an effect on protein-DNA interactions, protein-protein interactions, subcellular localization and protein stability.63 Acetyl-CoA is also a critical building block for cellular metabolism, and, as such, protein acetylation/deacetylation has been implicated as a critical regulatory mechanism for metabolism.64 Given the catabolic nature of autophagy, regulation of autophagy by acetylation is thus an attractive hypothesis. Protein acetylation/deacetylation was initially implicated in autophagy by our studies using the histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA).65,66 We found that SAHA induces autophagy in various cell types in a ULK-dependent manner. However, the role of SAHA in maintaining the acetylation status of ATG proteins was not determined. More recently, a systematic genetic analysis of histone acyltransferase (HAT) complexes in yeast identified Esa1, the catalytic subunit of HAT complex NuA4, to be required for autophagy. By a process of elimination, Atg3 was deemed to be the substrate of Esa1. Acetylation mutants of Atg3 could not bind to Atg8 and failed to reconstitute atg3∆ cells, indicating that the acetylation event is required for autophagy.62 In a separate study, TIP60, the mammalian homolog of Esa1, was reported to acetylate ULK1. Knockdown of TIP60 impairs starvation-induced autophagy62,67 and acetylation mutants of ULK1 cannot rescue LC3 conversion in ulk1−/− MEFs. Furthermore, TIP60 activation is regulated by glycogen synthase kinase 3 β (GSK3B), a protein whose activity is responsive to growth factor withdrawal through PI3K-AKT-MTOR signaling pathways,67 thus directly linking the acetylase TIP60 to nutrient sensing and ULK1-mediated autophagy.
Ubiquitination
Autophagy and proteasome-mediated protein degradation are considered to be the two major mechanisms for cellular protein turnover. Therefore, conceptually it is not a surprise to note that these two processes intimately communicate with each other.68-71 Furthermore, both autophagy and proteasome-mediated protein degradation are subjected to a common posttranslational regulatory mechanism, ubiquitination. For example, ubiquitination has been established as a critical event for autophagosomes to recognize and recruit specific cargos, including protein aggregates, mitochondria and invading pathogens.70,72
Recent evidence suggests that ubiquitin-mediated proteasomal degradation also targets the components of the ULK1 complex. There is evidence of direct ubiquitination of ULK1 in response to nerve growth factor in neurons, although the significance of this with respect to autophagy is unknown.73 Ubiquitination is also alluded to as part of the HSP90-CDC37 regulation of ULK1. Kundu and colleagues reported that treatment with the HSP90 antagonist 17AAG, leads to a decrease in ULK1 protein levels that can be rescued by cotreatment with the proteasomal inhibitor MG132.37 Interestingly, ULK2 is not an HSP90 client, suggesting that although these two proteins are functionally redundant, they may be subject to distinct regulation. Additionally, ATG13 protein level is stabilized by ULK1, ULK2 and C12orf44.35,37 In the absence of C12orf44, ATG13 protein levels decrease, and such decrease can be blocked by the proteasomal inhibitor MG132.35
Interestingly, it has been observed that upon amino acid starvation, activation of the ULK1 complex is accompanied by a concurrent decrease of ULK1 protein levels. The functional relevance of this protein level change is not clear. It is possible that the decrease in ULK1 is a feedback mechanism to ensure that autophagy can be effectively turned off once nutrients become available. Further study is needed to determine the biological role of the starvation-associated ULK1 decrease and whether it is through ubiquitin-mediated degradation. If so, this will represent an elegant mechanism through which a massive autophagic degradation is controlled by specific proteasomal degradation of ULK1.
Relaying the Nutrient Signals Downstream
Knockout mice of autophagy essential genes such as Atg3, Atg5 or Atg7 do not have gross developmental defects but die within a day after birth, because the animals are unable to survive the neonatal starvation period.74-76 This phenotype seems to be the hallmark of autophagy-specific and essential genes. While siRNA-mediated knockdown of ULK1 is sufficient to block starvation-induced autophagy in multiple cell types, ulk1−/− mice show a relatively mild autophagy phenotype, with delayed red blood cell maturation (which requires mitophagy), but are viable.77 This has been attributed to the redundancy between ULK1 and the closely-related ULK2. Indeed, ulk1−/−;ulk2−/− mice,19 as well as atg13−/− mice,49 have the same neonatal death phenotype as other Atg knockout mice. rb1cc1−/− mice die between embryonic day 13.5 and 16.5 due to defects in heart and liver development, reflecting the involvement of RB1CC1 in other processes outside of autophagy.54
The ULK1 kinase complex has been suggested to function in the initial stages of the canonical autophagy pathway. However, the exact functional relationship between the ULK1 complex and other ATG proteins is still not completely defined, and many controversial studies have been reported. Related to this issue, although the kinase activity of Atg1/ULK1 is required for autophagy, the protein substrate(s) of ULK1 that mediates its autophagic function has not been identified; and whether Atg1/ULK1 also has kinase-independent functions in autophagy is not clear. Therefore, to understand how the ULK1 complex relays upstream nutrient signals to the downstream autophagy pathway, these crucial questions need to be solved.
The epistasis of the ULK1 complex with other ATG complexes
In addition to the ULK1 complex, other ATG components that function upstream of LC3 conjugation to phosphatidylethanolamine (PE) include the PIK3C3/VPS34 complex and the ATG12–ATG5-ATG16L1 complex. As the ULK1 complex directly senses MTOR activity, if the autophagy “cascade” proceeds in a strictly linear fashion, it is logical to place the ULK1 complex at the most upstream position in the autophagy pathway, at least for amino acid starvation-induced autophagy. While mounting evidence supports this view, such as the recent imaging-based hierarchy study by Mizushima and colleagues,78 there are also conflicting results suggesting that the autophagy pathway may not be a simple linear process.
PIK3C3/VPS34 is a lipid kinase, and its enzymatic product phosphatidylinositol 3-phosphate (PtdIns3P) is required, directly or indirectly, for the recruitment of multiple autophagy components (WIPI1, ZFYVE1/DFCP1, ATG5 and LC3) to forming autophagosomes.79,80 Like the ULK1 complex, the PIK3C3 complex has been proposed to be a major point of regulation for autophagy induction. For example, the autophagic function of the PIK3C3 complex can be closely controlled through dynamic interaction of the PIK3C3 component BECN1 with its inhibitory binding partners, the BCL2 family of proteins including BCL2, BCL2L1/BCL-XL, and BCL2L11/BIM.81-83
As both the ULK1 and PIK3C3 complexes need to be activated to initiate autophagy, their regulation must be coordinated. It has been suggested that recruitment of the PIK3C3 complex to autophagic membranes is dependent on ULK1, thus placing the ULK1 complex upstream of the PIK3C3 complex. The major evidence for this claim is that (1) in rb1cc1−/− cells, starvation fails to induce the PIK3C3 complex to form punctate structures; and (2) inhibition of the kinase activity of PIK3C3 cannot completely prevent starvation-induced ULK1 translocation.78 Recent work lends mechanistic insights into ULK1-PIK3C3 communication: in response to nutrient starvation, AMBRA1, a component of the PIK3C3 complex, is reported to be phosphorylated in a ULK1-dependent manner.36 This phosphorylation releases the AMBRA1-PIK3C3 complex from dynein and the microtubule network, freeing the complex to translocate to autophagy initiation sites on the ER (it should be noted that AMBRA1 is not the only link between ULK1/Atg1 and cytoskeletal motors: in Drosophila, Atg1 can activate myosin II to help drive autophagosome formation; and in mammalian cells this interaction appears to regulate ATG9 trafficking84). Albeit a reasonable explanation for the ULK1/Atg1-PIK3C3 connection, because AMBRA1 does not have a functional counterpart in yeast, it is unlikely to be the universal mediator for these two autophagy complexes.
Although current evidence supports a unidirectional signaling flow from the ULK1 complex to the PIK3C3 complex, this model is likely to be oversimplified, and additional crosstalk should be considered. Particularly, experimental outcomes using the pharmacological agent wortmannin should be interpreted with caution. In addition to PIK3C3, wortmannin can inhibit class I PI3 kinases as well, and thus might have an impact on autophagy and the behavior of the ULK1 complex via multiple mechanisms. Furthermore, wortmannin treatment often results in the formation of large amounts of intracellular vesicles. Therefore, what exactly is the nature of ULK1 puncta upon wortmannin treatment? Also, can knockout of Atg14 or genes encoding other components of the PIK3C3 complex recapitulate the effect of wortmannin, i.e., inhibiting starvation-induced LC3 conjugation but not ULK1 puncta formation?
The functional relationship between the ULK1 and ATG5 complexes is equally complicated. For example, Rb1cc1 knockout can block ATG5 puncta formation and conversely Atg5 knockout can also block ULK1 puncta formation.21 A somewhat puzzling observation is that starvation could induce ULK1 puncta formation in atg5−/− cells only in the presence of wortmannin.78 This observation led to the model that ULK1 is the first complex recruited to the autophagosome formation site, preceding the PIK3C3 complex and ATG5. The requirement for wortmannin is attributed to the potential transient nature of ULK1 localization on autophagic membranes, which may be stabilized by blocking the immediate downstream event, the function of the PIK3C3 complex.78 On the other hand, alternative possibilities should be considered here: if indeed ULK1 localization on the autophagic membrane is so transient and can only be observed in atg5−/− cells in the presence of wortmannin, why can it be readily observed in wild-type cells in the absence of wortmannin? And again, what exactly is the nature of ULK1 puncta upon wortmannin treatment?
It is possible that all these puzzling observations of imaging analysis are the outcome of limited optical resolution. That is, even if there is absolute autophagic membrane translocation of a certain ATG protein, when the membrane structure is too small (for example, at the very early phase of phagophore nucleation), it will not be possible to monitor such translocation events via microscopy. For the phagophore to grow in size upon starvation, the biochemical conjugation of LC3 to PE is required. Starvation-stimulated LC3 conjugation is dependent on the ULK1 complex, PIK3C3 complex and ATG5, thus explaining the interdependence of the “translocation” of these three complexes in imaging-based analyses. Regardless, given the likely upstream requirement of ULK1 for localization of other autophagy components and the apparent fact that ULK1 is a mediator of multiple upstream kinases, it is clear that the prime function of ULK1 may be as a node to convert incoming signals into autophagosomes.
To address the epistatic relationship between the ULK1 complex and other upstream ATG complexes, Jiang and colleagues have recently found that RB1CC1 interacts directly with ATG16L1, providing a functional link between the ULK complex and the ATG5 complex.85 Interestingly, this interaction appears to be transient (stabilized by ATG3-ablation) and is required for ULK1 complex-dependent autophagy such as that induced by amino acid starvation, but is dispensable for ULK1 complex-independent autophagy (ULK1 complex-independent autophagy will be discussed below). Therefore, this interaction is a specific route through which the ULK1 complex relays signals to downstream autophagy machinery.
The ULK1 complex and ATG9 cycling
Atg1/ULK1 has been implicated in Atg9/ATG9 cycling in both yeast and mammalian cells, as reported independently by the Klionsky, Tooze and Ohsumi groups.16,86,87 In yeast, Atg9 cycles between the PAS and peripheral sites. Atg1 is required for Atg9 cycling and it is thought to be recruited to the PAS through indirect interaction with Atg17.87 The role of ULK1 in ATG9 cycling in mammalian cells is not as well defined. While earlier reports showed that knockdown of ULK1 blocks starvation-induced redistribution of ATG9,16 recent work based mostly on confocal imaging suggests that ATG9 localizes to membrane structures adjacent to early autophagy markers such as ULK1, ZFYVE1 and WIPI2, and is recruited independently of ULK1.88 In a model for Parkinson disease, ATG9-positive structures can still be recruited to damaged mitochondria in rb1cc1−/− MEFs. Likewise, ULK1-positive punctate structures can be observed in atg9−/− MEFs, indicating that recruitment of ATG9 and ULK1 to membrane structures is independent of each other.89 Similarly, independent recruitment of ATG9 and ULK1 is observed in Salmonella-induced xenophagy in atg9−/− and rb1cc1−/− MEFs.90 It is noteworthy that GFP-LC3 and GFP-ATG5 but not GFP-WIPI or GFP-ULK1 can still be recruited to Salmonella-containing vacuoles in rb1cc1−/− MEFs. However, the growth of Salmonella is not suppressed in rb1cc1−/− MEFs, indicating a failure to complete xenophagy to achieve bacterial clearance in these cells.
Potential role of the ULK1 complex in downstream events
While ULK1 is traditionally viewed as orchestrating early events in autophagosome formation, an Atg1-Atg8 interaction was recently reported in two independent yeast studies, suggesting a role for Atg1 in late stages of autophagy as well. The Atg8 interaction targets Atg1 to the vacuole for degradation. Expression of an Atg1 mutant that cannot interact with Atg8 results in an autophagy defect and reduces the accumulation of autophagic bodies in vacuolar protease-deficient cells, indicating that the interaction promotes production of fully formed autophagosomes.28,91 In addition, Kraft et al. showed that ULK1 also interacts with mammalian ATG8s and a binding-defective mutant of ULK1 shows reduced recruitment to autophagosomes. However, the authors did not show that this interaction results in lysosomal turnover of ULK1 or that it is essential for functional autophagy.28 Prior to these studies, mammalian ULK1 was reported to interact with the LC3-related proteins GABARAPL2/GATE16 and GABARAP, and to a lesser extent LC3 in a yeast two-hybrid screen of a human fetal brain cDNA library. While the interaction was validated by co-immunoprecipitation experiments in cells, the functional involvement of this interaction in autophagy was not addressed.39,92 Taken together, it is possible that ULK1 may have a role in later stages of the autophagy pathway in the mammalian system as well. Technically, to confirm such a role of ULK1 in cells could prove to be difficult, unless distinct mutants of ULK1 can be created to precisely dissect the upstream and downstream functions of this enzyme.
The kinase substrate(s) of ULK1
Although the protein kinase activity of Atg1/ULK1 has been demonstrated to be essential for its autophagy function, the relevant substrate(s) has yet to be identified. This substrate should meet the following criteria: its phosphorylation by ULK1 is enhanced upon amino acid starvation and blocking such phosphorylation (for example by mutating the phosphorylation site) will prevent ULK1-dependent autophagy. The identity of this substrate and the mechanism by which it mediates the autophagy activity of ULK1 might reveal how the ULK1 complex relays upstream signals to the downstream autophagy pathway, including the puzzling relationship between the ULK1 complex and PIK3C3.
To date, it has been shown that ULK1 can phosphorylate itself as well as its regulatory proteins ATG13 and RB1CC1.20,22,30 Autophosphorylation in the activation loop of Atg1 correlates with enhanced Atg1 kinase activity in yeast.93,94 Mutation of these sites disrupts starvation-induced autophagy without affecting Atg1 complex formation or localization at the PAS (corresponding sites in ULK1 have not been identified). Therefore it is most likely that an additional substrate(s) of Atg1/ULK1 accounts for its autophagic function. Large scale phospho-proteomic studies95 and Atg1/ULK1 consensus site mapping96 will help the search for this critical component. It is also likely that ATG13 or RB1CC1 is required for recruiting a ULK1 substrate(s) in a phosphorylation (by MTOR and/or ULK1)-regulated manner. Similarly, other autophagic proteins that have been shown to interact with the ULK1 complex (such as LC3 and ATG16L1) would be attractive candidate substrates as well.
The potential kinase-independent autophagic function of ULK1
Although the kinase activity of Atg1/ULK1 is indispensable for its autophagic function, Atg1/ULK1 might also possess additional kinase-independent functions in autophagy. In yeast, Atg9 cycling is disrupted in atg1∆ mutants but is unaffected by loss of Atg1 kinase activity, suggesting a kinase-independent function for the Atg1 complex.86 Also supporting this possibility is the observation that atg1∆ yeast fail to recruit Atg17 and Atg8 to the PAS, but yeast cells reconstituted with kinase-dead Atg1 mutants show abnormal accumulation of Atg17 and Atg8 at the PAS, suggesting that Atg1 may act as a scaffold to recruit Atg proteins to the PAS, with kinase activity being required only for subsequent steps.97 In mammalian cells, kinase-dead ULK1 cannot rescue starvation-induced autophagy in ulk1−/− cells. A kinase-dead ULK1 mutant can still interact with ATG13 and RB1CC120 but is unable to efficiently recruit downstream components, such as WIPI and ATG5.21 While the kinase-dead ULK1 complex does not form prominent punctate structures like its wild-type counterpart,21 it may still translocate from the cytoplasm onto an autophagosome precursor membrane structure that would be hard to detect if membrane elongation of the autophagosome cannot proceed without ULK1 kinase activity.
Nonautophagic Functions of the ULK1 Complex Components
ULK1
Before their roles in autophagy were characterized, ULK1 and RB1CC1 had been studied in other contexts. As its name implies, ULK1 was named due to its homology to UNC-51 in C. elegans.14,98 The C. elegans unc-51 mutants are mostly paralyzed, reflecting a defect in axonal elongation, and they have aberrant accumulation of enlarged vesicles and other membranous structures in a subset of neuronal cells.99,100 The Drosophila homolog of UNC-51 is also reported to be important for neuronal development through mediation of vesicular transport in axons.101,102 In mice, a role for the mammalian homologs ULK1 and ULK2 in neuronal development has been shown for several neuronal populations, mostly in tissue culture systems.38,73,103 ULK1 can be detected in cerebellar granule cells and has a punctate staining pattern along axons and growth cones of dorsal root ganglion neurons, indicating recruitment to membranous structures.73,103 While it is currently unclear why neuronal defects have not been reported in ulk1−/− mice (possibly due to redundancy), the role of ULK1 in neuronal development appears to be highly conserved, as murine ULK1 can be used to rescue the unc-51 defect in C. elegans.103 In all three model systems, ULK1 kinase activity is required to fulfill its neuronal functions, with the kinase dead (K46R) ULK1 mutant having dominant negative effects on neurite extension.99,101-103
There is currently no evidence that autophagy is required for axon elongation during neuronal development; an uncoordinated phenotype has not been reported for knockouts of other autophagy essential genes in C. elegans.104 Likewise neuronal development is rarely specifically mentioned in Drosophila or mouse genetic knockout models addressing Atg gene function in the context of autophagy. No neuronal functions have been documented for other members of the autophagic ULK1 complex (ATG13, RB1CC1 or C12orf44) either. Certainly autophagy is important in maintaining homeostasis in neurons, as exemplified by the mouse models for neurodegenerative diseases that utilize conditional and neural-specific knockout of Atg genes such as Atg5, Atg7 and Rb1cc1.105-107 On the other hand, the requirement of autophagy in neural development, if any, remains murky. It is possible that the role of ULK1 in neuronal development represents a specialized function involving novel interacting partners, and may be cell type-specific. Further studies are needed to clarify whether this documented neuronal role of ULK1 is because of its autophagy function.
RB1CC1/FIP200
Aside from its role in autophagy, RB1CC1 has been implicated in a diverse range of cellular processes. The functions of RB1CC1 have been reviewed elsewhere and are performed mainly through switching among its numerous interacting partners which include PTK2B, TSC1, MAP3K5, TRAF2, TP53 and the E3 ubiquitin ligase RNF111.108 Most interacting partners of RB1CC1 were discovered by yeast two-hybrid screens.
RB1CC1 has been reported to possess nuclear functions and its name (RB1-inducible coiled-coil 1) was derived due to its ability to regulate the retinoblastoma tumor suppressor protein RB1 as well as CDKN2A/p16 through direct transcriptional activation.109,110 As mentioned for ULK1, the other reported functions of RB1CC1 may give us insights into the molecular mechanisms and role of RB1CC1 in the ULK1 complex and autophagy. For instance, RB1CC1 interacts with CTNNB1/β-catenin and can mediate its degradation independent of the classical APC destruction complex.111 It was also found to enhance proteasomal degradation of several negative regulators of TGFB/TGFβ signaling by acting as a cofactor for the E3 ubiquitin ligase RNF111.112 RB1CC1 may thus exert similar scaffolding functions to bind degradation factors for ULK1, which is also degraded during starvation. RB1CC1 can also bind to protein phosphatase 1 (PP1),113 although the biological function of this interaction has not been studied. One possible function may be to recruit phosphatases to act upon one or all of the ULK1 complex components, which are regulated via phosphorylation-dephosphorylation.
ULK1-Independent Autophagy
LC3 conversion still occurs in atg13−/−, rb1cc1−/− and ulk1−/−;ulk2−/− MEFs,19,21,49 suggesting that the ULK1 complex is not essential for activation of the LC3 conjugation machinery and that there are other ULK1/ULK2-independent ways for setting off the autophagy cascade (Fig. 5). However, it should be noted that LC3 conjugation and delivery to the lysosome is not the sole purvey of autophagosomes, given that LC3 conjugation can occur on nonautophagsosomal structures such as phagosomes and entotic vacuoles (detailed below). Recent work has shown that overnight glucose deprivation-induced LC3 conversion that is dependent on ATG5 does not require ULK1 or ULK2.19 The authors attributed this to the accumulation of ammonia as a result of glutaminolysis (glutamine degradation) within the cells, and they were able to recapitulate the effect by treating cell cultures with ammonium chloride. Prior to and consistent with their discovery, ammonia derived from glutamine degradation was reported to induce autophagy in an MTOR-independent manner.114 It should also be noted that glucose deprivation has been reported to stimulate autophagy in a ULK1-dependent manner.48 Unlike the ammonium scenario, this is a typical bioenergetic response involving MTOR suppression plus AMPK activation, which subsequently activates the ULK1 complex. Seemingly contradictory with each other, glucose starvation might induce autophagy via both mechanisms, depending on exact biological contexts, such as growth conditions and cell types.
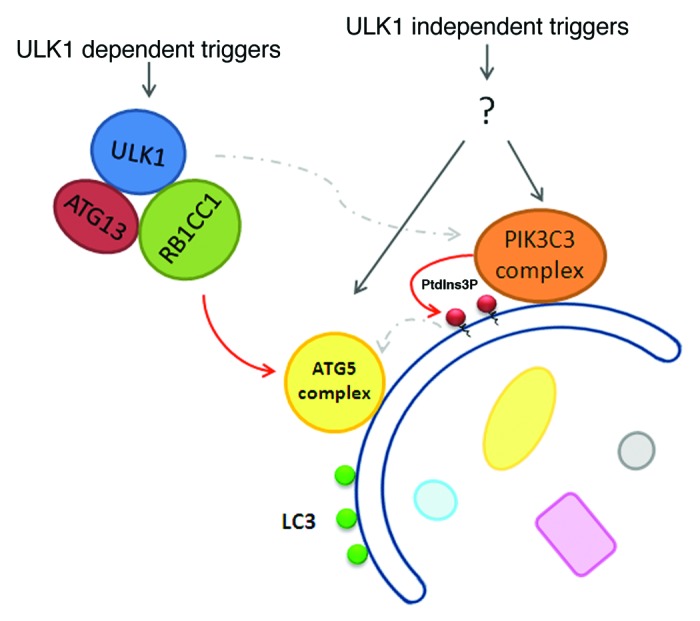
Figure 5. ULK1-dependent and -independent autophagy . The role of the ULK1 complex in amino acid starvation-induced autophagy is well established. There are, however, triggers for autophagy that can feed into the downstream machinery to trigger the autophagic cascade and LC3 conjugation in a ULK1 complex-independent manner. Because the ULK1 complex can directly communicate with the ATG5 complex and likely functions upstream of the PIK3C3 complex, triggers of ULK1-independent autophagy pathways might also signal through both the ATG5 and PIK3C3 complexes, as depicted in the figure.
Several other MTOR-independent autophagy inducers have been reported in the literature but did not address ULK1 dependency directly. These include amino sugars such as glucosamine and mannosamine, which were reported to induce LC3 conversion without dephosphorylation of MTOR substrates.115 Lithium, which is studied more in the context of neurodegenerative diseases, also induces LC3 conversion and degradation of mutant HTT/huntingtin without MTOR inactivation.116,117 Typically these MTOR-independent autophagy inducers require long incubation times before significant LC3 conversion can be detected. If such MTOR-independent autophagy is indeed also independent of the ULK1 complex, it is possible then that a major “purpose” of the ULK1 complex is to trigger a more rapid autophagy response.
In DT40 chicken cells, knockout of ATG13, but not ULK1/ULK2, abrogates autophagy upon amino acid starvation. Furthermore, only ATG13 splice variants that can interact with RB1CC1 can reconstitute autophagy, suggesting that the vital function of ATG13 in starvation-induced autophagy is independent of ULK1/ULK2 but dependent on RB1CC1 in chicken cells.118 However, in this study and in contrast to mammalian cells, inhibition of TOR with rapamycin or Torin1, fails to induce autophagy in wild-type DT40 cells. Since knockout of ULK1/ULK2 is sufficient to disrupt amino acid starvation-induced autophagy in mammalian cells, the regulation of autophagy might differ even among different vertebrates. Alternatively, it is possible that chicken cells may express another ULK1/ULK2 homolog that is able to complement the ULK1/ULK2 deletion.
Strikingly, the autophagy machinery is also utilized in certain cellular processes that are obviously not canonical autophagy.119 One such example is the membrane trafficking process phagocytosis, which, like autophagy, leads to lysosomal degradation of its specific cargo. However in phagocytosis, extracellular particles in the form of cell debris or pathogens are engulfed to form an internal phagosome which then undergoes maturation and degradation.120 Surprisingly, transient recruitment of GFP-LC3 to phagosomes was detected in macrophages when fed with beads coated with toll-like-receptor ligands.121 This LC3-associated-phagocytosis (LAP) is dependent on PIK3C3 complex activity, ATG5 and ATG7. It was later found to be independent of ULK1, as ulk1−/− macrophages display similar levels of LC3 lipidation in response to phagocytosis of dead cells.122 Another macro-endocytic process known as entosis is similar to phagocytosis but involves the engulfment of a live epithelial cell by another epithelial cell, resulting in the formation of an entotic vacuole. Morphologically, entosis is characterized by the formation of “cell-in-cell” structures and can be found in human cancers, although its significance is currently not known.123 The majority of cells that are engulfed by entosis undergo nonapoptotic cell death that is mediated by lysosomal fusion to the entotic vacuole.123,124 Acidification of the entotic vacuole is preceded by LC3 recruitment that is dependent on PIK3C3, ATG5 and ATG7, but not RB1CC1.124 The same study indicates that recruitment of LC3 to micropinosomes is not affected by RB1CC1 knockdown either, thus representing another process in which the autophagy machinery can be recruited to membranes independent of the ULK1 complex. This suggests that the ULK1 complex may be important in the recruitment of the autophagy machinery in the generation of de novo, double-membrane autophagosomes, but not in the recruitment of the machinery to pre-formed, single-membrane vesicular structures. However, these new findings also raise the question of how the canonical ATG proteins are recruited to exert nonautophagic functions.
ULK1 and Cancer
The relationship between autophagy and cancer is complicated, as autophagy has been attributed with both tumor-suppressive and cancer-promoting functions. In general, autophagy is a cell-survival mechanism and is thought to act as a tumor suppressor by preventing the accumulation of damaged and/or toxic components that, if left to persist, could lead to genomic instability and cancer. However, once a tumor has formed, this cell-survival trait of autophagy becomes detrimental as it enables tumor cells to survive energy deprivation and other stresses, or to cope with the damaging effects of chemotherapeutics. Therefore, inhibition of autophagy may prove beneficial in treating certain kinds of cancer. In order to inhibit autophagy, a target in the pathway is needed, and the ULKs are attractive in this aspect—being kinases, they are eminently druggable. Targeting the other kinase of the pathway, the lipid kinase PIK3C3, might lead to severe side effects because of its essential function in endocytosis.
One of the main uses of a ULK-targeted agent in cancer treatment might be in combination therapy with other therapeutics. The lack of clinical impact of MTOR inhibitors as an anti-cancer treatment may be due in part to the fact that they upregulate tumor-protective autophagy by activating ULK1.125 Furthermore, a plethora of the anticancer agents currently used can induce autophagy; thus, combining them with ULK1-targeting might be more effective therapeutically. However, it should be emphasized that whether autophagy functions to bolster or impair treatment is not always clear and is under extensive study. For example, in human osteosarcoma U2OS cells, sublethal treatments with the topoisomerase drugs etoposide and camptothecin can induce autophagy. This DNA-damage induced autophagy is promoted by the activation of the tumor suppressor TP53, through the direct transcriptional activation of ULK1 and ULK2.126 The upregulation in the expression level of ULK1 and ULK2 allows for sustained autophagy that leads to an increase in nonapoptotic cell death. The transcriptional upregulation of ULK1 can also proceed via TP53-independent pathways, which would also presumably allow for sustained autophagy in cells deficient in TP53.126 Despite this autophagy-stimulating nuclear-dependent function of TP53, cytoplasmic TP53 is reported to abrogate the induction of autophagy. Upon starvation, TP53 is hyperacetylated at K382 and binds RB1CC1, thus inhibiting conversion of LC3-I to LC3-II. A single conservative mutation to arginine can inhibit the interaction between TP53 and RB1CC1.127 Presumably this interaction titrates RB1CC1 away from the ULK1 complex, in which RB1CC1 is known to stabilize and stimulate ULK1 kinase activity.
While the potential role of ULK1-targeting in cancer treatment should be specifically determined case by case, the potential value of ULK1 as a prognostic marker for certain cancers should also be explored. Immunohistochemical analysis of a tissue microarray containing nonmetastatic invasive breast cancer tissue and matched adjacent normal tissue suggests that low expression of ULK1 is an adverse marker for progression of disease and increased risk of metastasis for patients with breast cancer.128 Conversely, a similar study in esophageal squamous cell carcinoma suggests that higher ULK1 expression is correlated with poor patient prognosis.129 As we try to ascertain the prognostic value of ULK1 in cancer, it will be important to remember that (1) autophagy may be independent of ULK1, (2) ULK1 exhibits redundancies with ULK2, and (3) measuring the protein levels of ULK1 is not a valid substitute readout for its kinase activity.
Conclusion
The autophagic function of Atg1/ULK1 is highly conserved from yeast to mammals, yet it is becoming evident that the regulation of ULK1 in mammalian cells differs from its yeast counterpart Atg1. Increasing reports show that the ULK1 complex is regulated by a variety of signaling pathways through a myriad of posttranslational modifications. One scenario is that different combinations of these modifications allow for a graded autophagic stimulation in response to a changing cellular environment. Much remains to be studied about the ULK1 complex and its role in autophagy. The relationship between ULK1 and ATG9 needs to be clarified. How does the ULK1 complex recruit other downstream autophagy players? What are the substrates of the ULK1/ULK2 kinase complex that are essential for autophagy? Does mammalian ULK1 play a role in later stages of autophagosome formation? What is the molecular nature underlying ULK1 puncta formation? Is it a clustering event of the ULK1 complex on membranes or a translocation of cytosolic ULK1 to membranous structures, and what is the membrane “receptor” of the complex? Addressing these questions would provide insights into the molecular mechanisms of an important regulatory event in the autophagy cascade. Furthermore, developing small molecule modulators of ULK1/ULK2 could be important therapeutically.
Acknowledgements
We thank members of X.J. lab for critical discussion. This work is supported in part by a Geoffrey Beene Cancer Research fund, a Goodwin Experimental Therapeutic Center fund, and NIH R01 CA166413.
Glossary
Abbreviations:
- AMPK
AMP-activated protein kinase
- Atg
autophagy-related
- CTD
C-terminal domain
- MEFs
mouse embryonic fibroblasts
- MTOR
mechanistic target of rapamycin
- MTORC1
MTOR complex 1
- PKA
protein kinase A
- ULK1
Unc-51 like kinase-1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/23323
References
- 1.Chen Y, Klionsky DJ. The regulation of autophagy - unanswered questions. J Cell Sci. 2011;124:161–70. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 3.Todde V, Veenhuis M, van der Klei IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta. 2009;1792:3–13. doi: 10.1016/j.bbadis.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 5.Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42:731–43. doi: 10.1016/j.molcel.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong AS, Cheung ZH, Ip NY. Molecular machinery of macroautophagy and its deregulation in diseases. Biochim Biophys Acta. 2011;1812:1490–7. doi: 10.1016/j.bbadis.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–74. doi: 10.1016/0014-5793(93)80398-E. [DOI] [PubMed] [Google Scholar]
- 10.Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275–80. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- 12.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene. 1997;192:245–50. doi: 10.1016/S0378-1119(97)00084-X. [DOI] [PubMed] [Google Scholar]
- 14.Kuroyanagi H, Yan J, Seki N, Yamanouchi Y, Suzuki Y, Takano T, et al. Human ULK1, a novel serine/threonine kinase related to UNC-51 kinase of Caenorhabditis elegans: cDNA cloning, expression, and chromosomal assignment. Genomics. 1998;51:76–85. doi: 10.1006/geno.1998.5340. [DOI] [PubMed] [Google Scholar]
- 15.Chan EYW, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–74. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 16.Young ARJ, Chan EYW, Hu XW, Köchl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 17.Lee EJ, Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy. 2011;7:689–95. doi: 10.4161/auto.7.7.15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan J, Kuroyanagi H, Tomemori T, Okazaki N, Asato K, Matsuda Y, et al. Mouse ULK2, a novel member of the UNC-51-like protein kinases: unique features of functional domains. Oncogene. 1999;18:5850–9. doi: 10.1038/sj.onc.1202988. [DOI] [PubMed] [Google Scholar]
- 19.Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011;108:11121–6. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan EYW, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–71. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan J-L, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell. 2005;16:2544–53. doi: 10.1091/mbc.E04-08-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabeya Y, Noda NN, Fujioka Y, Suzuki K, Inagaki F, Ohsumi Y. Characterization of the Atg17-Atg29-Atg31 complex specifically required for starvation-induced autophagy in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2009;389:612–5. doi: 10.1016/j.bbrc.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 27.Kawamata T, Kamada Y, Kabeya Y, Sekito T, Ohsumi Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol Biol Cell. 2008;19:2039–50. doi: 10.1091/mbc.E07-10-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J. 2012;31:3691–703. doi: 10.1038/emboj.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–13. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meijer WH, van der Klei IJ, Veenhuis M, Kiel JAKW. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy. 2007;3:106–16. doi: 10.4161/auto.3595. [DOI] [PubMed] [Google Scholar]
- 32.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheong H, Yorimitsu T, Reggiori F, Legakis JE, Wang C-W, Klionsky DJ. Atg17 regulates the magnitude of the autophagic response. Mol Biol Cell. 2005;16:3438–53. doi: 10.1091/mbc.E04-10-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5:973–9. doi: 10.4161/auto.5.7.9296. [DOI] [PubMed] [Google Scholar]
- 35.Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009;5:649–62. doi: 10.4161/auto.5.5.8249. [DOI] [PubMed] [Google Scholar]
- 36.Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–68. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell. 2011;43:572–85. doi: 10.1016/j.molcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–58. doi: 10.1101/gad.1151204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okazaki N, Yan J, Yuasa S, Ueno T, Kominami E, Masuho Y, et al. Interaction of the Unc-51-like kinase and microtubule-associated protein light chain 3 related proteins in the brain: possible role of vesicular transport in axonal elongation. Brain Res Mol Brain Res. 2000;85:1–12. doi: 10.1016/S0169-328X(00)00218-7. [DOI] [PubMed] [Google Scholar]
- 40.McKnight NC, Jefferies HB, Alemu EA, Saunders RE, Howell M, Johansen T, et al. Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J. 2012;31:1931–46. doi: 10.1038/emboj.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mack HID, Zheng B, Asara JM, Thomas SM. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy. 2012;8:1197–214. doi: 10.4161/auto.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeh YY, Shah KH, Herman PK. An Atg13 protein-mediated self-association of the Atg1 protein kinase is important for the induction of autophagy. J Biol Chem. 2011;286:28931–9. doi: 10.1074/jbc.M111.250324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, et al. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010;30:1049–58. doi: 10.1128/MCB.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Budovskaya YV, Stephan JS, Deminoff SJ, Herman PK. An evolutionary proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 2005;102:13933–8. doi: 10.1073/pnas.0501046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci U S A. 2009;106:17049–54. doi: 10.1073/pnas.0903316106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suttangkakul A, Li F, Chung T, Vierstra RD. The ATG1/ATG13 protein kinase complex is both a regulator and a target of autophagic recycling in Arabidopsis. Plant Cell. 2011;23:3761–79. doi: 10.1105/tpc.111.090993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. 2011;108:4788–93. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dorsey FC, Rose KL, Coenen S, Prater SM, Cavett V, Cleveland JL, et al. Mapping the phosphorylation sites of Ulk1. J Proteome Res. 2009;8:5253–63. doi: 10.1021/pr900583m. [DOI] [PubMed] [Google Scholar]
- 52.Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 2011;7:737–47. doi: 10.4161/auto.7.7.15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7:1212–21. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gan B, Peng X, Nagy T, Alcaraz A, Gu H, Guan J-L. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol. 2006;175:121–33. doi: 10.1083/jcb.200604129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gan B, Melkoumian ZK, Wu X, Guan KL, Guan J-L. Identification of FIP200 interaction with the TSC1-TSC2 complex and its role in regulation of cell size control. J Cell Biol. 2005;170:379–89. doi: 10.1083/jcb.200411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7:696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- 57.Blankson H, Holen I, Seglen PO. Disruption of the cytokeratin cytoskeleton and inhibition of hepatocytic autophagy by okadaic acid. Exp Cell Res. 1995;218:522–30. doi: 10.1006/excr.1995.1187. [DOI] [PubMed] [Google Scholar]
- 58.Samari HR, Møller MT, Holden L, Asmyhr T, Seglen PO. Stimulation of hepatocytic AMP-activated protein kinase by okadaic acid and other autophagy-suppressive toxins. Biochem J. 2005;386:237–44. doi: 10.1042/BJ20040609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bánréti A, Lukácsovich T, Csikós G, Erdélyi M, Sass M. PP2A regulates autophagy in two alternative ways in Drosophila. Autophagy. 2012;8:623–36. doi: 10.4161/auto.19081. [DOI] [PubMed] [Google Scholar]
- 60.Yorimitsu T, He C, Wang K, Klionsky DJ. Tap42-associated protein phosphatase type 2A negatively regulates induction of autophagy. Autophagy. 2009;5:616–24. doi: 10.4161/auto.5.5.8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McEwan DG, Dikic I. The Three Musketeers of Autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 2011;21:195–201. doi: 10.1016/j.tcb.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, et al. Function and molecular mechanism of acetylation in autophagy regulation. Science. 2012;336:474–7. doi: 10.1126/science.1216990. [DOI] [PubMed] [Google Scholar]
- 63.Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17:666–72. doi: 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- 64.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13:270–6. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- 65.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101:18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A. 2012;109:6561–5. doi: 10.1073/pnas.1204429109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science. 2012;336:477–81. doi: 10.1126/science.1217032. [DOI] [PubMed] [Google Scholar]
- 68.Korolchuk VI, Menzies FM, Rubinsztein DC. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy. 2009;5:862–3. doi: 10.4161/auto.8840. [DOI] [PubMed] [Google Scholar]
- 69.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–27. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–8. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 71.Gao Z, Gammoh N, Wong PM, Erdjument-Bromage H, Tempst P, Jiang X. Processing of autophagic protein LC3 by the 20S proteasome. Autophagy. 2010;6:126–37. doi: 10.4161/auto.6.1.10928. [DOI] [PubMed] [Google Scholar]
- 72.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–41. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 73.Zhou X, Babu JR, da Silva S, Shu Q, Graef IA, Oliver T, et al. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc Natl Acad Sci U S A. 2007;104:5842–7. doi: 10.1073/pnas.0701402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 75.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19:4762–75. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–21. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:6. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 81.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 82.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 83.Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell. 2012;47:359–70. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang HW, Wang YB, Wang SL, Wu MH, Lin SY, Chen GC. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J. 2011;30:636–51. doi: 10.1038/emboj.2010.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gammoh N, Florey O, Overholtzer M, Jiang X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat Struct Mol Biol. 2012 doi: 10.1038/nsmb.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6:79–90. doi: 10.1016/S1534-5807(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 87.Sekito T, Kawamata T, Ichikawa R, Suzuki K, Ohsumi Y. Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells. 2009;14:525–38. doi: 10.1111/j.1365-2443.2009.01299.x. [DOI] [PubMed] [Google Scholar]
- 88.Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23:1860–73. doi: 10.1091/mbc.E11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci. 2012;125:1488–99. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- 90.Kageyama S, Omori H, Saitoh T, Sone T, Guan JL, Akira S, et al. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell. 2011;22:2290–300. doi: 10.1091/mbc.E10-11-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nakatogawa H, Ohbayashi S, Sakoh-Nakatogawa M, Kakuta S, Suzuki SW, Kirisako H, et al. The autophagy-related protein kinase Atg1 interacts with the ubiquitin-like protein Atg8 via the Atg8 family interacting motif to facilitate autophagosome formation. J Biol Chem. 2012;287:28503–7. doi: 10.1074/jbc.C112.387514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yeh YY, Wrasman K, Herman PK. Autophosphorylation within the Atg1 activation loop is required for both kinase activity and the induction of autophagy in Saccharomyces cerevisiae. Genetics. 2010;185:871–82. doi: 10.1534/genetics.110.116566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kijanska M, Dohnal I, Reiter W, Kaspar S, Stoffel I, Ammerer G, et al. Activation of Atg1 kinase in autophagy by regulated phosphorylation. Autophagy. 2010;6:1168–78. doi: 10.4161/auto.6.8.13849. [DOI] [PubMed] [Google Scholar]
- 95.Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–84. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 96.Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, et al. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal. 2010;3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheong H, Nair U, Geng J, Klionsky DJ. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:668–81. doi: 10.1091/mbc.E07-08-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yan J, Kuroyanagi H, Kuroiwa A, Matsuda Y, Tokumitsu H, Tomoda T, et al. Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. Biochem Biophys Res Commun. 1998;246:222–7. doi: 10.1006/bbrc.1998.8546. [DOI] [PubMed] [Google Scholar]
- 99.Ogura K, Wicky C, Magnenat L, Tobler H, Mori I, Müller F, et al. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev. 1994;8:2389–400. doi: 10.1101/gad.8.20.2389. [DOI] [PubMed] [Google Scholar]
- 100.McIntire SL, Garriga G, White J, Jacobson D, Horvitz HR. Genes necessary for directed axonal elongation or fasciculation in C. elegans. Neuron. 1992;8:307–22. doi: 10.1016/0896-6273(92)90297-Q. [DOI] [PubMed] [Google Scholar]
- 101.Toda H, Mochizuki H, Flores R, 3rd, Josowitz R, Krasieva TB, Lamorte VJ, et al. UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 2008;22:3292–307. doi: 10.1101/gad.1734608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mochizuki H, Toda H, Ando M, Kurusu M, Tomoda T, Furukubo-Tokunaga K. Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the Drosophila brain. PLoS One. 2011;6:e19632. doi: 10.1371/journal.pone.0019632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 1999;24:833–46. doi: 10.1016/S0896-6273(00)81031-4. [DOI] [PubMed] [Google Scholar]
- 104.Meléndez A, Neufeld TP. The cell biology of autophagy in metazoans: a developing story. Development. 2008;135:2347–60. doi: 10.1242/dev.016105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liang CC, Wang C, Peng X, Gan B, Guan JL. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem. 2010;285:3499–509. doi: 10.1074/jbc.M109.072389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 107.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 108.Gan B, Guan J-L. FIP200, a key signaling node to coordinately regulate various cellular processes. Cell Signal. 2008;20:787–94. doi: 10.1016/j.cellsig.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ochi Y, Chano T, Ikebuchi K, Inoue H, Isono T, Arai A, et al. RB1CC1 activates the p16 promoter through the interaction with hSNF5. Oncol Rep. 2011;26:805–12. doi: 10.3892/or.2011.1329. [DOI] [PubMed] [Google Scholar]
- 110.Ikebuchi K, Chano T, Ochi Y, Tameno H, Shimada T, Hisa Y, et al. RB1CC1 activates the promoter and expression of RB1 in human cancer. Int J Cancer. 2009;125:861–7. doi: 10.1002/ijc.24466. [DOI] [PubMed] [Google Scholar]
- 111.Choi JD, Ryu M, Ae Park M, Jeong G, Lee JS. FIP200 inhibits β-catenin-mediated transcription by promoting APC-independent β-catenin ubiquitination. Oncogene. 2012 doi: 10.1038/onc.2012.262. [DOI] [PubMed] [Google Scholar]
- 112.Koinuma D, Shinozaki M, Nagano Y, Ikushima H, Horiguchi K, Goto K, et al. RB1CC1 protein positively regulates transforming growth factor-beta signaling through the modulation of Arkadia E3 ubiquitin ligase activity. J Biol Chem. 2011;286:32502–12. doi: 10.1074/jbc.M111.227561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Meiselbach H, Sticht H, Enz R. Structural analysis of the protein phosphatase 1 docking motif: molecular description of binding specificities identifies interacting proteins. Chem Biol. 2006;13:49–59. doi: 10.1016/j.chembiol.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 114.Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010;3:ra31. doi: 10.1126/scisignal.2000911. [DOI] [PubMed] [Google Scholar]
- 115.Shintani T, Yamazaki F, Katoh T, Umekawa M, Matahira Y, Hori S, et al. Glucosamine induces autophagy via an mTOR-independent pathway. Biochem Biophys Res Commun. 2010;391:1775–9. doi: 10.1016/j.bbrc.2009.12.154. [DOI] [PubMed] [Google Scholar]
- 116.Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–11. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 118.Alers S, Löffler AS, Paasch F, Dieterle AM, Keppeler H, Lauber K, et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy. 2011;7:1423–33. doi: 10.4161/auto.7.12.18027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol. 2012;22:374–80. doi: 10.1016/j.tcb.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sanjuan MA, Milasta S, Green DR. Toll-like receptor signaling in the lysosomal pathways. Immunol Rev. 2009;227:203–20. doi: 10.1111/j.1600-065X.2008.00732.x. [DOI] [PubMed] [Google Scholar]
- 121.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–7. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 122.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 123.Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–79. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 124.Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13:1335–43. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huang S, Yang ZJ, Yu C, Sinicrope FA. Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J Biol Chem. 2011;286:40002–12. doi: 10.1074/jbc.M111.297432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011;18:1598–607. doi: 10.1038/cdd.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Mariño G, Galluzzi L, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle. 2011;10:2763–9. doi: 10.4161/cc.10.16.16868. [DOI] [PubMed] [Google Scholar]
- 128.Tang J, Deng R, Luo RZ, Shen GP, Cai MY, Du ZM, et al. Low expression of ULK1 is associated with operable breast cancer progression and is an adverse prognostic marker of survival for patients. Breast Cancer Res Treat. 2012;134:549–60. doi: 10.1007/s10549-012-2080-y. [DOI] [PubMed] [Google Scholar]
- 129.Jiang S, Li Y, Zhu YH, Wu XQ, Tang J, Li Z, et al. Intensive expression of UNC-51-like kinase 1 is a novel biomarker of poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Sci. 2011;102:1568–75. doi: 10.1111/j.1349-7006.2011.01964.x. [DOI] [PubMed] [Google Scholar]