

Abstract
Lasonolide A (LSA) is a natural product with high and selective cytotoxicity against mesenchymal cancer cells, including leukemia, melanomas and glioblastomas. Here, we reveal that LSA induces rapid and reversible premature chromosome condensation (PCC) associated with cell detachment, plasma membrane smoothening and actin reorganization. PCC is induced at all phases of the cell cycle in proliferative cells as well as in circulating human lymphocytes in G0. It is independent of Cdk1 signaling, associated with cyclin B downregulation and induced in cells at LSA concentrations that are three orders of magnitude lower than those required to block phosphatases 1 and 2A in vitro. At the epigenetic level, LSA-induced PCC is coupled with histone H3 and H1 hyperphosphorylation and deacetylation. Treatment with SAHA reduced LSA-induced PCC, implicating histone deacetylation as one of the PCC effector mechanisms. In addition, PCC is coupled with topoisomerase II (Top2) and Aurora A hyperphosphorylation and activation. Inhibition of Top2 or Aurora A partially blocked LSA-induced PCC. Our findings demonstrate the profound epigenetic alterations induced by LSA and the potential of LSA as a new cytogenetic tool. Based on the unique cellular effects of LSA, further studies are warranted to uncover the cellular target of lasonolide A (“TOL”).
Keywords: Histone H3 phosphorylation, histone H3 deacetylation, chromosome condensation, Maturation Promoting Factor, topoisomerase II, Aurora kinase
Introduction
Lasonolide A (LSA, Fig. 1) was originally isolated from the deep-water marine sponge Forcepia sp.1 LSA has also recently been synthesized.2-5 Its potential as novel anticancer agent stems from its low nanomolar growth inhibitory concentration among the NCI-60 human tumor cell lines and its tissue specificity.6 The cellular target of LSA is presently unknown. Recently, LSA has been shown to affect murine thymus lymphoma EL-4.Il-2 cell adhesion and to induce the detachment of human pancreatic cancer cells, as it activates protein kinase C (PKC), mitogen-activated protein kinases (MAPK) Erk1/2 and p38.6
Figure 1. Structure of Lasonolide A
During normal cell cycle progression, as cells enter mitosis, they detach and undergo extensive chromosome condensation to allow mitotic segregation. Topoisomerase II (Top2) is essential for decatenating daughter DNA molecules and for mitotic chromosome condensation and segregation.7-9 The phenotype of temperature-sensitive Top2 mutants in S. pombe includes extended chromosomes with defective condensation.9 Top2 depletion also induces mitotic catastrophe, failure of cell division and cell death.8 Aurora kinase is another key enzyme during mitosis. It phosphorylates condensins and is essential for their chromosome association and functions.10-12 Aurora kinase also promotes chromosome condensation by phosphorylating histone H3 at serines 10 and 28.12 Hence, epigenetic modifications are coordinated with chromosome condensation.13
Chromosome condensation outside mitosis is known as premature chromosome condensation (PCC). PCC is used for cytogenetic studies. It can be induced by (1) virus-mediated fusion between mitotic and interphase cells; (2) chemically mediated cell fusion, for example by polyethylene glycol (PEG); (3) chemical inducers, primarily phosphatase inhibitors such as okadaic acid, calyculin A and fostreicin.14-18 In the latter case, activation of the maturation/mitosis promoting factor (MPF) is a key mediator of PCC, as Cdk1 (p34cdc2) bound to cyclin B1 is activated by tyrosine dephosphorylation by cdc25c.19 Cdc25 itself is regulated by auto-phosphorylation and dephosphorylation by protein phosphatases PP1 or PP2A. PP1 and PP2A inhibitions lead to cdc25 activation, followed by activated MPF, which promotes premature mitotic entry (reviewed in ref.20). Because of their dependency on MPF (cyclin B1-cdk1), the phosphatase inhibitors preferentially induce PCC in G2-phase.
Here, we present the unusual morphological changes induced by LSA in human cells and focus on a previously unnoticed nuclear modification: the massive, rapid and reversible chromosome condensation induced by LSA at all phases of the cell cycle. We differentiate LSA from the known PCC inducer okadaic acid and identify key biochemical and epigenetic components of LSA-induced PCC.
Results
Lasonolide A induces rapid, extensive and reversible premature chromosome condensation
To observe the changes in cellular chromatin architecture after LSA treatment, we stained cellular DNA with propidium iodide (PI) in Burkitt lymphoma CA46 cells. Within 1 h treatment at low 25 nM concentrations, LSA modified PI staining and the overall nuclear shape (Fig. 2A). Nuclei became circular instead of the lobular shape of untreated cells, as chromatin and chromosome condensations were induced by LSA (Fig. 2A). At 100 nM LSA, 97.5% of the nuclei exhibited condensed chromatin or chromosomes (Fig. 2A) together with an overall rounded nuclear shape. Time-course experiments showed that chromosome condensation was induced within 30 min exposure (Fig. 2B), and that the effects of LSA on chromosome and chromatin condensation were reversible. As shown in Figure 2C, chromosome and chromatin condensations reserved within 2 h after LSA removal.

Figure 2. Lasonolide A induces rapid and reversible premature chromosome condensation at nanomolar concentrations. A-D. Rapid, extensive and reversible induction of premature chromosome condensation (PCC) by Lasonolide A (LSA) in Burkitt’s lymphoma CA46 cells. After LSA treatment, cells were cytospun and stained with propidium iodide (PI). Representative images are shown. (A) Concentration response for PCC induction. Cells were treated with increasing concentrations of LSA for 1 h. (B) Time-course of PCC induction. Cells were exposed to LSA (100 nM) for the indicated times. (C) Reversibility of LSA-induced PCC upon drug removal. After 1 h treatment with 100 nM LSA, cells were cultured in drug-free medium for 1, 2, 4 or 24 h. (D). LSA induced PCC in all phases of the cell cycle. Cells treated with 100 nM LSA for 1 h were subjected to FACS with PI staining (upper panel). Red dots represent the FACS location of four representative cells in different cell cycle phases. Middle: representative nuclear morphologies for isolated cells (left) at different cell cycle phases. Bottom: chromosome spread by hypotonic treatment. Representative morphologies of spread chromosomes at different cell cycle phases are shown. (E). Induction of chromatin condensation in non-replicating human primary lymphocytes treated with 200 nM LSA for 1 h. PI staining was used to stain nuclear DNA. The arrows indicate the nuclei with condensed chromatin, and the numbers and percentage indicate the ratio of positive lymphocytes with condensed chromatin.
Testing of other human cells showed that LSA induced chromosome condensation not only in CA46 cells, but also in breast, colon cancer and leukemia cells (Table 1). In general, suspension cell lines such as leukemia and lymphoma cell lines were more sensitive than attached epithelial cancer cell lines. In the attached cell lines, PCC was associated with cell detachment. As LSA was removed by centrifugation in drug-free medium, the cells reattached and their nuclear staining normalized. Taken together, our results reveal that LSA induces chromosome and chromatin condensation in a rapid, extensive and reversible way.
Table 1. Premature chromosome condensation (PCC) induced by Lasonolide A in different cell lines.
| Tumor | Cell Line | Efficiency |
|
|---|---|---|---|
| Untreated | Treated | ||
| Burkitt’s lymphoma |
CA46 |
3.7% |
97.9% |
| Burkitt’s lymphoma |
Ramos |
2.2% |
94.8% |
| Burkitt’s lymphoma |
Daudi |
2.2% |
36.7% |
| Promyelocytic leukemia |
HL60 |
3.3% |
86.7% |
| Breast cancer |
MDA-MD-231 |
2.4% |
13.2% |
| Breast cancer |
MCF-7 |
1.6% |
12% |
| Colon cancer |
HCT-116 |
2.2% |
13.1% |
| Colon cancer | HT-29 | 2.7% | 18% |
Cells were treated with 100 nM Lasonolide A for 1 h. PCC efficiency was determined as the percentage of cells with condensed chromosomes by immunofluorescence microscopy after nuclear staining with propidium iodide.
Lasonolide A induces premature chromosome condensation at all phases of the cell cycle
Next we asked whether LSA-induced chromatin condensation was dependent on a specific phase in the cell cycle. As 1-h LSA treatment produced no significant change in cell cycle (Fig. 2D, top panel), we determined the cell cycle phase of individual cells treated with LSA based on their PI content determined by FACS analysis. Figure 2D (middle panel) shows that cells condensed their chromosomes at all phases of the cell cycle in response to LSA.
LSA-induced chromosome condensation was further examined by chromosome spreading. Cells in different phases of the cell cycle showed different morphology, with univalent or bivalent chromosomes that were well-spread (Fig. 2D, bottom panel). To further establish that LSA induces chromosome condensation independently of the cell cycle, the effects of LSA were determined on quiescent human peripheral blood lymphocytes. LSA-treated lymphocytes (200 nM, 1 h) showed 35.5% of the cells with condensed chromatin (Fig. 2E). These results demonstrate that LSA is a potent inducer of premature chromosome condensation (PCC) in all phases of the cell cycle and is effective even in quiescent G0 cells.
Cellular morphological and structural changes induced by LSA in association with premature chromosome condensation
To further characterize the nuclear changes induced by LSA, we performed electron microscopy. Extensive morphological and structural changes were induced by LSA besides PCC induction. Transmission electron microscopy (Fig. 3A) confirmed that nuclei lost their lobulated contour (1). They also lost their condensed peripheral chromatin regions (3), while the overall chromatin was condensed with enlarged inter-chromosomal space (2). We also noted the appearance of an outer membrane structure around the nuclear envelope (4) whose significance is unclear. Scanning electron microscopy (Fig. 3B) showed marked smoothening of the plasma membrane (5), consistent with the transmission electron microscopy images (Fig. 3A).

Figure 3. Electron microscopy image of Lasonolide A-treated cells. CA46 cells were treated with 100 nM LSA for 1 h. (A). Representative images obtained by transmission electron microscopy (Hitachi H700 microscope). Examples of morphological changes are indicated on the images: 1, inhibition of nuclear lobulation; 2, chromatin condensation; 3, disappearance of condensed perinuclear chromatin; 4, appearance of an outer membrane surrounding the nuclear envelope; 5, smoothening of the plasma membrane. (B). Representative images obtained by scanning electron microscopy (Hitachi H700 microscope). Note the cell surface smoothening (5).
We also looked at the cellular skeleton networks. Microtubule fibers that cross the cell body and the tubulin network node in the center of cells were removed by LSA (Fig. S1A). LSA also re-arranged the actin network by inducing the disappearance of the outer actin stress fibers (Fig. S1B), while the α-tubulin and actin filaments were aggregated around the cell membrane. Together, these morphological studies demonstrate that LSA induces a broad range of cellular changes in association with PCC. In the rest of the study, we will focus on the pathways that are connected with the induction of PCC.
Lasonolide A is a more potent inducer of premature chromosome condensation than okadaic acid in cells, but a weaker phosphatase inhibitor in biochemical assays
Next, we compared LSA to the classical PCC inducer, okadaic acid (OA).21 Under conditions where 100 nM LSA induced PCC in 99.5% of CA46 cells, 1 μM OA induced PCC in only 31% of the cells (Fig. 4A). Therefore, LSA is a markedly more potent PCC inducer than OA.

Figure 4. Lasonolide A is a more potent and more efficient inducer of PCC than okadaic acid, but a weaker phosphatase inhibitor in vitro. (A) Comparison between LSA and okadaic acid (OA) with regard to their PCC induction potencies. CA46 cells were treated with 100 nM LSA or 1 µM OA for 1 h. Representative PI staining fluorescence images are shown and the ratio of cells with condensed chromosomes are indicated below the images. (B) Effects of LSA and OA on phosphatase type 2A (PP2A) activity in vitro. (C) Same for phosphatase type 1 (PP1).
As OA is a known phosphatase inhibitor,21 we tested the effects of LSA on phosphatases in vitro. Purified phosphatase type 1 (PP1) and phosphatase type 2A (PP2A) were incubated with LSA, and phosphatase activities were determined. As shown in Figure 4B and C, LSA was a relatively weak inhibitor of PP1 or PP2A in vitro. Its IC50s were 2.6 ± 1.1 μM for PP2A and 102.2 ± 30.9 μM for PP1, which is more than two orders of magnitude higher than OA (20.9 ± 5.0 nM for PP2A and 0.28 ± 0.05 μM for PP1). Taken together, these results demonstrate that LSA is a much weaker phosphatase inhibitor but a more potent PCC inducer than OA.
Lasonolide A induces premature chromosome condensation independently of MPF
During normal cell cycle progression, chromosome condensation is triggered as cells enter mitosis by the dephosphorylated (active) mitosis-promoting factor (MPF, i.e., Cdk1/cyclin B1 complex).22 To investigate whether LSA-induced PCC is mediated through MPF signaling, Cdk1 and cyclin B1 were measured in LSA-treated cells. Figure 5 shows no significant change of Cdk1 level. Unexpectedly, the level of cyclin B1 was reduced by LSA (Fig. 5A and B). Furthermore, inactivation of Cdk1/cyclin B1 pathway with Cdk-specific inhibitor roscovitin23 or flavopiridol24 did not influence LSA-induced PCC (Fig. 5C). These data demonstrate that LSA-induced PCC is independently of Cdk1/cyclin B1 activation.

Figure 5. Lasonolide A-induced PCC is independent of cyclin B1/Cdk1. A. Concentration-dependent reduction of cyclin B1. CA46 cells were treated for 1 h with the indicated LSA concentrations. Protein levels of cyclin B1 and Cdk1 were detected by western blotting. Representative western blots are shown. (B) Time-dependent reduction of cyclin B1. Cells were treated with 100 nM LSA for 1, 2, 4, 8 and 24 h. Protein levels of cyclin B1 and Cdk1 were detected by western blotting. Representative western blots are shown. (C) Cyclin B1/Cdk1 inactivation does not interfere with LSA-induced PCC. Cells were pretreated with the Cdk inhibitors, roscovitine (15 µM) or flavopiridol (0.5 µM) for 1 h. Cells were then treated with 100 nM LSA for 1 h. Nuclear DNA was stained with PI and cells were examined by confocal microscopy. Representative images show that neither roscovitine nor flavopiridol affects LSA-induced PCC.
Lasonolide A induces histone hyperphosphorylation and H3 deacetylation
Because histones are modified during mitotic entry and chromatin condensation,13 we examined histone phosphorylation in LSA-treated cells. Figure 6 shows rapid histone H3 hyperphosphorylation at Ser28, Ser10 and Thr3 in response to LSA (Fig. 6A‒D). Phosphorylations at Ser28 or Thr3 reversed to basal level 1 h after LSA removal, while phosphorylations at Ser10 was more persistent (Fig. 6B and C). Histones H125 and H2B (at Ser14)26 were also hyperphosphorylated in LSA-treated cells. On the other hand, LSA did not induce γ-H2AX (histone H2AX phosphorylation at Ser139) (Fig. 6E). These data demonstrate that LSA induces rapid histone hyperphosphorylation in association with PCC.

Figure 6. Lasonolide A induces histone phosphorylation. (A) CA46 cells were treated with 100 nM LSA for the indicated times. Western blotting was used to detect phosphorylation levels for histone H3. (B) Reversibility of the LSA-induced H3 phosphorylations upon drug removal. After 1 h treatment with 100 nM LSA, cells were cultured in drug-free medium for the indicated times. Western blotting was used to detect phosphorylation levels. (C) Kinetics of histone H3 phosphorylations during and after LSA treatments. Data from western blots as shown in (A and B) were quantified. (D) Immunofluorescence images of H3 phosphorylation (Ser 28). Cells were treated with 100 nM LSA for 1 h. Representative images are shown. Green, p-H3-Ser28; red, nuclear DNA. (E) Induction of histones H1 and H2B phosphorylations and lack of H2AX phosphorylation in LSA-treated cells.
We also determined histone H3 acetylation in response to LSA. Figure 7 shows that LSA induces histone H3 deacetylation at Arg9 (Fig. 7A and B). Suberoylanilide hydroxamic acid (SAHA), a potent histone deacetylases (HDAC) inhibitor used to treat cutaneous T-cell lymphoma,27 was used to study further LSA-induced histone deacetylation. SAHA enhanced histone H3 acetylation in the absence of LSA, but LSA reduced SAHA-induced H3 hyperacetylation (Fig. 7B and C). Under conditions where LSA induced PCC in about 97.2% of the cells (100 nM, 1 h) by itself, SAHA reduced LSA-induced PCC to 61.3% (Fig. 7D and E). Taken together, these data demonstrate that histone H3 deacetylation is required for chromosome condensation by LSA.
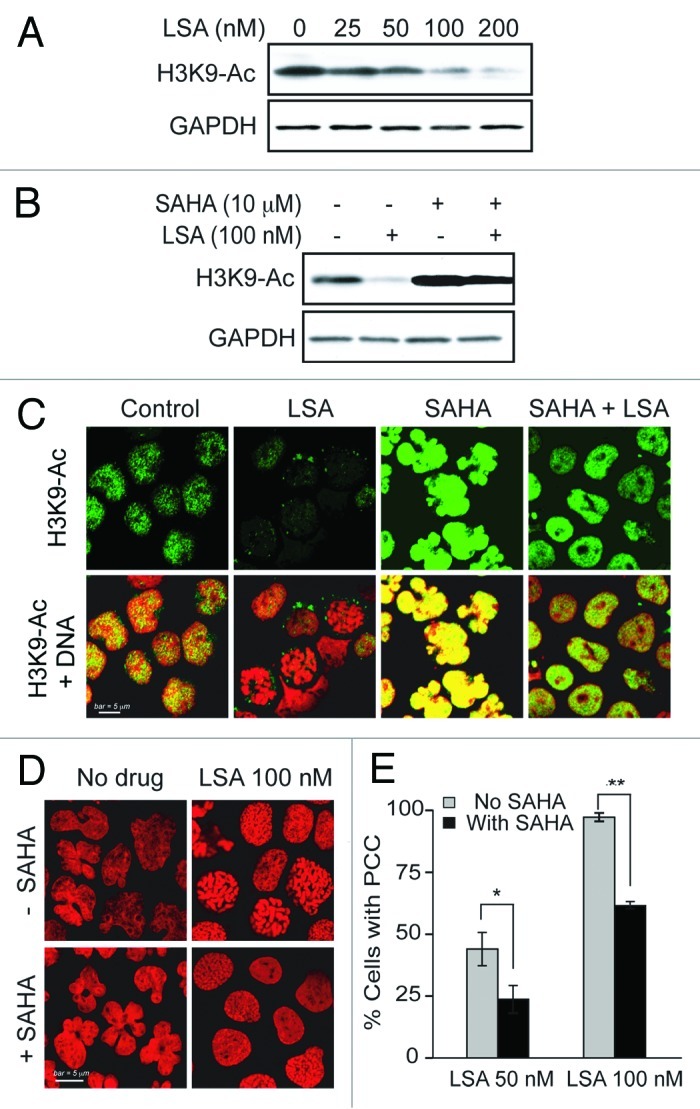
Figure 7. Histone H3 deacetylation is involved in Lasonolide A-induced PCC. (A) Reduction of histone H3 acetylation (at Lysine 9, H3K9-Ac) by LSA. CA46 cells were treated with the indicated LSA concentrations for 1 h. Western blotting was used to measure H3 acetylation level at Lysine 9. (B) Cells were treated with 100 nM LSA for 1 h in the absence or presence of 4 h pretreatment of 10 µM suberoylanilide hydroxamic acid (SAHA). Representative western blots are shown. (C) Immunofluorescence detection for H3 acetylation at Lysine 9. Representative images show reduction of H3 acetylation by LSA and antagonistic effects of SAHA. (D) Attenuation of LSA-induced PCC by enhanced acetylation. Cells were pre-treated with 10 µM SAHA for 4 h and then treated together with LSA for 1 h. Nuclear DNA was stained with PI and examined by confocal microscopy. Representative images are shown. (E) The ratio of cells with condensed chromosomes were quantified and plotted in histograms based on three independent experiments. Standard t-test were used for statistical analyses, *p < 0.05; **p < 0.01.
Lasonolide A induces hyperphosphorylation and activation of Topoisomerase II
One of the key functions of Top2 is to decatenate DNA loops for chromosome condensation and segregation in mitosis.7-9 Moreover, Top2α bears the MPM-2 phospho-epitope.28 To investigate whether Top2 is involved in the induction of PCC by LSA, we measured the overall Ser/Thr phosphorylation patterns by using MPM-2 antibodies, which recognize phospho-antigens of Top2α and other proteins that are phosphorylated specifically in mitosis.28,29 MPM-2 signals with the molecular size of Top2 (170–180 kDa) were markedly enhanced within minutes of LSA treatment (Fig. 8A‒C). MPM-2 induction was detectable within 15 min after LSA addition and was 16-fold above control at 1 h (Fig. 8A and B, left panel). The LSA-induced Top2 hyperphosphorylation reversed within minutes after removal of LSA (Fig. 8A and B, right panel). These results indicate that LSA induces rapid and reversible Top2α hyperphosphorylation.

Figure 8. Lasonolide A induces Top2 hyperphosphorylation and activation. (A‒C) Rapid and reversible induction of Top2 phosphorylation by LSA using the MPM-2 antibody. (A) Left: CA46 cells were treated with 100 nM LSA for the indicated times. Right: Reversibility of the LSA-induced hyperphosphorylation on the MPM-2 epitope upon drug removal. After 1 h treatment with 100 nM LSA, cells were cultured in drug-free medium for the indicated times. Western blotting was used to detect phosphorylation levels with a MPM-2 antibody. (B) Kinetics of LSA-induced hyperphosphorylation using the MPM-2 antibody. Data from western blots as shown in (A) were quantified. (C) Representative images of MPM-2 induction in LSA-treated cells (100 nM LSA, 1 h). (D and E) Activation of Top2 in LSA-treated cells. Cells were treated with LSA (100 nM) for 1 h. Nuclear extracts (0.5 μg) from untreated or LSA-treated cells were incubated with 1 μg kinetoplast DNA (kDNA) at 37°C to detect the decatenation activity of Top2. kDNA was incubated with nuclear lysates (0.5 μg) for the indicated times (D). Nuclear lysates (μg) diluted serially were incubated with kDNA for 30 min (E). DNA products were separated on agarose gels containing ethidium bromide. LM, XhoI-cut linear kDNA marker; DM, Top2-decatenated DNA marker. (F and G) Involvement of Top2 in LSA-induced PCC. Cells were pretreated with the Top2 inhibitors ICRF 187 [50 μg/mL (F) or etoposide VP-16, 100 μM (G)] for 15 min and then exposed together with LSA for an additional hour. Nuclei were stained with PI and representative confocal images are shown.
To investigate the modulation of Top2 activity by LSA, the decatenation activity of Top2 was determined.30 Nuclear extracts were prepared from LSA-treated CA46 cells and incubated with kinetoplast DNA (kDNA). Decatenation was enhanced in the samples from LSA-treated cells (Fig. 8D and E), demonstrating Top2 activation by LSA. To further study the involvement of Top2, the effect of the Top2 inhibitors ICRF 187 and etoposide (VP-16)30 were determined on LSA-induced PCC. The nuclear shape change and chromosome condensation could still be observed in cells co-treated with Top2 inhibitors. However, the pattern of chromosome condensation was significantly changed (Fig. 8F and G). Instead of the well-defined and organized pattern of chromosome condensation induced by LSA (Figs. 1, 3, 8F and G), PI staining of nuclei co-treated with LSA and Top2 inhibitors showed only partial condensation (Fig. 8F and G). Taken together, these data demonstrate that LSA induces Top2 hyperphosphorylation and activation and suggest that Top2 is a determinant of LSA-induced PCC.
Aurora A is activated with the induction of PCC by Lasonolide A
Aurora kinase is a key player in mitosis.10-12 To investigate the involvement of Aurora A in LSA-induced chromosome condensation, we used specific antibodies to detect the phosphorylation of Aurora A at Thr288 (activation site of Aurora A). Aurora A was quickly activated by Lasonolide A (within 1 h; Fig. 9A and B), without change in total Aurora A protein levels. This activation coincided with the hyperphosphorylation of histone H3 at serines 10 and 28, which are known sites for Aurora A-mediated phosphorylation (Fig. 6A). The activation of Aurora A was reversible upon LSA removal (Fig. 9C). To detect the functional effect of Aurora kinases on the LSA-induced PCC, the Aurora A inhibitor ZM447439 and the Aurora B inhibitor Hesperin were used to pretreat cells before LSA treatment. Both inhibitors effectively blocked LSA-induced chromosome condensation (Fig. 9D), indicating that the Aurora kinases are involved in LSA-induced PCC.

Figure 9. Aurora A is activated and involved in PCC induction by Lasonolide A. (A and B) Induction of Aurora A phosphorylation at Threonine 288 by LSA. (A) CA46 cells were treated with the indicated concentrations of LSA for 1 h. Cell lysates from cells treated with nocodazole (20 h) are shown as positive control. Western blotting was used to measure Aurora A phosphorylation. (B) Rapid induction of Aurora A phosphorylation by LSA. Western blotting was used to detect Aurora A phosphorylation levels at the indicated times. (C) Reversibility of Aurora A phosphorylation upon LSA removal. LSA was removed after 1 h treatment and cells were cultured in drug-free medium for 1, 2, 4 h. (D) Inhibition of Aurora A attenuated LSA-induced PCC. Cells were pretreated with 2 μM ZM447439 (Aurora A inhibitor) or 100 nM hesperidin (Aurora B inhibitor) for 30 min before adding 100 nM LSA for an additional hour. PI was used to stain nuclear DNA. Representative images are shown.
Discussion
The present study reveals the potency of LSA as PCC inducer. Notably, LSA-induced PCC is rapid, independent of the classical MPF (cyclin-Cdk) pathway and reversible upon LSA removal. PCC makes chromosomes condense outside of mitosis. Because normal mitosis is brief, and therefore only detectable in a small fraction of dividing cells, drug-induced PCC is broadly used to study chromosome architecture and loci in cytogenetics. The classical PCC-inducing drugs are phosphatase inhibitors, including okadaic acid, calyculin A and fostreicin.14-16 Calyculin A can induce PCC at 50 nM and achieve 10–20% PCC index after 30 min exposure in both suspension cells and adherent cells.16-18 Okadaic acid is active at 100 nM selectively in unattached suspension cell types.15,31 By contrast, here we show that LSA induces PCC efficiently in a broad range of cancer cells and in normal unstimulated human peripheral blood lymphocytes (Fig. 2E), under conditions where okadaic acid is inactive. In the case of okadaic acid and calyculin A, PCC induction can only be achieved in normal lymphocytes when the cells are induced to replicate with phytohaemagglutinin (PHA) stimulation32 or additional adenosine triphosphate (ATP) and Cdk1/cyclin B kinase.33 Thus, from a practical standpoint, LSA could provide a rapid, simple and efficient way to induce PCC in quiescent human peripheral blood lymphocytes and can be used in biodosimetry. More broadly, our findings reveal the potential use of LSA as a new tool for cytogenetic research.
The molecular effectors driving LSA-induced PCC remain to be fully elucidated. LSA-induced PCC is linked to epigenetic modifications including histone hyperphosphorylation and deacetylation. Histone phosphorylation during mitosis is critical for proper chromosome condensation, compaction and chromosome segregation, and post-translational modifications at serines 10 and 28 of histone H3 serve as markers of mitotic chromosome condensation.34-36 Histone hyperphosphorylation reflects the massive, rapid and yet reversible chromatin re-organization induced by LSA. Histone H3 phosphorylations at serines 10 and 28 can be produced by multiple protein kinases,12,37 among which Aurora kinase B is the most prevalent for chromosome condensation. Moreover, the histone H3 threonine 3 kinase, Haspin is phosphorylated by Aurora B during mitosis.38 Therefore, it is plausible that activation of Aurora kinases plays key role in LSA-induced PCC and histone hyperphosphorylation. Parallel to histone hyperphosphorylation, LSA reduced histone acetylation during PCC (Fig. 7). Hyperphosphorylation and deacetylation might be coordinated during chromosome condensation, as histone H3 phosphorylation at serine 10 can block acetylation at lysine 9.39 Accordingly, the clinical histone deacetylase (HDAC) inhibitor, SAHA has been shown to induce chromatin decondensation.40 Our finding that SAHA reduced LSA-induced PCC while enhancing the acetylation of H3 suggests that histone deacetylation is important for chromosome condensation.
Consistent with the fact that LSA activates normal mitotic condensation pathways, we found that LSA activated Top2 and Aurora A kinase while inducing PCC. Moreover, the fact that both Top2 and Aurora kinase inhibitors reduced LSA-induced PCC supports the functional implication of both Top2α and Aurora kinase in LSA-induced PCC. Top2 is essential for mitotic chromosome organization, condensation and segregation.7-9,17,18,30,41 Top2α can be linked to Aurora B, as Top2α is one of the substrates of Aurora B, and Top2α-depletion reduces Aurora kinase B activity.42,43 Unexpectedly, we found no evidence that MPF is required for LSA-induced PCC. On the contrary, our experiments show that cyclin B is downregulated, and the potent cdk inhibitor, roscovitin, is inactive against LSA-induced PCC. These results suggest that LSA induces PCC differently from the classical PCC inducers, okadaic acid and calyculin A, by activating Aurora kinases and Top2α independently of cdk’s.
LSA also induces the hyperphosphorylation of a broad range of cytoplasmic proteins and activates several protein kinases including PKC, MAP kinase (p38 and ERK1/2) and AKT,6 which may account for the cytoplasmic and membranes effects of LSA (see Fig. 3). Together with LSA-induced activation of Aurora kinases and Top2α (see Figs. 8 and 9), these findings demonstrate that LSA activates multiple signal transduction pathways that control the plasma membrane, cytoskeleton, signal transduction kinases6 and chromosome condensation. It is therefore likely that LSA targets an essential cellular node that regulates both chromatin and the cytoskeleton (Fig. 10A). Because of the selectivity of LSA [stereospecificity,5 rapid and reversible effects (see Figs. 2, 6 and 9)], it is plausible that LSA has a selective cellular target, which we propose to refer to as target(s) of LSA (“TOL”).

Figure 10. Summary of Lasonolide A-induced cellular effects and proposed target of Lasonolide A (TOL) associated with premature chromosome condensation (See Discussion for details). (A) Summary of LSA-induced PCC-associated cellular events. (B) Hypothetical protein phosphatase(s) as TOL, regulating PCC, cell rounding and attachment and, ultimately, cell death upon prolonged exposure to LSA.
Because of the extensive hyperphosphorylations induced by LSA and its chromosome condensing effects that overlap with the phosphatase inhibitor okadaic acid and calyculin A, we favor the possibility that “TOL” might be a phosphatase(s) that dephosphorylate(s) key cellular effectors (Fig. 10B). Blocking “TOL” would lead to the hyperphosphorylation and activation of multiple downstream cell signal pathways that are involved LSA-induced events, including PCC and cell rounding upon short drug exposure and cell death upon extended exposure (Fig. 10). Whether LSA acts as a direct phosphatase inhibitor or upstream from phosphatases by targeting signal transduction (for instance G-proteins) remains to be firmly established. If LSA acts on phosphatases, its activity might require whole phosphatase complex(es), since LSA is less inhibitory on purified PP2A and PP1 than OA (see Fig. 4). LSA may therefore act, like many other natural products, as an interfacial inhibitor by stalling a phosphatase(s) complex, possibly by binding at the interface of its constitutive functional subunits.44,45
The discovery of “TOL” will be critical not only for the rational use of LSA as a cytogenetic research tool, but also possibly as a novel antitumor agent. If LSA turns out to target a key phosphatase network, it would become a potentially valuable anticancer drug, as targeting protein phosphatases is presently a rational, yet challenging task.46,47
Materials and Methods
Drugs and chemicals
Lasonolide A was obtained by total chemical synthesis as reported.4,48 The purity of the compound was more than 99%. Okadaic acid (495604) was purchased from Calbiochem Chemical Co. Roscovitine (sc-24002) and Flavopiridol (sc-202157) were from Santa Cruz Biotech. Inc. ICRF 187 (D1446), etoposide (VP 16, E1383), nocodazole (M-1404) were from Sigma Chemical Co. Suberoylanilide hydroxamic acid (SAHA) was from chemical and biology core facility (NIDDK, NIH). Lasonolide A was prepared at 5 mM in DMSO. Okadaic acid sodium salt (OA) was dissolved at 100 mM in distilled water and then filtrated with 0.22 μm filter. Roscovitine and flavopiridol were dissolved at 20 mM in DMSO. SAHA was dissolved at 10 mM in DMSO. Etoposide (VP-16) was dissolved at 100 mM in DMSO. Drug aliquots were stored as stock solutions at –20°C. All drugs were diluted to desired concentrations in full medium immediately prior to each experiment. The final DMSO concentrations did not exceed 0.1%.
Cell culture
Burkitt’s lymphoma CA46 cells were obtained from the Developmental Therapeutics Program (DTP, National Cancer Institute) and cultured in RPMI 1640 medium (Life Inc. 11875) supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 100 μg/mL of streptomycin, 100 units/mL of penicillin and 2 mM L-glutamine in a humidified atmosphere (5% CO2) at 37°C. Human peripheral lymphocytes were obtained from the Blood Bank at the NIH and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum.
Electron microscopy
For transmission electron microscopy (TEM), cells were harvested, pelleted and fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 2 h at room temperature. After rinsing with cacodylate buffer, samples were post-fixed in 1% osmium tetroxide for 1 h. Then the cells were stained with 0.5% uranyl acetate, dehydrated in graded ethanol and propylene oxide and infiltrated in equal volume of epoxy resin and propylene oxide overnight. Samples were embedded in epoxy resin and cured in 55°C oven for 48 h. Cured blocks were thin-sectioned and stained in uranyl acetate and lead citrate before examination by electron microscopy (Hitachi H700 microscope). For scanning electron microscopy (SEM), cells were fixed by using 4% formaldehyde and 2% glutaraldehyde in 0.1 M cacodylate buffer and then filtered. The filters with the attached cells were washed using sodium cacodylate buffer (0.1 M). Samples were post-fixed in 1% OsO4 (osmium tetroxide in cacodylate buffer) for 1 h, washed again by using sodium cacodylate buffer and dehydrated. Filters were mounted onto 25-mm aluminum stubs and coated. Samples were imaged in a Hitachi S-3000N VPSEM at low voltage (4 kV) to obtain optimum surface information. All TEM and SEM analyses were performed by Electron Microscopy Facility, NCI, NIH.
Cell cycle analysis
Cells were harvested and washed twice with phosphate buffered saline (PBS). Pellets were suspended in 200 µl PBS and fixed with 1 ml ice-cold 70% ethanol. After overnight storage at -20°C, cells were centrifuged and pellets washed twice in PBS. To determine DNA content, 500 µl staining solution containing 10 µg/ml propidium iodide (Sigma, 81845) and 100 μg/mL RNase A (Sigma, R6513) in PBS were added to the pellet. Cells were analyzed with a FACScan flow cytometer (BD Biosciences) with CellQuest software (BD Biosciences).
Chromosome morphology
After drug treatment, CA46 cells were cytospun (800 rpm, 8 min) to glass slides and were fixed for 20 min with 4% paraformaldehyde in phosphate buffered saline (PBS, pH 7.4). After two washes with PBS and incubation for 20 min in 70% ethanol, followed by washing with PBS, the cells were stained with 20 μg/mL propidium iodide with 100 μg/mL RNase A for 15 min in the dark. Finally, slides were washed with PBS three times and mounted with Vectashield anti-fade mounting media (Vector Laboratories, Inc., H-1000). Images were taken using a Nikon Eclipse TE-300 confocal microscope.
Chromosome spread assay
Cells were incubated in 1mL hypotonic solution (10 mM Tris‐HCl pH 7.4, 40 mM glycerol, 20 mM NaCl, mM CaCl2, 0.5 mM MgCl2) at 37°C for 15 min. Cells were cytospun at 1,800 rpm for 8 min and fixed with 4% paraformaldehyde for 20 min. Procedure for chromosome morphology was as above.
In vitro phosphatase assay
Phosphatase activity in vitro was determined by measuring the generation of free PO4 from the phosphopeptide RRA(pT)VA, using the molybdate-malachite green-phosphate complex assay (Promega, V2460). The phosphatase assay was performed in a PP2A- or PP1-specific reaction buffer (PP2A: 50 mM imidazole, pH 7.2, 0.2 mM EGTA, 0.02% 2-mercaptoethanol, 0.1 mg/mL of bovine serum albumin; PP1: 50 mM TRIS-HCl, pH 7.0, 0.1 mM Na2EDTA, 5 mM dithiothreitol, 0.01% Brij 35) using 100 μM phosphopeptide substrate and 0.03 Units of PP2A (Millipore, 14–111) or PP1 (Biolabs, P0754). LSA or OA were co-incubated with PP1 and PP2A for 60 min at room temperature. Then molybdate dye was added, and the amount of free phosphate generated was determined from the optical density at 600 nm (VERSAmax, Molecular Device) using a standard curve for free phosphate. Phosphatase activity was defined as picomoles of free PO4 generated per minute per microgram of protein. The phosphatase activity without drug was defined as 100%.
Topoisomerase II activity assay
The effect of LSA on the Top2 activity in vitro was measured by the ATP-dependent decatenation of kinetoplast DNA (kDNA). The kDNA decatenation assay was performed according to the manufacturer’s recommendations: Topo II assay kit (Topogen, 1001–1). Briefly, LSA-treated CA46 cells were harvested and swollen in cold TEMP buffer (10 mM TRIS-HCl, pH 7.5, 1 mM EDTA, 4 mM MgCl2, 0.5 mM PMSF) on ice for 10 min. Nuclear pellets were spun down at 1,500 x g for 10 min at 4°C, resuspended in 30 μl TMP buffer (TEMP buffer without MgCl2), mixed with 30 μl 1 M NaCl, vortexed, and left on ice for 60 min. After centrifugation at 15,000 x g for 15 min, the supernatant containing Top2 activity was aliquoted, and protein concentration determined. The reaction buffer consisting of 50 mmol/L TRIS-HCl, pH 7.7, 120 mmol/L KCl, 10 mmol/L MgCl2, 1 mmol/L ATP, 0.5 mmol/L DTT, 0.5 mmol/L EDTA and 30 µg/mL bovine serum albumin was mixed with 0.1 µg kDNA and 0.5 μg cell lysate from LSA-treated cells in a total volume of 15 µL. After incubation at 37°C for 30 min, reactions were terminated by adding 10% SDS (1 µL). DNA samples were subjected to electrophoresis in a 1% agarose gel in 1x TAE buffer at 4 V/cm for 2 h.
Western blots
Protein levels were determined by western blotting with indicated specific primary antibodies: cyclin B1 (Santa Cruz Biotechnology, sc245), Cdk1 (Santa Cruz Biotechnology, sc-54), MPM-2 (Millipore, 05–368), phospho-H3 (Ser28) (Cell Signaling, 9713), phospho-H3 (Ser10) (Cell Signaling, 9710), phospho-H1(Millipore, 05–1324), phosphorylated-Aurora A (Thr288) (Cell Signaling, 3079), GAPDH (Cell signaling, 2118), phosphorylated-H2AX (γ-H2AX, Millipore, 05–636), acetylated-H3 (H3K9, Millipore,06–942), phospho-H3 (Thr3) (Millipore, 07–424) and Histone H3 (Millipore, 06–755), phosphorylated H2B-Ser14 (Santa Cruz Biotechnology, SC-3167), Aurora A (BD Bioscience, 2914). Shown were the representative data from repeated experiments.
Immunofluorescence assays
CA46 cells were cytospun to slides at 800 rpm for 8 min. Immunofluorescence assays were processed as previously.49 Briefly, the slides were fixed for 20 min with 4% paraformaldehyde in PBS (pH 7.4), and washed twice with PBS. After incubation for 20 min with 70% ethanol and washing with PBS, the cells were incubated in blocking buffer (8% bovine serum albumin in PBS) for 1 h before incubation for 2 h with primary antibodies. Slides were incubated for an additional 1 h with the Alex488-conjugated secondary antibody (Alexa Fluor® 488 donkey anti-goat IgG, Molecular Probes, Invitrogen, A-11055). After three washes in PBS, cells were stained with 0.5 μg/mL propidium iodide with 100 μg/mL RNase A (Sigma) for 15 min in the dark. Finally, slides were washed three times with PBS and mounted with Vectashield anti-fade mounting media (Vector Laboratories, Inc., H-1000). Images were taken using a Nikon Eclipse TE-300 confocal microscope.
Statistical analyses
All the data are represented as mean values ± SD. The significance of differences between means was assessed by Student’s t-test, with p < 0.05 and p < 0.01 considered statistically significant.
Supplementary Material
Acknowledgments
Our studies are supported by the Center for Cancer Research, National Cancer Institute Intramural Program. We wish to thank Ernest Hamel, Toxicology and Pharmacology Branch, Developmental Therapeutics Program, National Cancer Institute, Frederick, MD for bringing LSA to our attention and providing initial drug samples. We also wish to thank Kiyung S. Lee, Laboratory of Metabolism, CCR-NCI, Bethesda for discussions and for suggesting or providing the Aurora and Plk inhibitors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interests were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22768
Reference
- 1.Horton PA, Koehn FE, Longley RE, Mcconnell OJ. Lasonolide-a, a New Cytotoxic Macrolide from the Marine Sponge Forcepia Sp. J Am Chem Soc. 1994;116:6015–6. doi: 10.1021/ja00092a081. [DOI] [Google Scholar]
- 2.Dalgard JE, Rychnovsky SD. Enantioselective synthesis of the C18-C25 segment of lasonolide A by an oxonia-cope prins cascade. Org Lett. 2005;7:1589–91. doi: 10.1021/ol050270s. [DOI] [PubMed] [Google Scholar]
- 3.Yoshimura T, Yakushiji F, Kondo S, Wu X, Shindo M, Shishido K. Total synthesis of (+)-lasonolide A. Org Lett. 2006;8:475–8. doi: 10.1021/ol0527678. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh AK, Gong G. Enantioselective total synthesis of macrolide antitumor agent (-)-lasonolide A. Org Lett. 2007;9:1437–40. doi: 10.1021/ol0701013. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh AK, Ren GB. Stereoselective synthesis of both tetrahydropyran rings of the antitumor macrolide, (-)-lasonolide A. J Org Chem. 2012;77:2559–65. doi: 10.1021/jo202631e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isbrucker RA, Guzmán EA, Pitts TP, Wright AE. Early effects of lasonolide a on pancreatic cancer cells. J Pharmacol Exp Ther. 2009;331:733–9. doi: 10.1124/jpet.109.155531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuvier O, Hirano T. A role of topoisomerase II in linking DNA replication to chromosome condensation. J Cell Biol. 2003;160:645–55. doi: 10.1083/jcb.200209023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez RE, Lim CU, Cole K, Bianchini CH, Schools GP, Davis BE, et al. Effects of conditional depletion of topoisomerase II on cell cycle progression in mammalian cells. Cell Cycle. 2011;10:3505–14. doi: 10.4161/cc.10.20.17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uemura T, Ohkura H, Adachi Y, Morino K, Shiozaki K, Yanagida M. DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell. 1987;50:917–25. doi: 10.1016/0092-8674(87)90518-6. [DOI] [PubMed] [Google Scholar]
- 10.Lipp JJ, Hirota T, Poser I, Peters JM. Aurora B controls the association of condensin I but not condensin II with mitotic chromosomes. J Cell Sci. 2007;120:1245–55. doi: 10.1242/jcs.03425. [DOI] [PubMed] [Google Scholar]
- 11.Hagstrom KA, Holmes VF, Cozzarelli NR, Meyer BJ. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev. 2002;16:729–42. doi: 10.1101/gad.968302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol. 2001;152:669–82. doi: 10.1083/jcb.152.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J Cell Sci. 1998;111:3497–506. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- 14.Guo XW, Th’ng JP, Swank RA, Anderson HJ, Tudan C, Bradbury EM, et al. Chromosome condensation induced by fostriecin does not require p34cdc2 kinase activity and histone H1 hyperphosphorylation, but is associated with enhanced histone H2A and H3 phosphorylation. EMBO J. 1995;14:976–85. doi: 10.1002/j.1460-2075.1995.tb07078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh S, Schroeter D, Paweletz N. Okadaic acid overrides the S-phase check point and accelerates progression of G2-phase to induce premature mitosis in HeLa cells. Exp Cell Res. 1996;227:165–9. doi: 10.1006/excr.1996.0262. [DOI] [PubMed] [Google Scholar]
- 16.Tosuji H, Mabuchi I, Fusetani N, Nakazawa T. Calyculin A induces contractile ring-like apparatus formation and condensation of chromosomes in unfertilized sea urchin eggs. Proc Natl Acad Sci U S A. 1992;89:10613–7. doi: 10.1073/pnas.89.22.10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miura T, Blakely WF. Optimization of calyculin A-induced premature chromosome condensation assay for chromosome aberration studies. Cytometry A. 2011;79:1016–22. doi: 10.1002/cyto.a.21154. [DOI] [PubMed] [Google Scholar]
- 18.Prasanna PG, Blakely WF. Premature chromosome condensation in human resting peripheral blood lymphocytes for chromosome aberration analysis using specific whole-chromosome DNA hybridization probes. Methods Mol Biol. 2005;291:49–57. doi: 10.1385/1-59259-840-4:049. [DOI] [PubMed] [Google Scholar]
- 19.Moreno S, Hayles J, Nurse P. Regulation of p34cdc2 protein kinase during mitosis. Cell. 1989;58:361–72. doi: 10.1016/0092-8674(89)90850-7. [DOI] [PubMed] [Google Scholar]
- 20.Gotoh E, Durante M. Chromosome condensation outside of mitosis: mechanisms and new tools. J Cell Physiol. 2006;209:297–304. doi: 10.1002/jcp.20720. [DOI] [PubMed] [Google Scholar]
- 21.Yamashita K, Yasuda H, Pines J, Yasumoto K, Nishitani H, Ohtsubo M, et al. Okadaic acid, a potent inhibitor of type 1 and type 2A protein phosphatases, activates cdc2/H1 kinase and transiently induces a premature mitosis-like state in BHK21 cells. EMBO J. 1990;9:4331–8. doi: 10.1002/j.1460-2075.1990.tb07882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreno S, Hayles J, Nurse P. Regulation of the cell cycle timing of mitosis. J Cell Sci Suppl. 1989;12:1–8. doi: 10.1242/jcs.1989.supplement_12.1. [DOI] [PubMed] [Google Scholar]
- 23.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–36. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 24.Senderowicz AM. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest New Drugs. 1999;17:313–20. doi: 10.1023/A:1006353008903. [DOI] [PubMed] [Google Scholar]
- 25.Halmer L, Gruss C. Effects of cell cycle dependent histone H1 phosphorylation on chromatin structure and chromatin replication. Nucleic Acids Res. 1996;24:1420–7. doi: 10.1093/nar/24.8.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de la Barre AE, Angelov D, Molla A, Dimitrov S. The N-terminus of histone H2B, but not that of histone H3 or its phosphorylation, is essential for chromosome condensation. EMBO J. 2001;20:6383–93. doi: 10.1093/emboj/20.22.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escargueil AE, Larsen AK. Mitosis-specific MPM-2 phosphorylation of DNA topoisomerase IIalpha is regulated directly by protein phosphatase 2A. Biochem J. 2007;403:235–42. doi: 10.1042/BJ20061460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taagepera S, Dent P, Her JH, Sturgill TW, Gorbsky GJ. The MPM-2 antibody inhibits mitogen-activated protein kinase activity by binding to an epitope containing phosphothreonine-183. Mol Biol Cell. 1994;5:1243–51. doi: 10.1091/mbc.5.11.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–33. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balakrishnan S, Shirsath K, Bhat N, Anjaria K. Biodosimetry for high dose accidental exposures by drug induced premature chromosome condensation (PCC) assay. Mutat Res. 2010;699:11–6. doi: 10.1016/j.mrgentox.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Kanda R, Eguchi-Kasai K, Hayata I. Phosphatase inhibitors and premature chromosome condensation in human peripheral lymphocytes at different cell-cycle phases. Somat Cell Mol Genet. 1999;25:1–8. doi: 10.1023/B:SCAM.0000007135.12486.e3. [DOI] [PubMed] [Google Scholar]
- 33.Prasanna PG, Blakely WF. Premature chromosome condensation in human resting peripheral blood lymphocytes for chromosome aberration analysis using specific whole-chromosome DNA hybridization probes. Methods Mol Biol. 2005;291:49–57. doi: 10.1385/1-59259-840-4:049. [DOI] [PubMed] [Google Scholar]
- 34.Xu D, Bai J, Duan Q, Costa M, Dai W. Covalent modifications of histones during mitosis and meiosis. Cell Cycle. 2009;8:3688–94. doi: 10.4161/cc.8.22.9908. [DOI] [PubMed] [Google Scholar]
- 35.Wang F, Dai J, Daum JR, Niedzialkowska E, Banerjee B, Stukenberg PT, et al. Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science. 2010;330:231–5. doi: 10.1126/science.1189435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polioudaki H, Markaki Y, Kourmouli N, Dialynas G, Theodoropoulos PA, Singh PB, et al. Mitotic phosphorylation of histone H3 at threonine 3. FEBS Lett. 2004;560:39–44. doi: 10.1016/S0014-5793(04)00060-2. [DOI] [PubMed] [Google Scholar]
- 37.Baek SH. When signaling kinases meet histones and histone modifiers in the nucleus. Mol Cell. 2011;42:274–84. doi: 10.1016/j.molcel.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 38.Wang F, Ulyanova NP, van der Waal MS, Patnaik D, Lens SM, Higgins JM. A positive feedback loop involving Haspin and Aurora B promotes CPC accumulation at centromeres in mitosis. Curr Biol. 2011;21:1061–9. doi: 10.1016/j.cub.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edmondson DG, Davie JK, Zhou J, Mirnikjoo B, Tatchell K, Dent SY. Site-specific loss of acetylation upon phosphorylation of histone H3. J Biol Chem. 2002;277:29496–502. doi: 10.1074/jbc.M200651200. [DOI] [PubMed] [Google Scholar]
- 40.Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem. 2004;92:223–37. doi: 10.1002/jcb.20045. [DOI] [PubMed] [Google Scholar]
- 41.Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer. 2009;9:327–37. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrison C, Henzing AJ, Jensen ON, Osheroff N, Dodson H, Kandels-Lewis SE, et al. Proteomic analysis of human metaphase chromosomes reveals topoisomerase II alpha as an Aurora B substrate. Nucleic Acids Res. 2002;30:5318–27. doi: 10.1093/nar/gkf665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coelho PA, Queiroz-Machado J, Carmo AM, Moutinho-Pereira S, Maiato H, Sunkel CE. Dual role of topoisomerase II in centromere resolution and aurora B activity. PLoS Biol. 2008;6:e207. doi: 10.1371/journal.pbio.0060207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pommier Y, Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat Rev Drug Discov. 2012;11:25–36. doi: 10.1038/nrd3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bollen M, Gerlich DW, Lesage B. Mitotic phosphatases: from entry guards to exit guides. Trends Cell Biol. 2009;19:531–41. doi: 10.1016/j.tcb.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Aceto N, Bentires-Alj M. Targeting protein-tyrosine phosphatases in breast cancer. Oncotarget. 2012;3:514–5. doi: 10.18632/oncotarget.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Labbé DP, Hardy S, Tremblay ML. Protein tyrosine phosphatases in cancer: friends and foes! Prog Mol Biol Transl Sci. 2012;106:253–306. doi: 10.1016/B978-0-12-396456-4.00009-2. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh AK, Gong G. Total synthesis of potent antitumor agent (-)-lasonolide A: a cycloaddition-based strategy. Chem Asian J. 2008;3:1811–23. doi: 10.1002/asia.200800164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011;39:3607–20. doi: 10.1093/nar/gkq1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.