

Abstract
Ursolic acid (UA) is a pentacyclic triterpenoid with promising cancer chemopreventive properties. A better understanding of the mechanisms underlying anticancer activity of UA is needed for further development as a clinically useful chemopreventive agent. Here, we found that both endoplasmic reticulum (ER) stress and autophagy were induced by UA in MCF-7 human breast cancer cells. Surprisingly, ER stress was identified as an effect rather than a cause of UA-induced autophagy. Autophagy-dependent ER stress protected the cells from UA-induced apoptosis through EIF2AK3-mediated upregulation of MCL1. Activation of MAPK1/3 but not inhibition of MTOR pathway contributed to UA-induced cytoprotective autophagy in MCF-7 cells. Our findings uncovered a novel cellular mechanism involved in the anticancer activity of UA, and also provided a useful model to study biological significance and mechanisms of autophagy-mediated ER stress.
Keywords: ursolic acid, MCF-7, ER stress, EIF2AK3, autophagy, MCL1, cytoprotection, apoptosis
Introduction
The endoplasmic reticulum (ER) is an important organelle found in eukaryotic cells. One of the main functions of ER is protein folding. The status of protein folding in ER is tightly monitored and regulated by a primitive, evolutionarily conserved signaling pathway, collectively termed the unfolded protein response (UPR) or ER stress response.1 Three ER resident transmembrane proteins ATF6 (activating transcription factor 6), ERN1 (endoplasmic reticulum to nucleus signaling 1) and EIF2AK3 (eukaryotic translation initiation factor 2-α kinase 3) are the stress sensors of ER. Activation of these sensors results in increase of ER protein-folding capacity and decrease of ER protein load. The final outcome of UPR is mitigation of ER stress. However, prolonged activity of the UPR could lead to cell death.2
Autophagy is generally defined as a lysosome-dependent mechanism to degrade intracellular components including damaged or obsolete organelles and proteins.3 ER stress as one important inducer of autophagy is well established.4 EIF2AK3-EIF2S1 or the ERN1-MAPK8/9/10 pathway has been recognized as the crucial mediator of ER stress-induced autophagy.5-7 Growing evidence suggests that both ER stress response and autophagy might play an important role in modulation of anticancer effects of chemopreventive agents.8-12
Terpenoids such as the monoterpene d-Limonene, the sesquiterpene farnesol, the diterpene vitamin A or retinol and the triterpenoids asiatic acid, celastrol and ursolic acid, are among the various chemopreventive agents that have shown promise for inhibiting breast cancer in preclinical models.13-15 Ursolic acid (UA) is a pentacyclic triterpenoid found in apple, rosemary and holy basil. Previous studies have shown that UA is effective against various types of cancer both in vitro and in vivo.16,17 Purported mechanisms include cell cycle arrest and apoptosis induction through inhibition of AKT,18 STAT3,19 NFKB120 and activation of MAPK8/9/1021 or TP5322 signaling pathways. However, the role of ER stress response in UA-induced apoptosis has not been addressed.
In the present study with MCF-7 human breast cancer cells, we discovered that UA induced ER stress response, evidenced by increased expression of ER chaperone HSPA5 and activation of ER resident kinase EIF2AK3-ERN1 pathways. We demonstrated that ER stress was an effect rather than a cause of autophagy induction. Autophagy-dependent ER stress suppressed UA-induced apoptosis through upregulation of MCL1. Finally we identified MAPK1/3 pathway activation, rather than MTOR pathway inhibition, as a mechanism of autophagy induction by UA. Our findings revealed a novel cellular mechanism triggered by UA, compromising its apoptotic activity.
Results
UA activated UPR in MCF-7 human breast cancer cells
MCF-7 cells were treated with various concentrations of UA for 24 h, and then overall growth inhibitory effects were measured by crystal violet staining and apoptosis was detected by Annexin V staining of externalized phosphatidylserine in apoptotic cells, respectively. As shown in Figure 1A and B, UA treatment decreased, in a dose-dependent manner, cell number and increased apoptosis. Significant apoptosis was observed at concentrations of 30 μM and above. Treatment with 20 μM UA for 24 h decreased cell number and only slightly increased apoptosis. Cell cycle distribution analysis showed that cells treated with 20 μM UA treatment were arrested at G1 (data not shown). We next analyzed changes of ER stress markers by UA at sub-lethal and lethal doses by western blotting. As shown in Figure 1C, UA treatment in the concentration range of 10 to 20 μM, significantly increased protein abundance of ER chaperone HSPA5, the ER resident kinase EIF2AK3 and ERN1, and their phosphorylation levels. In line with EIF2AK3 and ERN1 induction, phosphorylation of EIF2S1 and expressions of ATF4 and XBP1s were significantly induced in response to UA treatment. These parameters were decreased with onset of apoptosis at 25 μM or higher UA. The data suggested that ER stress was induced by UA at relatively low concentrations in MCF-7 cells.

Figure 1. Ursolic acid (UA) induced cytoprotective EIF2AK3 activation in MCF-7 human breast cancer cells. UA induced cell number reduction (A) and apoptosis (B) in a dose-dependent manner. MCF-7 cells were treated with various concentrations of UA for 24 h, and then overall inhibitory effects and apoptosis were measured by crystal violet staining and annexin V/FITC staining, respectively. (C) Unfolded protein response induced by UA. MCF-7 cells were treated with various concentrations of UA for 24 h and then ER stress associated markers were analyzed by western blotting. (D) Effects of EIF2AK3 inactivation by RNAi on UA-induced apoptosis. The cells were transfected with 50 nmol/L of EIF2AK3 siRNA using siPORT™ NeoFX™ Transfection Agent. At 24 h post-transfection, the cells were treated with 20 μM for 24 h and apoptosis induction was assessed by sub-G1 analysis (n = 3, **p < 0.01). (The blots shown are representative of three independent experiments).
To determine the biological function of UPR, we tested effects of knockdown of these ER stress-related proteins by siRNA on apoptosis induction by UA in MCF-7 cells. As shown in Figure 1D, EIF2AK3 silencing led to a dramatically enhanced apoptosis induction by UA compared with that of nontargeting siRNA control (22.2% vs 4.1%). But no significant changes were found when HSPA5 or ERN1 was silenced (data not shown). These results indicated that neither EIF2AK3 activation nor HSPA5 or ERN1 induction inhibited apoptosis induction in response to UA treatment in MCF-7 cells.
UA induced autophagy in MCF-7 human breast cancer cells
Because ER stress is a well-known inducer of autophagy, we asked whether autophagy was induced in response to UA exposure in MCF-7 cells. We analyzed conversion of LC3-I to LC3-II, a biochemical marker of autophagy, by western blotting. As shown in Figure 2A, treatment with UA at concentrations of 15 to 25 μM led to a significant increase of LC3-II level. Interestingly, at relatively high concentrations (30 μM and above), UA-induced LC3-II almost disappeared. To verify the UA-induced autophagy, immunofluorescence staining was employed to analyze distribution patterns of LC3. As shown in Figure 2B, a diffuse localization of LC3 fluorescence was observed in control cells, whereas a punctated pattern of LC3 fluorescence was detected in UA-treated cells. Quantitative analysis of LC3-punctate cells showed that the number of cells with LC3 puncta in UA-treated cells was significantly increased compared with the untreated control (Fig. 2C). Furthermore, transmission electron microscopy (TEM) revealed many autophagic vacuoles in UA-treated cells (Fig. 2D). A higher magnification image clearly showed the presence of autophagic vacuoles containing partially degraded cytoplasmic material (Fig. 2D). These data together supported the concept that there was induction of autophagy by UA in MCF-7 cells at relatively low concentrations, in close association with ER stress response.

Figure 2. UA induced cytoprotective autophagy in MCF-7 human breast cancer cells. (A) UA caused an increased LC3-I to LC3-II conversion analyzed by western blotting (24 h). (B) Puncta distribution of LC3 induced by UA detected by immunofluorescence staining (24 h). (C) Quantitative analysis of LC3 puncta in cells in UA-treated and untreated cells. (D) TEM analysis of autophagic vacuoles in UA-treated and untreated cells (12 h). E. Increase of autophagosome formation was involved in UA-induced autophagy in MCF-7 cells. The cells were treated with 20 μM UA for 24 h in the presence or absence of 100 nM BAF (added 2 h before cell harvest) and then LC3 was analyzed by western blotting. (F and G) Effects of autophagy inhibitor 3-MA (F) or WORT (G) on UA-induced LC3-I to LC3-II conversion and PARP1 cleavage examined by western blotting. (H) Effects of autophagy inhibitor 3-MA or WORT on UA-induced apoptosis. The cells were treated with 20 μM UA in the presence or absence of 3-MA (3 mM) or WORT (1 μM) for 24 h and then apoptosis was assessed, measured by sub-G1 analysis (n = 3, **p < 0.01). (The blots shown are representative of three independent experiments).
To determine whether autophagy induced by UA is due to increased formation of autophagosome or decreased its degradation, we measured autophagy flux using bafilomycin A1, an inhibitor of autophagosome degradation. As shown in Figure 2E, bafilomycin A1 treatment led to accumulation of LC3-II whereas UA treatment significantly increased LC3-II levels either in the presence or absence of bafilomycin A1, suggesting that increase of autophagosome formation contributed to UA-induced autophagy in MCF-7 cells.
To examine biological significance of autophagy induction by UA, we tested the effects of autophagy inhibitors 3-MA or WORT on apoptosis induction by UA in MCF-7 cells. As shown in Figure 2F and G, UA-induced conversion of LC3-I to LC3-II was significantly attenuated in the presence of 3-MA or WORT. Under such conditions, cleavage of PARP1 (Fig. 2F and G) and apoptosis (Fig. 2H) by UA treatment were dramatically increased compared with that in the absence of 3-MA (35.4% vs 4.9%) or WORT (28.7% vs 4.9%). Taken together, these results suggested that exposure to low-level UA induced cytoprotective autophagy against its induction of apoptosis in MCF-7 cells.
ATG5 and BECN1 were involved in UA-induced autophagy in MCF-7 cells
To investigate molecular mediators in UA-induced autophagy, we analyzed changes of BECN1 (a component of the autophagy-specific PtdIns3K complex)23 and ATG5 (involved in elongation of the autophagic phagophore by the formation of the ATG12-ATG5 complex during autophagy)24 in response to UA in MCF-7 cells. The cells were exposed to various concentrations of UA for 24 h, protein levels of BECN1 and ATG5 were assessed by western blotting. As shown in Figure 3A, UA caused a peak increase of BECN1 (~25 μM) and ATG5 (~15 μM). This special dose-response pattern is generally consistent with that of autophagy induction and onset of apoptosis at the higher UA exposure levels (Fig. 2A). To determine the role of BECN1 and ATG5 in UA-induced autophagy, we used siRNA to knock down these two ATG genes simultaneously and examined autophagy induction. As shown in Figure 3B, basal and UA-induced BECN1 and ATG5 were significantly suppressed by their specific siRNAs. Under such conditions, UA-induced LC3-I to LC3-II conversion was significantly attenuated with an increase of PARP1 cleavage and apoptosis (Fig. 3C). These data supported involvement of ATG5 and BECN1 in UA-induced autophagy, and a prosurvival role of autophagy against UA-mediated cell death.
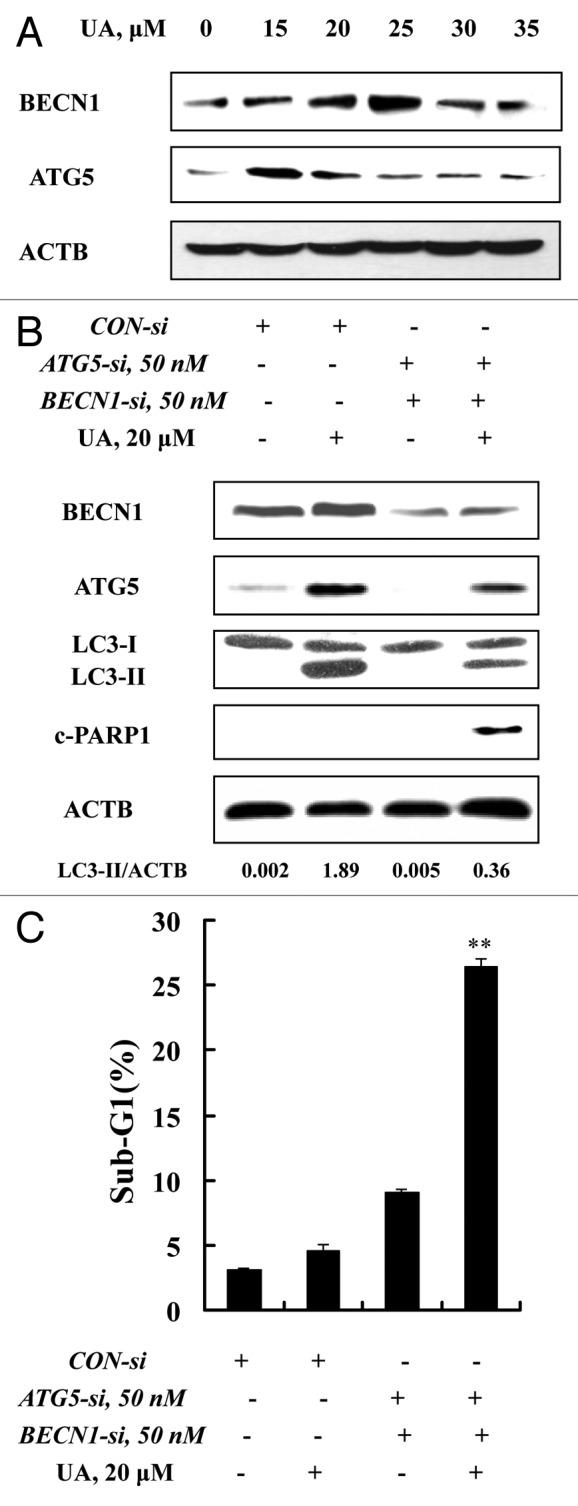
Figure 3. ATG5 and BECN1 were involved in UA-induced autophagy in MCF-7 cells. (A) Effects of UA treatment on BECN1 and ATG5 expression. MCF-7 cells were treated with various concentrations of UA for 24 h, and then expression of BECN1 and ATG5 was analyzed by western blotting. (B) Effects of BECN1 and ATG5 knockdown on UA-induced LC3-I to LC3-II conversion. The cells were transfected simultaneously with 50 nmol/L of BECN1 and 50 nmol/L of ATG5 siRNAs using siPORT™ NeoFX™ Transfection Agent. After 24 h transfection, the cells were treated with 20 μM UA for 24 h. LC3 was analyzed by western blotting. (C) Effects of BECN1 and ATG5 knockdown on UA-induced apoptosis measured by sub-G1 analysis (n = 3, **p < 0.01). (The blots shown are representative of three independent experiments).
ER stress was an effect rather than a cause of UA-induced autophagy
Having found activation of both ER stress and autophagy, we next questioned if ER stress was the cause of autophagy induction in response to UA in MCF-7 cells or vice versa. Since EIF2AK3 and ERN1 are the known mediators in ER stress-induced autophagy,5-7 we examined autophagy induction when either EIF2AK3 or ERN1 was silenced. As shown in Figure 4A, transfection of siRNA targeting EIF2AK3 caused a significant downregulation of both basal and UA-induced EIF2AK3, and inhibited EIF2S1 phosphorylation, but did not affect UA-induced LC3-I to LC3-II conversion. Similar results were also found when ERN1 was silenced (Fig. 4B). The results indicated that neither EIF2AK3 nor ERN1 was likely to play a critical role in UA-induced autophagy. When autophagy was inhibited by 3-MA, the key ER stress markers EIF2S1 phosphorylation and HSPA5 expression, measured by western blotting, were dramatically diminished. Furthermore, similar results were found when autophagy was inhibited by another inhibitor WORT (Fig. 4D) or when BECN1 and ATG5 were silenced by a siRNA approach (Fig. 4E). These results suggested ER stress as a downstream event of autophagy in response to UA.

Figure 4. ER stress was an effect rather than a cause of UA-induced autophagy. (A and B) Effects of EIF2AK3 (A) or ERN1 (B) knockdown on UA-induced LC3-I to LC3-II conversion. The cells were transfected with 50 nmol/L of EIF2AK3 siRNA or 50 nmol/L of ERN1 siRNA using siPORT™ NeoFX™ Transfection Agent. At 24 h post-transfection, the cells were treated with 20 μM for 24 h and then EIF2AK3, p-EIF2S1, ERN1 and LC3 were assessed by western blotting. (C and D) Effects of autophagy inhibition by 3-MA (C) or WORT (D) on UA-induced HSPA5 expression and EIF2S1 phosphorylation. The cells were treated with 20 μM UA in the presence or absence of 3-MA or WORT for 24 h and were analyzed by western blotting. (E) Effects of autophagy inhibition by BECN1/ATG5 knockdown on UA-induced HSPA5 expression and EIF2S1 phosphorylation. The cells were simultaneously transfected BECN1 and ATG5 siRNAs using siPORT™ NeoFX™ Transfection Agent. After 24 h transfection, the cells were treated with 20 μM UA for 24 h and were assessed by western blotting. (F) Time-course analysis of key parameters of ER stress, autophagy and apoptosis induced by UA in MCF-7cells. The cells were treated with UA for the indicated time and then LC3, EIF2S1 phosphorylation and PARP1 cleavage were assessed by western blotting. (G) UA induces autophagy-mediated ER stress in MDA-MB-231 cells. The cells were treated with UA for the indicated time and then LC3 and EIF2S1 phosphorylation were assessed by western blotting (left); The cells were pretreated with 3-MA for 1 h and further treated with UA for 6 h and western blotting was used to analyze LC3 and EIF2AK3-EIF2S1 phosphorylation (middle) and sub-G1 analysis was used to assess apoptosis induction by UA in the presence or absence of 3-MA for 24 h (right, n = 3, **p < 0.01). (The blots shown are representative of three independent experiments).
To further confirm the tandem sequence of UA-induced autophagy, ER stress, and apoptosis, we next analyzed the time-course changes of key parameters of these events induced by UA in MCF-7 cells. As shown in Figure 4F, a significantly increased LC3-II level was detected as early as after 2 h of UA treatment, whereas the increase of EIF2S1 phosphorylation was observed at 6 h of the treatment. No cleavage of PARP1 was found during this time frame. These time-course results indicate that UA induced autophagy preceding ER stress, which preceded apoptosis. Moreover, similar time-course responses of autophagy and ER stress induced by UA were also found in MDA-MB-231 breast cancer cells (Fig. 4G, left). Consistent with the time-course changes, inhibition of autophagy by its inhibitor 3-MA led to a significant attenuation of UA-induced autophagy and ER stress (Fig. 4G, middle), followed by an increase of apoptosis (Fig. 4G, right). The data clearly suggested that autophagy was the inducer of UA-mediated ER stress and the cytoprotective effect of autophagy against UA-induced apoptosis was attributed to the activation of EIF2AK3.
Upregulation of MCL1 contributed to the prosurvival property of UA-induced, autophagy-dependent EIF2AK3 activation
To decipher mechanisms of the EIF2AK3-mediated antiapoptotic effect, we assessed changes of MCL1, which has been reported to be regulated by UPR.25 MCF-7 cells were treated with various concentrations of UA for 24 h and the protein level of MCL1 was analyzed by western blotting. As shown in Figure 5A, at concentrations of 15 and 20 μM, UA induced a significant increase of MCL1, whereas at concentrations of 25 μM and above, UA decreased MCL1 expression, with onset of PARP1 cleavage. We next determined the relationship between MCL1 induction and EIF2AK3 activation. When EIF2AK3 was inhibited by RNA interference, MCL1 induction by UA was nearly abolished (Fig. 5B) and PARP1 cleavage was dramatically increased, suggesting that upregulation of MCL1 is a downstream event of EIF2AK3 activation. Given the critical role of autophagy induction in UA-induced EIF2AK3 activation, we then examined the effect of autophagy inhibition by either RNA interference or a chemical inhibitor on MCL1 induction by UA. As shown in Figure 5C, inhibition of BECN1 and ATG5 by genetic approach led to abolishment of UA-induced upregulation of MCL1. Similar results were also found when autophagy was blocked by knockdown of ATG5 alone (Fig. 5D) or its inhibitor 3-MA (Fig. 5E). To critically determine biological significance of MCL1 induction by UA, we tested the effect of MCL1 silencing on UA-induced apoptosis. As shown in Figure 5F, apoptosis induction by UA was dramatically increased when MCL1 was inhibited by RNA interference. Collectively, EIF2AK3-mediated upregulation of MCL1 was responsible for the cytoprotective effect of autophagy induced by UA against its apoptosis potency in MCF-7 cells.

Figure 5. Upregulation of MCL1 contributed to the prosurvival property of UA-induced, autophagy-dependent EIF2AK3 activation. (A) Effects of UA treatments on MCL1 expression. The cells were treated with various concentrations of UA for 24 h and MCL1 expression was analyzed by western blotting. (B) Effects of EIF2AK3 inactivation by RNAi on MCL1 induction by UA. EIF2AK3 was silenced by siRNA and MCL1 expression was analyzed by western blotting. (C) Effects of BECN1 and ATG5 knockdown on MCL1 induction by UA. BECN1 and ATG5 were simultaneously silenced by a siRNA approach and MCL1 was measured by western blotting. (D) Effects of ATG5 knockdown on phosphorylation of EIF2AK3-EIF2S1 and MCL1 induction. ATG5 was silenced by siRNA approach and phosphorylation of EIF2AK3-EIF2S1 and MCL1 expression were measured by western blotting. (E) Effects of 3-MA on MCL1 induction by UA. The cells were treated with 20 μM UA in the presence or absence of 3-MA for 24 h, MCL1 was analyzed by western blotting. (E) Effects of MCL1 inhibition by siRNA on UA-induced apoptosis measured by sub-G1 analysis (n = 3, **p < 0.01). (The blots shown are representative of three independent experiments).
Activation of MAPK1/3 but not inhibition of MTOR pathway was involved in UA-induced autophagy
The above data ruled out ER stress as an inducer of UA-induced autophagy in MCF-7 cells. In the following investigations on mechanisms involved in autophagy induction by UA, we examined the role of the MTOR pathway, which is considered a classic regulator of autophagy.26 After the cells were treated with various concentrations of UA for 24 h, western blotting detected (Fig. 6A) that UA caused a dose-dependent decrease of AKT phosphorylation without affecting total level of AKT. In contrast, UA increased EIF4EBP1 and RPS6KB1/2 phosphorylation dose-dependently. To confirm these results, we compared with RAPA, a well-known inhibitor of MTOR, as a positive control side-by-side with UA on phosphorylation of EIF4EBP1 and RPS6KB1/2 in the same experiment (Fig. 6B). As expected, 1μM RAPA significantly suppressed phosphorylation of EIF4EBP1 and RPS6KB1/2, as opposed to the induction of EIF4EBP1 and RPS6KB1/2 phosphorylation by UA. The results suggested that UA-induced autophagy in MCF-7 cells did not involve MTOR inhibition.
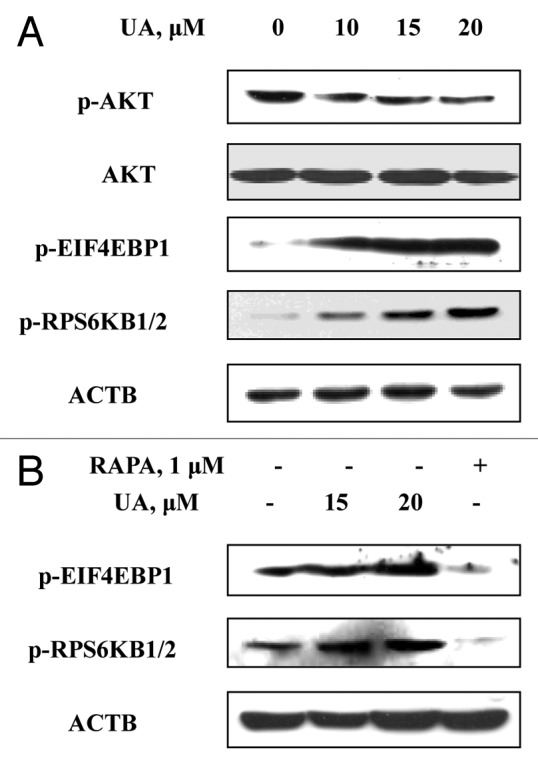
Figure 6. UA-induced autophagy was not associated with inhibition of the MTOR pathway. (A) Effects of UA treatment on phosphorylation of AKT, EIF4EBP1 and RPS6KB1/2. The cells were treated with various concentrations of UA for 24 h and were analyzed by western blotting. (B) Effects of UA treatments or RAPA on phosphorylation of EIF4EBP1 and RPS6KB1/2 measured by western blotting. (The blots shown are representative of three independent experiments).
We next determined the role of MAPKs in UA-induced autophagy in MCF-7 cells. As shown in Figure 7A, UA exposure for 24 h induced a concentration-dependent increase of total and phosphorylation levels of MAPK1/3. Similar changes of MAPK8/9 and its substrate JUN phosphorylation were also detected. However, no such changes were found with MAPK11/12/13/14. To assess the role of MAPK1/3 or MAPK8/9/10 activation in autophagy induction by UA, we tested the effects of MAPK1/3 or MAPK8/9/10 inhibitors on UA-induced LC3-I to LC3-II conversion. Inhibition of MAPK1/3 by PD98059 led to a significant attenuation of UA-induced LC3-II formation (Fig. 7B), whereas no significant change was found when MAPK8/9/10 activation was inhibited by SP600125 (Fig. 7C), ruling out MAPK8/9/10 involvement in autophagy induction by UA. In line with the critical role of MAPK1/3 activation in UA-induced autophagy, inhibition of MAPK1/3 activation led to a significant decrease of UA-induced EIF2S1 phosphorylation and MCL1 upregulation, followed by a dramatic increase of PARP1 cleavage (Fig. 7B). Last, we evaluated effect of MAPK1/3 inhibition on UA-induced apoptosis detected through sub-G1 analysis. As shown in Figure 7D, UA caused a dramatically enhanced apoptosis in the presence of the MAPK1/3 inhibitor PD98059 compared with that found in the absence of the inhibitor (23.8% vs 2.1%), consistent with a critical role of MAPK1/3 activation in UA-induced cytoprotective autophagy (Fig. 8).

Figure 7. Activation of MAPK1/3 contributed to UA-induced autophagy. (A) Effects of UA treatment on MAPKs. The cells were treated with various concentrations of UA for 24 h. The total and phosphorylation of MAPK1/3, MAPK8/9/10, MAPK11/12/13/14, the expression of BCL2 and DDIT3 were analyzed by western blotting. (B) Effects of MAPK1/3 inhibitor on UA-induced changes of key parameters of autophagy, ER stress and apoptosis. The cells were treated with 20 μM UA in the presence or absence of PD98059 for 24 h, phosphorylation of MAPK1/3, LC3-I to LC3-II conversion, EIF2S1 phosphorylation, MCL1 and PARP1 cleavage were analyzed by western blotting. (C) Effects of MAPK8/9/10 inhibitor on UA-induced LC3-I to LC3-II conversion. The cells were treated with 20 μM UA in the presence or absence of SP600125 for 24 h, LC3 was analyzed by western blotting. (D) Effects of MAPK1/3 inhibition by its inhibitor on UA-induced apoptosis. The cells were treated with 20 μM UA in the presence or absence of PD98059 for 24 h, then apoptosis was assessed by sub-G1 analysis (n = 3, **p < 0.01). (The blots shown are representative of three independent experiments).
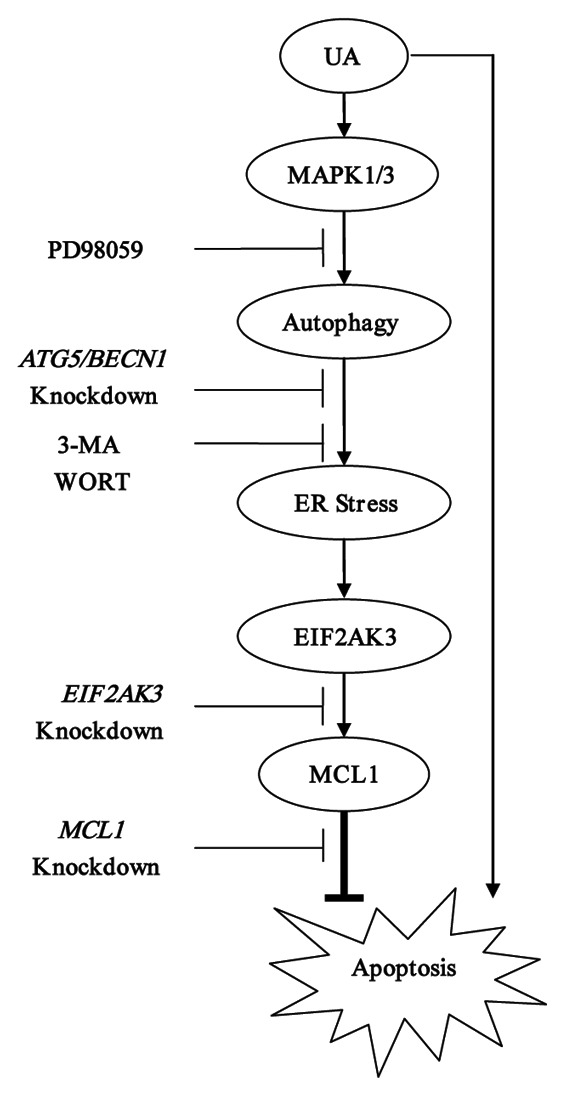
Figure 8. Signaling pathways underlying UA-induced cytoprotective autophagy in human MCF-7 breast cancer cells. UA at relatively low concentrations induced autophagy which in turn led to activation of UPR including EIF2AK3 pathway. Activation of EIF2AK3 suppressed UA-mediated apoptosis through upregulation of MCL1. UA-induced cytoprotective autophagy was attributed to MAPK1/3 activation but not associated with MTOR inhibition.
Discussion
Previous studies have shown that UA is a promising chemopreventive agent against various types of cancer including breast cancer. A better understanding of the mechanisms underlying anticancer activity of UA is critical for further development as a clinically useful chemopreventive agent. To this end, our current study demonstrated that autophagy-dependent ER stress response by UA compromised its apoptotic effect. Therefore, inhibition of autophagy or UPR (EIF2AK3) activation might improve the efficacy of UA against human breast cancer. Our findings uncovered a novel cellular mechanism that could be effectively targeted to boost up the anticancer activity of UA.
It has been shown that many chemotherapeutic drugs or chemopreventive agents induce both prosurvival signaling cascades that can mediate drug resistance and apoptosis signaling. Examples include MAPK1/3 activation by microtubule stabilizing drugs,27 MCL1 upregulation by a small molecule inhibitor of BCL2 family proteins ABT-73728 or tumor necrosis factor (ligand) superfamily member 10 (TNFSF10),29 cytoprotective autophagy induction by sulforaphane, a naturally occurring member of the isothiocyanate family of chemopreventive agents10 and AKT activation by penta-1,2,3,4,6-O-galloyl-β-D-glucose (PGG), a naturally occurring polyphonolic gallotannin compound.30 Activation of prosurvival signalings by these drugs or agents compromises their anticancer activity. Targeting these survival signaling pathways can dramatically improve the efficacy of these drugs or agents, at least in vitro. Therefore identification of such prosurvival signaling has important clinical ramifications.
In the current study, UA activated ER resident protein kinase EIF2AK3-EIF2S1 pathway which, paradoxically, has both pro- and antisurvival properties.31 Knocking down EIF2AK3 by siRNA resulted in a significant increase of UA-induced apoptosis in MCF-7 breast cancer cells (Fig. 1C), suggesting EIF2AK3-dependent signaling plays a prosurvival role that protects cells from UA-mediated apoptosis. In terms of mechanisms underlying the EIF2AK3-mediated prosurvival effect, we found that EIF2AK3 activation by UA treatment led to upregulation of MCL1, whereas inhibition of MCL1 by RNA interference dramatically increased UA-mediated lethality, suggesting MCL1 was a downstream target of EIF2AK3 activation to mediate an antiapoptotic action. MCL1 is transcriptionally regulated by transcription factor ATF4,25 a transcription factor that is regulated by EIF2AK3-EIF2S1 pathway,1 and our data revealed that ATF4 was indeed induced in response to UA treatment (Fig. 1C). We therefore postulated that EIF2AK3-mediated upregulation of MCL1 likely occurred through transcriptional activation of ATF4. It is noteworthy that MCL1 was found to be downregulated in U266 human multiple myeloma cells32 and U937 human leukemia cells.33 The possible explanation for this controversial action is the dose-response pattern. At the concentrations of 15 and 20 μM, UA caused a dose-dependent increase of MCL1 expression, whereas at the concentrations of 25 μM and above, UA became a negative regulator of MCL1 in MCF-7 cells (Fig. 5A), coinciding with appearance of PARP1 cleavage. We speculated that MCL1 may be regulated by multiple mechanisms in UA-treated cells. At relatively low UA concentrations, EIF2AK3-mediated, upregulation action played a predominant role, leading to MCL1 induction, whereas at high UA concentrations, autophagy-mediated EIF2AK3 activation was not sustainable and the inhibiting mechanism (such as STAT3 or NFKB1 inhibition by UA) became predominant, leading to an overall MCL1 reduction. This special dose-response feature closely tracked with UA-mediated cytoprotective autophagy response, further supporting a link between MCL1 induction and UA-induced autophagy-dependent EIF2AK3 activation.
As mentioned above, ER stress is known to cause autophagy, but less is known of the reverse relationship. Indeed treatment MCF-7 cells with UA induced autophagy specific marker LC3-I to LC3-II conversion and its puncta distribution, suggesting autophagy was induced in response to UA in MCF-7 cells. However, knockdown of either EIF2AK3 or ERN1, critical mediators of ER stress, did not affect UA-induced LC3 conversion. More interestingly, blockade of autophagy by either inhibitor or genetic approach nearly abolished UA-induced HSPA5 upregulation and EIF2S1 phosphorylation. Our time-course results further supported that autophagy induced by UA occurred earlier than ER stress events. Collectively, the data suggested that ER stress was the effect rather than cause of UA-induced autophagy. Our findings established an unconventional relationship between autophagy induction and the ER stress response. An autophagy-mediated EIF2AK3-MCL1 signaling cascade might provide a mechanistic explanation for the prosurvival role of autophagy in some model systems. The biological significance and molecular mechanisms of autophagy-dependent ER stress need to be further investigated.
In terms of mechanisms of UA-induced autophagy in MCF-7 cells, our data clearly ruled out the classical MTOR pathway, which was contradictory to a recent report that UA-induced autophagy is associated with inhibition of the MTOR signaling pathway in PC-3 prostate cancer cells.34 The possible interpretations for this difference include cell type and UA concentrations. UA induced an obvious autophagic response in PC-3 cells at concentrations of 30 μM and above, which was well correlated with inhibition of phosphorylation of RPS6KB1/2 and EIF4EBP1. In our current study, a significant autophagy induction in MCF-7 cells was detected at concentrations of 25 μM and below. Further studies on these differential responses are clearly needed. Our data supported a role of MAPK1/3, but not MAPK8/9/10 and MAPK11/12/13/14 as a critical mediator of UA-induced cytoprotective autophagy in MCF-7 cells, which is consistent with the prosurvival role of MAPK1/3 activation in UA-induced cell death in MCF-7 cells.
In summary, treating MCF-7 human breast cancer cells with a sublethal dose of UA induced an autophagy-mediated ER stress, which compromised UA-induced apoptosis through EIF2AK3-mediated upregulation of MCL1. Activation of MAPK1/3 but not inhibition of MTOR was responsible for autophagy induction by UA in MCF-7 cells. Our findings revealed a novel signaling cascade involved in compromising the UA-induced apoptotic effect and also provided a useful model to study autophagy-mediated ER stress.
Materials and Methods
Chemicals and reagents
Ursolic acid (UA) (U6753), 3-methyladenine (3-MA) (M9281), rapamycin (R0395) and bafilomycin A1 (B1793) were purchased from Sigma-Aldrich. Antibodies specific for MCL1 (CST, 4572), HSPA5 (CST, 3183), EIF2AK3 (CST, 3192), ERN1 (CST, 3294), MAPK1/3 (CST, 4695), MAPK8/9/10 (CST, 9258), MAPK11/12/14 (CST, 8690), AKT (CST, 9272), phospho-EIF2S1 (CST, 3597), phospho-MAPK1/3 (CST, 4094), phospho-MAPK8/9/10 (CST, 4668), phospho-JUN (CST, 5464), phospho-MAPK11/12/13/14 (CST, 4511), phospho-AKT (CST, 9271), phospho-RPS6KB1/2 (CST, 9205), BECN1 (CST, 3738), ATG5 (CST, 8540), BCL2 (CST, 2870), DDIT3 (CST, 2895) and cleaved poly (ADP-ribose)polymerase(PARP1; p89) (CST, 9548) were purchased from Cell Signaling Technology. Antibodies for phospho-EIF2AK3 (sc-32577) and ATF4 (sc-22800) were purchased from Santa Cruz Biotecnology. Antibody for phospho-ERN1 (ab124945) was purchased from Abcam. Antibodies for EIF2S1 (Bioworld, BS3651) and phospho-EIF4EBP1 (BS4746) were purchased from Bioworld. Antibody for XBP1s (647502) was purchased from Biolegend. Antibodies for LC3 (MBL, PM036) and CASP7 (M053-3) were purchased from MBL International Corporation. Wortmannin (681676), MAPK1/3 inhibitor PD98059 (513000) and MAPK8/9/10 inhibitor SP600125 (420119) were purchased from Calbiochem.
Cell culture and treatments
MCF-7 and MDA-MB-231 cell lines were obtained from the American Type Culture Collection. The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo, SH30022.01B) supplemented with 10% fetal bovine serum without antibiotics. At 24 to 48 h after plating when cells were at 50–60% confluence, the medium was changed before starting the treatment with UA and/or other agents.
Crystal violet staining
For the evaluation of overall inhibitory effect of UA on cell growth, the cells were treated with various concentrations of UA for 24 h. After treatment, the culture medium was removed and the cells were fixed in 1% glutaraldehyde solution in PBS for 15 min. The fixed cells were stained with 0.02% aqueous solution of crystal violet for 30 min. After washing with PBS, the stained cells were solubilized with 70% ethanol. The absorbance at 570 nm with the reference filter 405 nm was evaluated using a microplate reader (Thermo).
Apoptosis evaluation
Apoptosis was assessed by three methods. The first was sub-G1 analysis by flow cytometry. The second one was Annexin V staining of externalized phosphatidylserine. The third method was immunoblot analysis of PARP1 cleavage.
Autophagy detection
Autophagy induction was determined by three methods. The first method was western blotting for conversion of the microtubule-associated protein 1 light chain 3 (LC3)-I to LC3-II. The second one was immunofluorescence staining for LC3 localization as reported previously.10 The third was transmission electron microscope analysis of autophagic vacuoles.
Western blotting
The cell lysate was prepared in ice-cold radioimmuno-precipitation assay buffer. Cell lysates were resolved by electrophoresis and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, IPVH00010). The blot was then probed with a primary antibody followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies (MBL, 330 and 458). The signal was visualized by enhanced chemiluminescence (Fisher/Pierce, 32106) and recorded on an X-ray film (Estman Kodak Company, XBT-1).
RNA interference
siRNAs targeting EIF2AK3 (sc-36213), ERN1(sc-40705), HSPA5 (sc-29338), BECN1 (sc-29797) and MCL1 (sc-35877) were purchased from Santa Cruz Biotechnologies. siRNAs targeting ATG5 and nontargeting siRNA were synthesized by Integrated DNA Technologies. The cells were transfected with 50 nmol/L of specific or nontargeting siRNA using siPORT™ NeoFX™ Transfection Agent (Ambion, AM4510) for 24 h and then were used for subsequent experiments.
Statistical analysis
Data are presented as mean ± SD for triplicates. These data were analyzed with ANOVA with appropriate post-hoc comparison among means. p < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by grants from Ministry of Science and Technology of China (2012BAD33B09), National Natural Science Foundation of China (NSFC, 31071533, 30972172) and Chinese Universities Scientific Fund (2009-2-11). We thank Prof. Junxuan Lu for kind help in revising the English language of the manuscript. We also thank the Lab for Bio-imaging of the Core Facilities of the Institute of Biophysics, CAS, for our electron microscopy work and we are grateful to Ms. Lei Sun for her help of making EM samples and analyzing EM images.
Glossary
Abbreviations:
- PtdIns3K
phosphatidylinositol 3-kinase
- MAPKs
mitogen-activated protein kinases
- ATF4
activating transcription factor 4 (tax-responsive enhancer element B67)
- ATF6
activating transcription factor 6
- STAT3
signal transducer and activator of transcription 3 (acute-phase response factor)
- TP53
tumor protein p53
- BCL2
B-cell CLL/lymphoma 2
- MCL1
myeloid cell leukemia sequence 1 (BCL2-related)
- NFKB1
nuclear factor of kappa light polypeptide gene enhancer in B-cells 1
- MTOR
mechanistic target of rapamycin
- ERN1
endoplasmic reticulum to nucleus signaling 1
- XBP1s
spliced X-box binding protein 1
- EIF2AK3
eukaryotic translation initiation factor 2-alpha kinase 3
- EIF2S1
eukaryotic translation initiation factor 2, subunit 1 alpha, 35 kDa
- DDIT3
DNA-damage-inducible transcript 3
- HSPA5
heat shock 70 kDa protein 5 (glucose-regulated protein, 78 kDa)
- EIF4EBP1
eukaryotic translation initiation factor 4E binding protein 1
- RPS6KB1/2
ribosomal protein S6 kinase, 70 kDa, polypeptide 1/2
- LC3
microtubule-associated protein 1 light chain 3
- PARP1
poly (ADP-ribose) polymerase 1
- ATG5
autophagy-related 5
- BECN1
Beclin 1, autophagy related
- CASP7
caspase 7, apoptosis-related cysteine peptidase
- ACTB
actin, beta
- UA
ursolic acid
- TNFSF10
tumor necrosis factor (ligand) superfamily, member 10
- PGG
penta-1,2,3,4,6-O-galloyl-beta-D-glucose
- RAPA
rapamycin
- 3-MA
3-methyladenine
- WORT
wortmannin
- BAF
bafilomycin A1
- TEM
transmission electron microscopy
- ER
endoplasmic reticulum
- UPR
unfolded protein response
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/22805
References
- 1.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 2.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moretti L, Cha YI, Niermann KJ, Lu B. Switch between apoptosis and autophagy: radiation-induced endoplasmic reticulum stress? Cell Cycle. 2007;6:793–8. doi: 10.4161/cc.6.7.4036. [DOI] [PubMed] [Google Scholar]
- 5.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–9. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 6.Tallóczy Z, Jiang W, Virgin HW, 4th, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–5. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeh TC, Chiang PC, Li TK, Hsu JL, Lin CJ, Wang SW, et al. Genistein induces apoptosis in human hepatocellular carcinomas via interaction of endoplasmic reticulum stress and mitochondrial insult. Biochem Pharmacol. 2007;73:782–92. doi: 10.1016/j.bcp.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 9.Wang FM, Galson DL, Roodman GD, Ouyang H. Resveratrol triggers the pro-apoptotic endoplasmic reticulum stress response and represses pro-survival XBP1 signaling in human multiple myeloma cells. Exp Hematol. 2011;39:999–1006. doi: 10.1016/j.exphem.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006;66:5828–35. doi: 10.1158/0008-5472.CAN-06-0139. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura Y, Yogosawa S, Izutani Y, Watanabe H, Otsuji E, Sakai T. A combination of indol-3-carbinol and genistein synergistically induces apoptosis in human colon cancer HT-29 cells by inhibiting Akt phosphorylation and progression of autophagy. Mol Cancer. 2009;8:100. doi: 10.1186/1476-4598-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puissant A, Auberger P. AMPK- and p62/SQSTM1-dependent autophagy mediate Resveratrol-induced cell death in chronic myelogenous leukemia. Autophagy. 2010;6:655–7. doi: 10.4161/auto.6.5.12126. [DOI] [PubMed] [Google Scholar]
- 13.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–69. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 14.Rabi T, Gupta S. Dietary terpenoids and prostate cancer chemoprevention. Front Biosci. 2008;13:3457–69. doi: 10.2741/2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabi T, Bishayee A. Terpenoids and breast cancer chemoprevention. Breast Cancer Res Treat. 2009;115:223–39. doi: 10.1007/s10549-008-0118-y. [DOI] [PubMed] [Google Scholar]
- 16.Bishayee A, Ahmed S, Brankov N, Perloff M. Triterpenoids as potential agents for the chemoprevention and therapy of breast cancer. Front Biosci. 2011;16:980–96. doi: 10.2741/3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kowalczyk MC, Walaszek Z, Kowalczyk P, Kinjo T, Hanausek M, Slaga TJ. Differential effects of several phytochemicals and their derivatives on murine keratinocytes in vitro and in vivo: implications for skin cancer prevention. Carcinogenesis. 2009;30:1008–15. doi: 10.1093/carcin/bgp069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Achiwa Y, Hasegawa K, Udagawa Y. Regulation of the phosphatidylinositol 3-kinase-Akt and the mitogen-activated protein kinase pathways by ursolic acid in human endometrial cancer cells. Biosci Biotechnol Biochem. 2007;71:31–7. doi: 10.1271/bbb.60288. [DOI] [PubMed] [Google Scholar]
- 19.Pathak AK, Bhutani M, Nair AS, Ahn KS, Chakraborty A, Kadara H, et al. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res. 2007;5:943–55. doi: 10.1158/1541-7786.MCR-06-0348. [DOI] [PubMed] [Google Scholar]
- 20.Shishodia S, Majumdar S, Banerjee S, Aggarwal BB. Ursolic acid inhibits nuclear factor-kappaB activation induced by carcinogenic agents through suppression of IkappaBalpha kinase and p65 phosphorylation: correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res. 2003;63:4375–83. [PubMed] [Google Scholar]
- 21.Zhang Y, Kong C, Zeng Y, Wang L, Li Z, Wang H, et al. Ursolic acid induces PC-3 cell apoptosis via activation of JNK and inhibition of Akt pathways in vitro. Mol Carcinog. 2010;49:374–85. doi: 10.1002/mc.20610. [DOI] [PubMed] [Google Scholar]
- 22.Hsu YL, Kuo PL, Lin CC. Proliferative inhibition, cell-cycle dysregulation, and induction of apoptosis by ursolic acid in human non-small cell lung cancer A549 cells. Life Sci. 2004;75:2303–16. doi: 10.1016/j.lfs.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 23.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 24.Tang Y, Chen Y, Jiang H, Nie D. Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ. 2011;18:602–18. doi: 10.1038/cdd.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu J, Dang N, Menu E, De Bryune E, Xu D, Van Camp B, et al. Activation of ATF4 mediates unwanted Mcl-1 accumulation by proteasome inhibition. Blood. 2012;119:826–37. doi: 10.1182/blood-2011-07-366492. [DOI] [PubMed] [Google Scholar]
- 26.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–18. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 27.MacKeigan JP, Collins TS, Ting JP. MEK inhibition enhances paclitaxel-induced tumor apoptosis. J Biol Chem. 2000;275:38953–6. doi: 10.1074/jbc.C000684200. [DOI] [PubMed] [Google Scholar]
- 28.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi S, Lee SH, Meng XW, Mott JL, Bronk SF, Werneburg NW, et al. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J Biol Chem. 2007;282:18407–17. doi: 10.1074/jbc.M610010200. [DOI] [PubMed] [Google Scholar]
- 30.Hu H, Chai Y, Wang L, Zhang J, Lee HJ, Kim SH, et al. Pentagalloylglucose induces autophagy and caspase-independent programmed deaths in human PC-3 and mouse TRAMP-C2 prostate cancer cells. Mol Cancer Ther. 2009;8:2833–43. doi: 10.1158/1535-7163.MCT-09-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahmani M, Davis EM, Crabtree TR, Habibi JR, Nguyen TK, Dent P, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. 2007;27:5499–513. doi: 10.1128/MCB.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pathak AK, Bhutani M, Nair AS, Ahn KS, Chakraborty A, Kadara H, et al. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res. 2007;5:943–55. doi: 10.1158/1541-7786.MCR-06-0348. [DOI] [PubMed] [Google Scholar]
- 33.Gao N, Cheng S, Budhraja A, Gao Z, Chen J, Liu EH, et al. Ursolic acid induces apoptosis in human leukaemia cells and exhibits anti-leukaemic activity in nude mice through the PKB pathway. Br J Pharmacol. 2012;165:1813–26. doi: 10.1111/j.1476-5381.2011.01684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin SW, Kim SY, Park JW. Autophagy inhibition enhances ursolic acid-induced apoptosis in PC3 cells. Biochim Biophys Acta. 2012;1823:451–7. doi: 10.1016/j.bbamcr.2011.10.014. [DOI] [PubMed] [Google Scholar]