

Abstract
The programmed cell death-1 (PD)-1 receptor (CD279) is a potent T cell inhibitor with a critical role in peripheral tolerance, but it can also compromise anti-viral and antitumor T cell responses. The effects of PD-1 on the cell cycle leading to inhibition of T cell expansion are poorly understood. Recently, we examined the effects of PD-1 on the molecular control of the cell cycle machinery and on TCR-activated signaling pathways that regulate these downstream outcomes. Our studies showed that PD-1 blocks cell cycle progression in the G1 phase. PD-1 did not alter the expression of G1 phase cyclins or cyclin-dependent kinases (Cdks) but, instead, suppressed the transcription of SKP2, the substrate recognition component of the SCFSkp2 ubiquitin ligase that leads p27kip1 to degradation and resulted in accumulation of p27kip1. Subsequently, T cells receiving PD-1 signals displayed impaired Cdk2 activation and failed to phosphorylate two critical Cdk2 substrates, the retinoblastoma gene product (Rb) and the TGFβ-specific transcription factor Smad3, leading to suppression of E2F target genes but enhanced Smad3 transactivation. These events resulted in upregulation of the Cdk4/6 inhibitor p15INK4B and repression of the Cdk-activating phosphatase Cdc25A. The suppressive effect of PD-1 on Skp2 expression was mediated by inhibition of both PI3K/Akt and Ras/MEK/Erk pathways and was only partially reversed by IL-2, which restored activation of MEK/Erk but not Akt. Thus, PD-1 targets Ras and PI3K/Akt signaling to inhibit transcription of Skp2 and to activate Smad3 as an integral component of a pathway that regulates blockade of cell cycle progression in T lymphocytes. Here, we discuss the detailed sequence of these signaling events and their implications in mediating cell-intrinsic and -extrinsic mechanisms that inhibit proliferation of T effector cells in response to PD-1-mediated signaling.
Keywords: Cdc25, Cdk2, PD-1, Smad3, T cells
The PD-1/PD-L Pathway in the Regulation of T cell Responses
Maintenance of peripheral tolerance is essential for homeostasis of the immune system. While central tolerance mechanisms result in deletion of the majority of self-reactive T cells, some T lymphocytes specific for self-antigens escape this process and circulate in the periphery.1 To control the development of autoimmunity, multiple mechanisms of peripheral tolerance have evolved, including T cell anergy, deletion and suppression by regulatory T cells (Treg).2 The pathway consisting of the programmed death-1 (PD-1) receptor (CD279) and its ligands, PD-L1 and PD-L2 (B7-DC; CD273), is a newer pathway in the B7-CD28 family and regulates the balance between stimulatory and inhibitory signals needed for effective immunity and the maintenance of self-tolerance and T cell homeostasis.3 PD-1, initially identified as a gene upregulated in a T cell hybridoma undergoing cell death,4 is upregulated on T cells upon activation via the T cell receptor. Besides T lymphocytes, PD-1 expression is induced upon activation of natural killer T (NKT) cells, B cells, monocytes and certain subsets of dendritic cell (DC).5 The ligands for PD-1, PD-L1 (also known as B7-H1) and PD-L2 (also known as B7-DC) have distinct expression patterns6-9 PD-L1 is constitutively expressed in low levels on APCs (DCs, macrophages and B cells) and is further upregulated upon activation. PD-L1 is also induced on activated T cells6,10 PD-L1 is expressed on a wide variety of nonhematopoietic cell types, including vascular endothelial cells, pancreatic islet cells and at sites of immune privilege including the placenta, testes and eye. In contrast, expression of PD-L2 is induced primarily on DCs and macrophages upon activation.5
Due to its cell-specific and tissue-specific distribution, PD-1 exerts its effects during the initial phase of activation and expansion of autoreactive T cells by attenuating self-reactive T cells during presentation of self-antigen by dendritic cells (DCs).11,12 PD-1 also inhibits the functions of self-reactive effector T cells against non-hematopoietic tissues and mediates tissue tolerance by suppressing tissue-reactive T cells and protecting against immune-mediated tissue damage.13,14 In contrast to its important beneficial role in maintaining T cell homeostasis, PD-1 mediates potent inhibitory signals after ligation with PD-1 ligands expressed on malignant tumors that prevent the expansion of T effector cells and have detrimental effects on antitumor immunity.15-18 Moreover, expression of PD-1 by “exhausted” virus-specific T cells that are characteristic of chronic viral infections prevents the proliferation and function of virus-specific T effector cells and clearance of the virus.19-22 Because PD-1 has a potent antiproliferative function, we sought to determine how PD-1 affects the molecular events of the cell cycle machinery, thereby leading to blockade of T cell proliferation.
Effects of PD-1 on the Molecular Components of the Cell Cycle Machinery
Cell cycle progression is a tightly regulated process that depends on the expression and activation of positive and negative regulators of the cell cycle machinery. Expression of D1-type cyclins occurs during entry into G1 phase, induction of cyclin E at the late G1 phase and expression of cyclin A at the S phase. Cyclins associate with specific cyclin-dependent kinases (Cdks), which provide enzymatic activity to the cyclin-Cdk holoenzyme complexes and are inhibited by Cdk inhibitors. D-type cyclins associate with Cdk4 and its homolog Cdk6, whereas cyclin E and cyclin A associate with Cdk2. p27kip1, a member of the Kip/Cip family of Cdk inhibitors is abundantly expressed in T cells and interacts with Cdk2. Through this interaction, p27kip1 inhibits activation of cyclin E-Cdk2 and cyclin A-Cdk2 complexes.23,24 Although p27kip1 also associates with cyclin D-Cdk4 (or cyclin D-Cdk6), this interaction does not induce inhibition of cyclin D-associated enzymatic activity, but instead functions to sequester p27kip1, thereby facilitating cyclin E-Cdk2 activation.25 p15INK4B, a member of the INK family of the cdk inhibitors, selectively suppresses the enzymatic activity of the cyclin-Cdk complexes operative at the G1 phase. In contrast to their inhibition by the Cdk inhibitors, Cdks are positively regulated by the tyrosine phosphatase Cdc25A, which reduces the extent of Cdk tyrosine phosphorylation that has a negative effect on their enzymatic activation.26 Cdks promote cell cycle progression, in part, by phosphorylating the transcription factor Rb and related pocket proteins, thereby reversing their ability to sequester E2F transcription factors, which then leads to the expression of E2F-regulated genes.23
We examined the effects of PD-1 on the regulation of cell cycle progression in primary human CD4+ T cells in response to growth signals mediated through stimulation of the T cell receptor (TCR)/CD3 complex and the CD28 costimulatory pathway.27 We determined that PD-1 signaling induced blockade of cell cycle progression in G1 phase by increasing the abundance of the Cdk inhibitors p27kip1 and p15INK4B while suppressing expression of the gene encoding the Cdk-activating phosphatase Cdc25A. We further investigated how PD-1 altered TCR-proximal signaling events lead to these specific outcomes on the molecular components of the cell cycle machinery. Our studies showed that ligation of PD-1 during the stimulation of T cells through TCR-CD3 and CD28 inhibited activation of the PI3K-Akt and Ras-MEK/ERK pathways and blocked the expression of the gene encoding Skp2, the substrate-recognition component of the SCFSkp2 ubiquitin ligase, which mediates degradation of p27kip1, resulting in accumulation of p27kip1 and inhibition of Cdk2. Because of the impaired activity of Cdk2, T cells stimulated through PD-1 not only displayed decreased phosphorylation of Rb, but also failed to phosphorylate the checkpoint inhibitor Smad3 on the Cdk2-specific site, leading to enhanced Smad3 transcriptional activity, which resulted in the increased abundance of p15INK4B and the abrogation of the Cdk-activating phosphatase Cdc25A.
Our results identified the mechanism by which PD-1 inhibits cell cycle progression but also revealed unexpected molecular targets via which PD-1 might affect the fate of T cells by regulating Cdk2. Cdk2 is involved in many signaling pathways and functional outcomes (Fig. 1). In conjunction with cyclin E, Cdk2 phosphorylates p27kip1, resulting in ubiquitin-targeted degradation of p27kip1, which regulates not only cell cycle progression, but also cell motility and migration.28 Cdk2 promotes phosphorylation of Rb on specific sites, thereby reversing its ability to sequester E2F.23 Cdk2-mediated phosphorylation of Rb also impacts the interactions of Rb with histone deacetylases and other chromatin remodeling proteins.29,30 Cdk2-cyclin E also phosphorylates a number of substrates, which affect histone gene expression, centrosome duplication and replication origin licensing.31,32 Cdk2 directly regulates expression of genes including NFκB, Sp1, p300/CBP and subunits of the RNA polymerase.33 Cdk2 also phosphorylates Smad3 and antagonizes its antiproliferative function induced by TGFβ, whereas impaired phosphorylation on the Cdk-specific sites renders Smad3 more effective in executing its antiproliferative function.34 Inhibition of Cdk2 activation by PD-1 may lead to differential phosphorylation of such Cdk2 substrates, resulting in a distinct program of gene expression, differentiation and function of T cells. Thus, due to altering such functions of Cdk2, PD-1 may regulate T cell tolerance and homeostasis by reprogramming transcriptional and epigenetic events independently of its role as an inhibitor of cell cycle progression.
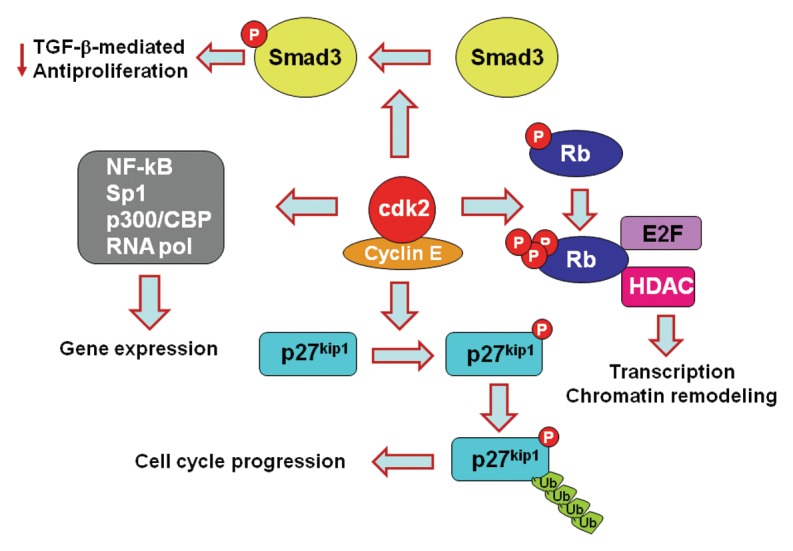
Figure 1. PD-1 affects multiple signaling pathways and functional outcomes by inhibiting Cdk2.
Synergistic Signaling Between PD-1 and TGFβ
Previous work indicated that PD-1 reduces the threshold of TGFβ-mediated signals, thereby synergizing with TGFβ to promote the conversion of naïve T cells into inducible Treg (iTreg) cells. Specifically, PD-1 can mediate the formation of iTregs in the presence of minimal amounts of TGFβ or even in the absence of exogenous TGFβ.35 On the basis of those findings and our results showing that PD-1 induced arrest at the G1 phase of the cell cycle, we postulated that PD-1 might promote TGFβ production or enhance the abundance of TGFβ receptors, thereby increasing the ability of CD4+ T cells to receive TGFβ signals. TGFβ signals are initiated through the type I and type II transmembrane serine/threonine kinase receptors. TGFβ binds and brings together the type I and type II receptors. In the resulting complex, the constitutively active TGFβ type II receptor phosphorylates and activates the type I receptor, which then transduces the signal to downstream components.36 Surprisingly, our studies revealed that PD-1 did not induce increases in TGFβ production or in the abundance of TGFβ receptors. Instead, PD-1 inhibited Cdk2-mediated phosphorylation of Smad3 and resulted in an enhanced Smad3 transactivation in a TGFβ-independent manner. Thus, PD-1 synergizes with TGFβ-mediated signaling at a level distal to the TGFβ receptor and regulates TGFβ-specific transcriptional events by directly regulating the function of Smad3.
Our findings provided molecular and biochemical explanation for the reduced threshold of TGFβ-mediated conversion of Treg cells when PD-1 signals are present and for the lack of iTreg cell conversion after adoptive transfer of wild-type, naïve T cells into PD-L1−/−PD-L2−/− Rag2 recipients, which developed massive lymphoproliferation.35 Our studies indicated that direct increase of Smad3 transactivation due to inhibition of Cdk-mediated phosphorylation of Smad3 is a central mechanism by which PD-1 can synergize with TGFβ to facilitate suppression of T effector cells via an intrinsic mechanism. In parallel, by synergizing with TGFβ signaling on naïve T cells, PD-1 promotes the differentiation of Treg cells, thereby facilitating suppression of T effector cells via an extrinsic mechanism (Fig. 2). These results indicate how PD-1 maintains T cell tolerance and immune quiescence via both T cell-intrinsic and -extrinsic mechanisms and provides a mechanistic understanding for the central role of PD-1 as a regulator of T cell tolerance and immune homeostasis in vivo.
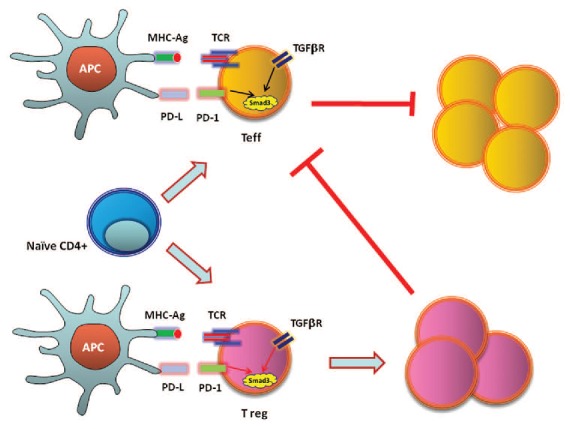
Figure 2. PD-1 inhibits T effector cells via cell intrinsic and extrinsic mechanisms by enhancing transactivation of Smad3.
PD-1 Promotes Ubiquitin-Dependent Degradation of CDC25A
A striking finding of our studies was that PD-1 abrogated Cdc25A both by suppressing mRNA and by promoting ubiquitin-dependent proteasomal degradation.27 Studies in normal epithelial cells and cancer cells lines have shown that Cdc25A expression is controlled by multilayered mechanisms. The transcription of the Cdc25A gene is positively regulated in an E2F-dependent manner and negatively regulated in a TGFβ/Smad-dependent manner.37,38 The abundance of Cdc25A protein is also regulated by ubiquitin-dependent proteasomal degradation. Two types of ubiquitin ligase (E3) complexes mediate ubiquitination of Cdc25A: the anaphase-promoting complex (APC) and the SCF complex.39,40 The APCcdh1 complex mediates degradation of Cdc25A from the exit of mitosis through the G1 phase of the cell cycle. Independently, the SCFβ-TrCP complex plays a critical role in Cdc25A degradation during proliferation and also in response to DNA damage. APC recognizes specific KEN box sequences of Cdc25A protein and induces phosphorylation-independent, ubiquitin-dependent degradation of Cdc25A. In contrast, phosphorylation of Cdc25A on serine 76 or serine 75 is a prerequisite for subsequent phosphorylation of serine 82, which is critical for β-TrCP binding.41-43 The checkpoint kinase Chk1 can phosphorylate serine 75, 76 and 123.41,44-46 The MAP kinase p38 can phosphorylate serine 75 and 123,47 while Smad3 has been reported to regulate phosphorylation of Cdc25A on serine 79 and serine 82, although the precise mechanism remains unclear.48
Very little is known about how Cdc25A is regulated by physiologic mechanisms involved in T cell responses. A previous study performed in T cells indicated that IL-3 or IL-7 withdrawal activates p38 MAPK resulting in phosphorylation of Cdc25A on serine 75 and serine 123, triggering its degradation.49 In that study, ubiquitin-targeted degradation of Cdc25A was considered the primary mechanism by which the abundance of the protein was controlled, but the effects of cytokine withdrawal on Cdc25A transcription were not addressed. Our recent findings revealed that in primary human CD4+ T cells, Cdc25A is regulated by both transcriptional and post-translational mechanisms.27 We determined that transcription of Cdc25A is upregulated during T cell activation via TCR/CD3 and CD28 and that PD-1 inhibits this event. Furthermore, PD-1 significantly increases ubiquitin-mediated degradation of Cdc25A, because inhibition of this pathway by the proteasome inhibitor MG132 resulted in comparable Cdc25A protein expression in T cells activated in the presence as in the absence of PD-1 signals. Thus, Cdc25A degradation is also an active mechanism via which PD-1 mediates cell cycle arrest. Further studies will be required to identify the specific ubiquitin ligase involved in ubiquitin-dependent proteasomal degradation of Cdc25A in response to PD-1. It is also important to determine whether PD-1 has a broader role in regulating ubiquitination and degradation of other signaling molecules involved in T cell activation by affecting the expression and function of ubiquitin ligases with established roles in the induction of T cell anergy and maintenance of immune homeostasis, including Cbl, Itch and Grail.50
In conclusion, our studies provided evidence that PD-1 targets the cell cycle machinery by inhibiting transcription of Skp2 and by activating Smad3. These findings also open new avenues to investigate the consequences of PD-1-mediated signals on T cell reprogramming by regulating cell cycle-independent functions of Cdk2 and Rb, which will have significant implications on the fate and function of T cells.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22135
References
- 1.Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol. 2002;2:11–9. doi: 10.1038/nri701. [DOI] [PubMed] [Google Scholar]
- 2.Li L, Boussiotis VA. Physiologic regulation of central and peripheral T cell tolerance: lessons for therapeutic applications. J Mol Med (Berl) 2006;84:887–99. doi: 10.1007/s00109-006-0098-5. [DOI] [PubMed] [Google Scholar]
- 3.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–66. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 7.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 8.Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–46. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 10.Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186:2772–9. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
- 11.Keir ME, Freeman GJ, Sharpe AH. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J Immunol. 2007;179:5064–70. doi: 10.4049/jimmunol.179.8.5064. [DOI] [PubMed] [Google Scholar]
- 12.Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. 2005;6:280–6. doi: 10.1038/ni1165. [DOI] [PubMed] [Google Scholar]
- 13.Reynoso ED, Elpek KG, Francisco L, Bronson R, Bellemare-Pelletier A, Sharpe AH, et al. Intestinal tolerance is converted to autoimmune enteritis upon PD-1 ligand blockade. J Immunol. 2009;182:2102–12. doi: 10.4049/jimmunol.0802769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. 2005;54:307–14. doi: 10.1007/s00262-004-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Currie AJ, Prosser A, McDonnell A, Cleaver AL, Robinson BW, Freeman GJ, et al. Dual control of antitumor CD8 T cells through the programmed death-1/programmed death-ligand 1 pathway and immunosuppressive CD4 T cells: regulation and counterregulation. J Immunol. 2009;183:7898–908. doi: 10.4049/jimmunol.0901060. [DOI] [PubMed] [Google Scholar]
- 17.Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–37. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545–52. doi: 10.1182/blood-2009-03-206672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe T, Bertoletti A, Tanoto TA. PD-1/PD-L1 pathway and T-cell exhaustion in chronic hepatitis virus infection. J Viral Hepat. 2010;17:453–8. doi: 10.1111/j.1365-2893.2010.01313.x. [DOI] [PubMed] [Google Scholar]
- 21.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–92. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 23.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 24.Russo AA, Jeffrey PD, Patten AK, Massagué J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–31. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 25.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–83. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–91. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5:ra46. doi: 10.1126/scisignal.2002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larrea MD, Wander SA, Slingerland JM. p27 as Jekyll and Hyde: regulation of cell cycle and cell motility. Cell Cycle. 2009;8:3455–61. doi: 10.4161/cc.8.21.9789. [DOI] [PubMed] [Google Scholar]
- 29.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–69. doi: 10.1016/S0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 30.Harbour JW, Dean DC. Chromatin remodeling and Rb activity. Curr Opin Cell Biol. 2000;12:685–9. doi: 10.1016/S0955-0674(00)00152-6. [DOI] [PubMed] [Google Scholar]
- 31.Hua XH, Newport J. Identification of a preinitiation step in DNA replication that is independent of origin recognition complex and cdc6, but dependent on cdk2. J Cell Biol. 1998;140:271–81. doi: 10.1083/jcb.140.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 33.Wells AD. Cyclin-dependent kinases: molecular switches controlling anergy and potential therapeutic targets for tolerance. Semin Immunol. 2007;19:173–9. doi: 10.1016/j.smim.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–31. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 35.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massagué J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 37.Vigo E, Müller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, et al. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol. 1999;19:6379–95. doi: 10.1128/mcb.19.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iavarone A, Massagué J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature. 1997;387:417–22. doi: 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- 39.Donzelli M, Squatrito M, Ganoth D, Hershko A, Pagano M, Draetta GF. Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 2002;21:4875–84. doi: 10.1093/emboj/cdf491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003;4:671–7. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donzelli M, Busino L, Chiesa M, Ganoth D, Hershko A, Draetta GF. Hierarchical order of phosphorylation events commits Cdc25A to betaTrCP-dependent degradation. Cell Cycle. 2004;3:469–71. doi: 10.4161/cc.3.4.770. [DOI] [PubMed] [Google Scholar]
- 42.Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, et al. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- 43.Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, et al. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–74. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–7. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 45.Sørensen CS, Syljuåsen RG, Falck J, Schroeder T, Rönnstrand L, Khanna KK, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–58. doi: 10.1016/S1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 46.Hassepass I, Voit R, Hoffmann I. Phosphorylation at serine 75 is required for UV-mediated degradation of human Cdc25A phosphatase at the S-phase checkpoint. J Biol Chem. 2003;278:29824–9. doi: 10.1074/jbc.M302704200. [DOI] [PubMed] [Google Scholar]
- 47.Kittipatarin C, Li WQ, Bulavin DV, Durum SK, Khaled AR. Cell cycling through Cdc25A: transducer of cytokine proliferative signals. Cell Cycle. 2006;5:907–12. doi: 10.4161/cc.5.9.2693. [DOI] [PubMed] [Google Scholar]
- 48.Ray D, Terao Y, Nimbalkar D, Chu LH, Donzelli M, Tsutsui T, et al. Transforming growth factor beta facilitates beta-TrCP-mediated degradation of Cdc25A in a Smad3-dependent manner. Mol Cell Biol. 2005;25:3338–47. doi: 10.1128/MCB.25.8.3338-3347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khaled AR, Bulavin DV, Kittipatarin C, Li WQ, Alvarez M, Kim K, et al. Cytokine-driven cell cycling is mediated through Cdc25A. J Cell Biol. 2005;169:755–63. doi: 10.1083/jcb.200409099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Macián F, García-Cózar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/S0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]