

Abstract
Neutrophil gelatinase-associated lipocalin (NGAL, a.k.a Lnc2) is a member of the lipocalin family and has diverse roles. NGAL can stabilize matrix metalloproteinase-9 from autodegradation. NGAL is considered as a siderocalin that is important in the transport of iron. NGAL expression has also been associated with certain neoplasias and is implicated in the metastasis of breast cancer. In a previous study, we examined whether ectopic NGAL expression would alter the sensitivity of breast epithelial, breast and colorectal cancer cells to the effects of the chemotherapeutic drug doxorubicin. While abundant NGAL expression was detected in all the cells infected with a retrovirus encoding NGAL, this expression did not alter the sensitivity of these cells to doxorubicin as compared with empty vector-transduced cells. We were also interested in determining the effects of ectopic NGAL expression on the sensitivity to small-molecule inhibitors targeting key signaling molecules. Ectopic NGAL expression increased the sensitivity of MCF-7 breast cancer cells to EGFR, Bcl-2 and calmodulin kinase inhibitors as well as the natural plant product berberine. Furthermore, when suboptimal concentrations of certain inhibitors were combined with doxorubicin, a reduction in the doxorubicin IC50 was frequently observed. An exception was observed when doxorubicin was combined with rapamycin, as doxorubicin suppressed the sensitivity of the NGAL-transduced MCF-7 cells to rapamycin when compared with the empty vector controls. In contrast, changes in the sensitivities of the NGAL-transduced HT-29 colorectal cancer cell line and the breast epithelial MCF-10A cell line were not detected compared with empty vector-transduced cells. Doxorubicin-resistant MCF-7/DoxR cells were examined in these experiments as a control drug-resistant line; it displayed increased sensitivity to EGFR and Bcl-2 inhibitors compared with empty vector transduced MCF-7 cells. These results indicate that NGAL expression can alter the sensitivity of certain cancer cells to small-molecule inhibitors, suggesting that patients whose tumors exhibit elevated NGAL expression or have become drug-resistant may display altered responses to certain small-molecule inhibitors.
Keywords: NGAL, Lcn2, lipocalins, siderocalins, targeted therapy, inhibitor sensitivity, EGFR, rapamycin, berberine, BCL-2, calmodulin kinase, breast cancer, colorectal cancer
Introduction
Identification of the signaling pathways that are critical for sensitivity to targeted therapy as well as conventional therapy is essential for improved cancer treatments. In fact, it has recently been demonstrated that in order for some targeted cancer therapy treatments to be effective, specific target genes need to be either mutated or overexpressed. Moreover, cells that are initially sensitive to targeted therapy often develop resistance. Certain types of cancers, namely melanoma,1-17 chronic myeloid leukemia (CML)18,19 and non-small cell lung cancer (NSCLC)20,21 have been intensively investigated for the mechanisms of sensitivity and resistance to small-molecule inhibitors. Thus, it is essential to understand why certain cancer patients are sensitive or develop resistance to various therapeutic approaches and whether the sensitivity results from intrinsic or extrinsic events.
A common phenomenon that occurs after treatment of cancer patients with chemotherapeutic drugs is drug resistance. The mechanisms behind these developments are many and include increased expression of drug transporters, amplification of critical survival genes, genetic mutations and deletions or increased activation of certain signal transduction pathways.22-26 Therefore, it is also important to understand how cancers become drug-resistant and whether or not their drug resistance can be reversed.
Over the past 35 y, many genes have been identified which can cause or contribute to the formation of cancer.27,28 These include two major classes of genes, the oncogenes29-33 and the tumor-suppressor genes such as retinoblastoma (RB),34-37 TP53,38-40 BRCA1,41,42 PTEN,43 TSC1 and TSC2.44-47 Many of these oncogenes and tumor suppressors are often critical regulators of cellular senescence.48-53 Moreover, microRNAs (miRNAs)54-60 and epigenetic modifications61-63 have been shown to play important roles in regulating cancer progression. Certain miRNAs may be induced by drugs such as metformin, which can control cancer growth.60 Some miRNAs may be regulated by epigenetic mechanism by tumor-suppressor genes such as BRCA1.63
In some cases, the genetic culprit involved in a particular cancer may be known [e.g., BCRABL in chronic myeloid leukemia,18,19 HER2 in certain forms of breast cancer,64-66 EGFR in certain lung cancers,25,27,28 BRAF in melanomas, thyroid cancers, non-small cell lung cancers and colorectal cancers (CRC)1-18,25,27,28]. However, in most cases, there are multiple genetic and epigenetic events occurring that can interact and result in a cancer cell capable of becoming metastatic and/or drug resistant. In addition, there are other important metabolic contributions by the tumor microenvironment that aid in the progression of the cancer cell as well as the development of sensitivity/resistance to various therapeutic approaches and the survival of cancer-initiating cells (CICs).67-69
One factor that may be important for cancer survival and metastasis is neutrophil gelatinase-associated lipocalin (NGAL). One of the genomic responses to common cancer treatments such as radiation and chemotherapy is the induction of NGAL expression.70-75 NGAL may act to stabilize MMP-9 and increase its ability to degrade the extracellular matrix, thereby promoting metastasis.
NGAL expression is regulated by the transcription factors NFκB, CEBP and others.76-79 Radiation and chemotherapy can induce reactive oxygen species (ROS) that result in NFκB activation80-83 and subsequent downstream NGAL transcription. In addition, the tumor microenvironment can alter intracellular NFκB activity.83 Chemo- and radiotherapy could result in the synthesis of NGAL in cancer cells, which may lead to the development of therapy-resistant cells. These cells can contribute to the reemergence and metastasis of the cancer, as increased NGAL expression may allow the cells to persist under conditions where therapy-sensitive cancer cells would not normally survive.
Cancer cells have increased demands for intracellular iron. NGAL is a member of the lipocalin family and, as such, is capable of serving as a siderocalin or molecule involved in the transport of iron and other molecules.84 Iron is essential for many key processes, including the rate-limiting step in DNA synthesis performed by ribonucleotide reductase.85 Iron (Fe2+) is also required for cells to progress through the cell cycle from G1 to S phase. Tumor cells have a high requirement for iron and express elevated levels of the transferrin receptor-1.86-89 Novel chelators of iron are being considered for cancer treatment.90 Iron chelators, such as Desferrioxamine (DFO), inhibit cellular iron transport and have been evaluated in various cancer clinical trials.91 Oxygen and iron concentrations may be altered in the tumor microenvironment due to drastic tumor growth.92-94 In order for a cancer cell to survive, invade and metastasize, it may have to have increased iron transport as well as elevated glycolysis.67-69,95,96 The role of iron transport in chemotherapeutic drug resistance of cancer cells is complex and may depend on the particular drug and cancer type investigated.97,98 Interestingly, some iron depletors have been shown to decrease resistance of certain cancer cells to chemotherapeutic drugs including doxorubicin.99,100
Increased levels of NGAL have been detected in the urine of patients with various types of cancer (i.e., brain, breast, colon, ovarian, pancreatic and prostate). Novel non-invasive urine-based tests could prove useful for the detection and/or prognosis of many cancer types.101-104 The role(s) of NGAL in chemotherapeutic drug resistance, invasion, cancer metastasis and sensitivity to targeted therapy have not been fully elucidated. Targeting NGAL could result in decreased cancer cell survival and tumor regression as well as improve the effectiveness of radiation and chemotherapy in cancer therapy. NGAL is considered by some scientists to possess characteristics of an oncogene. In some studies, NGAL has been shown to increase the mobility, invasion, metastasis and tumorigenesis of certain cancer cells (breast, CRC).105-108 Elevated expression of NGAL increases the invasiveness of certain cancer cell types, while inhibition of NGAL expression decreases their invasiveness and metastasis.72-75,105 Novel approaches to target MMP-9/NGAL are needed, as MMP-9 inhibitors have not performed well in clinical cancer trials,109 and NGAL has functions which are independent of MMP-9.
NGAL may exert many different effects that are important in invasion and metastasis. NGAL can stabilize MMP-9 at the cell surface,110-112 and this complex, in association with CD44, may promote the cleavage of E-Cadherin (E-Cad) into soluble(s) E-Cad thereby inducing epithelial-mesenchymal transition (EMT).112,113 Alternatively, NGAL may be important in the transport of inhibitors and natural products into cells or preventing their efflux from cells. In the following studies, we examined the effects of ectopic NGAL expression on the sensitivity of breast cancer and CRC to several small-molecule inhibitors targeting critical molecules in signal transduction pathways. Ectopic expression of NGAL increased the sensitivity of MCF-7 breast cancer cells to EGFR, CaM-K and Bcl-2 inhibitors as well as the natural plant product berberine. In contrast, ectopic expression of NGAL did not alter the sensitivity of the CRC line or the breast epithelial MCF-10A line to the various small-molecule inhibitors or natural products. We chose to examine the effects of ectopic NGAL expression on sensitivity to doxorubicin on two different types of cancers: breast cancer cells, which are generally sensitive to doxorubicin therapy, and CRC cells, which are considered resistant to doxorubicin therapy. Addition of suboptimal doses of some inhibitors lowered the IC50 of doxorubicin in MCF-7 breast cancer cells with the exception of rapamycin. In contrast, doxorubicin appeared to inhibit the effects of rapamycin on the normally sensitive MCF-7/NGAL but not MCF-7/pLXSN cells. These results could have clinical significance, as NGAL is often expressed at high levels in certain advanced cancer patients, and its expression is induced after chemo- and radiotherapy. NGAL expression could also alter the sensitivity of cancer and other patients to various small-molecule inhibitors and natural products such as berberine, which are used in traditional medicine.
Results
Effects of enforced NGAL expression on sensitivity to the EGFR inhibitor AG1478
MCF-10A, MCF-7 and HT-29 cells were infected with a retrovirus encoding NGAL or the empty retrovirus pLXSN. NGAL was detected in the supernatants from NGAL retrovirus-infected cells114,115 but not in the empty vector pLXSN virus-infected cells.115
We examined the effects of elevated NGAL expression on the sensitivity of all of the cell lines to the various inhibitors and doxorubicin by MTT assays. Graphs in Figure 1 represent the effects of varying concentrations of the EGFR inhibitor AG1478 (panels A, D and G) doxorubicin (panels B, E and H), and varying concentrations of doxorubicin with a constant concentration the EGFR inhibitor (panels C, F and I).

Figure 1. Sensitivity of NGAL- and pLXSN-infected cells and doxorubicin-resistant MCF-7/DoxR cells to the EGFR inhibitor AG1478, doxorubicin and the combination of doxorubicin and a constant dose of 250 nM AG1478. Cells were collected and seeded (2,000 cells/well) in 96-well plates. The following day, serial 2-fold dilutions of AG1478 (A, D and G), doxorubicin (B, E and H) or serial 2-fold dilutions of doxorubicin and a constant dose of 250 nM AG1478 (C, F and I) were added to the wells. Four days later, MTT assays were performed. (A–C) MCF-10A/pLXSN (solid squares), MCF-10A/NGAL (solid upright triangles), (D–F) MCF-7/pLXSN (solid squares), MCF-7/NGAL (solid upright triangles) and 25 nM doxorubicin-selected MCF-7/DoxR cells (solid downward triangles), (G–I) HT-29/pLXSN (solid squares), HT-29/NGAL (solid upward triangles). A hatched horizontal line is present at the 50% relative growth mark from which the IC50 can be calculated. A vertical arrow indicates the IC50. The statistical significance was determined by the unpaired t-test (***, p < 0.001). All the experiments in this figure were performed at the same time (set up on the same day). These experiments were repeated multiple times, and similar results were obtained.
Ectopic NGAL expression did not appear to alter the sensitivity of MCF-10A/NGAL cells to either the EGFR inhibitor AG1478 (panel A) or doxorubicin (panel B). Interestingly, a dose of the EGFR inhibitor at approximately the IC50 completely eliminated the growth of these cells, which are normally cultured in medium containing rEGF (panel C). These results demonstrate that the toxic effects of doxorubicin can be enhanced by the EGFR inhibitor in the breast epithelial MCF-10A line.
In contrast to the results observed with the breast epithelial MCF-10A/NGAL cells, ectopic NGAL expression did alter the sensitivity of MCF-7/NGAL cells to the EGFR inhibitor AG1478 (panel D) but not doxorubicin (panel E). In addition, the MCF-7/DoxR line was very sensitive to the EGFR inhibitor AG1478 (panel D) but was highly resistant to doxorubicin (panel E). Interestingly, a dose of the EGFR inhibitor reduced the concentration of doxorubicin required to reach the IC50 at least 10-fold in all the MCF-7 cells (panel F). The MCF-7/DoxR cells were included in these studies as they were derived directly from MCF-7 cells by selection in medium containing 25 nM doxorubicin for prolonged periods of time.114
NGAL expression did not significantly alter the sensitivity of HT29/NGAL cells to either the EGFR inhibitor AG1478 (panel G) or doxorubicin (panel H). In fact, the HT29 cells appeared to be very resistant to the effects of the EGFR inhibitor. A constant dose of 250 nM EGFR inhibitor did not produce any additive effects to the doxorubicin IC50 in HT29 cells (panel I), similar to what was seen in the other cell lines. The IC50s for the drug-treated cells are presented in Table 1.
Table 1. Effects of ectopic NGAL expression on the sensitivity of cells to doxorubicin, small-molecule inhibitors and the natural plant product berberine.1.
| Drug treatment | MCF-10A/pLXSN | MCF-10A/NGAL | MCF-7/pLXSN | MCF-7/NGAL | MCF-7/DoxR | HT-29/pLXSN | HT-29/NGAL |
|---|---|---|---|---|---|---|---|
| AG1478 (EGFR Inh) |
300 nM |
300 nM |
> 2,000 nM |
30 nM |
6 nM |
> 2,000 nM |
> 2,000 nM |
| ABT-737 (Bcl-2 Inh) |
400 nM |
300 nM |
1,000 nM |
1 nM |
1.5 nM |
600 nM |
600 nM |
| Rapamycin (mTORC1 Inh) |
0.2 nM |
0.2 nM |
0.15 nM |
0.2 nM |
> 100 nM |
> 100 nM |
> 100 nM |
| KN-93 (CaMK Inh) |
> 10,000 nM |
10,000 nM |
110 nM |
12 nM |
2,500 nM |
500 nM |
550 nM |
| Berberine |
> 2,000 nM |
> 2,000 nM |
2,000 nM |
12 nM |
> 2,000 nM |
> 2,000 nM |
2,000 nM |
| Doxorubicin |
30 nM |
25 nM |
80 nM |
60 nM |
2,000 nM |
40 nM |
40 nM |
| Dox + 250 nM AG1478 |
< 1nM |
< 1nM |
12 nM |
12 nM |
80 nM |
50 nM |
40 nM |
| Dox + 50 nM ABT-737 |
20 nM |
18 nM |
7 nM |
15 nM |
8 nM |
1.8 nM |
3 nM |
| Dox + 5 nM Rapa |
< 2nM |
< 2 nM |
1.5 nM |
70 nM |
2.5 nM |
28 nM |
28 nM |
| Dox + 250 nM KN-93 |
18 nM |
12 nM |
12 nM |
< 2 nM |
30 nM |
40 nM |
40 nM |
| Dox + 250 nM Ber | 28 nM | 20 nM | 70 nM | 10 nM | 8 nM | 50 nM | 50 nM |
1MTT analysis was performed with different unselected cancer lines and certain 25 nM doxorubicin resistant (DoxR). Determined by plating 2,000 cells/well in 96-well plates in phenol red-free RPMI , + 10% FBS. Serial 2-fold dilutions (n = 12 dilutions) of doxorubicin were dispensed into eight wells per each doxorubicin concentration after the first day. MTT analysis was performed after four additional days of incubation and results were normalized to untreated cells as described.114 All IC50s are estimated values derived from the graphs presented in Figures 1–5.
Effects of enforced NGAL expression on sensitivity to the Bcl-2 inhibitor ABT-737
The effects of varying concentrations of the Bcl-2 inhibitor ABT-737 (panels A, D and G), doxorubicin (panels B, E and H) or varying concentrations of doxorubicin and a constant concentration of ABT-737 (panels C, F and I) on growth in NGAL-expressing cells are presented in Figure 2. The results with the doxorubicin titrations are presented again to allow direct comparison with the results obtained with doxorubicin titrations combined with a constant dose of the Bcl-2 inhibitor.

Figure 2. Sensitivity of NGAL- and pLXSN-infected cells and doxorubicin-resistant MCF-7/DoxR cells to the Bcl-2 inhibitor ABT-737, doxorubicin and the combination of doxorubicin and a constant dose of 50 nM ABT-737. Cells were collected and seeded (2,000 cells/well) in 96-well plates. The following day, serial 2-fold dilutions of ABT-737 (A, D and G), doxorubicin (B, E and H) or serial 2-fold dilutions of doxorubicin and a constant dose of 50 nM ABT-737 (C, F and I) were added to the wells. Four days later, MTT assays were performed. (A–C) MCF-10A/pLXSN (solid squares), MCF-10A/NGAL (solid upright triangles), (D–F) MCF-7/pLXSN (solid squares), MCF-7/NGAL (solid upright triangles) and 25 nM doxorubicin-selected MCF-7/DoxR cells (solid downward triangles), (G–I) HT-29/pLXSN (solid squares), HT-29/NGAL (solid upward triangles). A hatched horizontal line is present at the 50% relative growth mark from which the IC50 can be calculated. A vertical arrow indicates the IC50. The statistical significance was determined by the unpaired t-test (***, p < 0.001). All the experiments in this figure were performed at the same time (set up on the same day). These experiments were repeated multiple times and similar results were obtained.
Overexpression of NGAL did not alter the IC50 of MCF-10A/NGAL cells to ABT-737; however, MCF-10A/NGAL cells were more sensitive to lower concentrations of the Bcl-2 inhibitor than MCF-10/pLXSN cells (panel A). The addition of the Bcl-2 inhibitor decreased the concentration of doxorubicin required to reach the IC50 in both MCF-10A/pLXSN and MCF-10A/NGAL by less than 2-fold (panel C).
In contrast to the results observed with the breast epithelial MCF-10A cells, NGAL expression did exhibit an effect on the sensitivity of MCF-7/NGAL cells to the Bcl-2 inhibitor ABT-737 (panel D) but not to doxorubicin only (panel E). MCF-7/pLXSN cells were at least 1,000-fold more resistant to the Bcl-2 inhibitor than either MCF-7/NGAL or MCF-7/DoxR cells. The MCF-7/DoxR line was also highly sensitive to the ABT-737 (panel D) but not to doxorubicin (panel E). Treatment with ABT-737 did significantly diminish the concentration of doxorubicin required to reach the IC50 in pLXSN- or NGAL-expressing cells (11- and 3.3-fold, respectively). Moreover, the IC50 for MCF-7/DoxR cells was considerably reduced (200-fold) when the Bcl-2 inhibitor was added (panel F).
In HT29 CRC cells, the effects of NGAL overexpression did not significantly alter the sensitivity of HT29/NGAL cells to either the Bcl-2 (panel G) or doxorubicin (panel H). However, a suboptimal dose of the Bcl-2 inhibitor did reduce the doxorubicin IC50 for both HT29/LXSN and HT29/NGAL cells greater than 30-fold (panel I).
Effects of enforced NGAL expression on sensitivity to the mTORC1 inhibitor rapamycin
Breast epithelial, breast cancer and CRC cell lines were exposed to varying concentrations of the mTORC1 inhibitor rapamycin (panels A, D and G) and varying concentrations of doxorubicin combined with a constant dose of rapamycin (panels C, F and I) to determine the effects of NGAL expression. Once again, the results with the doxorubicin titrations are presented (panels B, E and H) to allow for direct comparison of treated cells.
MCF-10A cells treated with rapamycin are presented in Figure 3 (panels A and C). Ectopic NGAL expression did not alter the IC50 of MCF-10A/NGAL cells to the mTORC1 inhibitor (panel A), although both MCF-10A/pLXSN and MCF-10A/NGAL cells were very sensitive to the mTORC1 inhibitor, with the IC50 for rapamycin at approximately 0.2 nM. Furthermore, no additive effects in sensitivity were seen with the addition of rapamycin when combined with doxorubicin (panel C).

Figure 3. Sensitivity of NGAL- and pLXSN-infected cells and doxorubicin-resistant MCF-7/DoxR cells to the mTORC1 inhibitor rapamycin, doxorubicin and the combination of doxorubicin and a constant dose of 5 nM rapamycin. Cells were collected and seeded (2,000 cells/well) in 96-well plates. The following day, serial 2-fold dilutions of rapamycin (A, D and G), doxorubicin (B, E and H) or serial 2-fold dilutions of doxorubicin and a constant dose of 5 nM rapamycin (C, F and I) were added to the wells. Four days later, MTT assays were performed. (A–C) MCF-10A/pLXSN (solid squares), MCF-10A/NGAL (solid upright triangles), (D–F) MCF-7/pLXSN (solid squares), MCF-7/NGAL (solid upright triangles) and 25 nM doxorubicin-selected MCF-7/DoxR cells (solid downward triangles), (G–I) HT-29/pLXSN (solid squares), HT-29/NGAL (solid upward triangles). A hatched horizontal line is present at the 50% relative growth mark from which the IC50 can be calculated. A vertical arrow indicates the IC50. The statistical significance was determined by the unpaired t-test (***, p < 0.001). All the experiments in this figure were performed at the same time (set up on the same day). These experiments were repeated multiple times and similar results were obtained.
Similar to MCF-10A cells, both MCF-7/pLXSN and MCF-7/NGAL cells were very sensitive to the mTORC1 inhibitor rapamycin (panel D). In contrast though, the doxorubicin-resistant MCF-7/DoxR cells were highly resistant to rapamycin. The combinatorial effects of different concentrations of doxorubicin and a constant dose of rapamycin were assayed, and as expected, the MCF-7/pLXSN cells were highly sensitive to the constant 5 nM dose of rapamycin in the presence of different concentrations of doxorubicin (panel F). Moreover, the dose of 5 nM rapamycin synergized with doxorubicin and lowered the doxorubicin concentration required to reach the IC50 greater than 10-fold in the doxorubicin-resistant MCF-7/DoxR cells. Interestingly, overexpression of NGAL appeared to prevent the effects of rapamycin when combined with different concentrations of doxorubicin in MCF-7/NGAL cells (panel F). These results suggest that doxorubicin neutralized the effects of rapamycin in MCF-7/NGAL cells.
Ectopic expression of NGAL did not alter the sensitivity of HT29/NGAL cells to either rapamycin (panel G) or doxorubicin (panel H). HT29 cells were exceedingly resistant to the mTORC1 inhibitor by itself as compared with the breast cell lines. A suboptimal dose of rapamycin was able to reduce the doxorubicin IC50 for HT29/LXSN and HT29/NGAL cells approximately 2-fold (panel I).
Effects of enforced NGAL expression on sensitivity to the CaM-K inhibitor KN-93
The CaM-K inhibitor KN-93 was also used to examine the potential effects NGAL overexpression may have on cell growth in response to chemotherapeutic drugs. Effects of KN-93 (panels A, D and G) and KN-93 in combination with varying concentrations of doxorubicin (panel C, F and I) were analyzed compared with the effects of doxorubicin alone (panels B, E and H).
Figure 4 (panels A, B and C) represents results with the breast epithelial MCF-10A cells. Ectopic NGAL expression appeared to slightly alter the sensitivity of MCF-10A cells to the CaM-K inhibitor, although a clear IC50 could not be established but was estimated to be close to 10,000 nM KN-93 in the MCF-10A/NGAL and greater than 10,000 nM in the MCF-10A/pLXSN cells (panel A). In general, both MCF-10A/NGAL and MCF-10A/LXSN cells were very resistant to the CaM-K inhibitor. These slight effects of KN-93 on MCF-10A/NGAL cells were similarly observed with the addition of varying concentrations of doxorubicin as the IC50s for both MCF-10A/pLXSN and MCF-10A/NGAL cells were marginally decreased (panel C).

Figure 4. Sensitivity of NGAL- and pLXSN-infected cells and doxorubicin-resistant MCF-7/DoxR cells to the CaM-K inhibitor KN-93, doxorubicin and the combination of doxorubicin and a constant dose of 250 nM KN-93. Cells were collected and seeded (2,000 cells/well) in 96-well plates. The following day, serial 2-fold dilutions of KN-93 (A, D and G), doxorubicin (B, E and H) or serial 2-fold dilutions of doxorubicin and a constant dose of 250 nM KN-93 (C, F and I) were added to the wells. Four days later, MTT assays were performed. (A–C) MCF-10A/pLXSN (solid squares), MCF-10A/NGAL (solid upright triangles), (D–F) MCF-7/pLXSN (solid squares), MCF-7/NGAL (solid upright triangles) and 25 nM doxorubicin-selected MCF-7/DoxR cells (solid downward triangles), (G–I) HT-29/pLXSN (solid squares), HT-29/NGAL (solid upward triangles). A hatched horizontal line is present at the 50% relative growth mark from which the IC50 can be calculated. A vertical arrow indicates the IC50. The statistical significance was determined by the unpaired t-test (***, p < 0.001). All the experiments in this figure were performed at the same time (set up on the same day). These experiments were repeated multiple times and similar results were obtained.
Both MCF-7/pLXSN and MCF-7/NGAL were very sensitive to the CaM-K inhibitor KN-93 and had IC50s of approximately 110 nM and 12 nM, respectively (Fig. 4, panel D). In contrast, the doxorubicin-resistant MCF-7/DoxR was more resistant to KN-93, and an IC50 of approximately 2,500 nM was observed. The effects of combining different concentrations of doxorubicin with a constant dose of KN-93 were assessed. The MCF-7/NGAL cells were highly sensitive to the constant dose of KN-93 in the presence of different concentrations of doxorubicin (panel F). Moreover, the dose of 250 nM KN-93 synergized with doxorubicin and lowered the doxorubicin IC50 in both MCF-7/pLXSN and the doxorubicin-resistant MCF-7/DoxR cells.
As seen in panel G of Figure 4, ectopic NGAL did not significantly alter the sensitivity of HT29/NGAL cells to the CaM-K inhibitor KN-93, and the HT29/pLXSN and HT29/NGAL cells had IC50s of approximately 500 nM and 600 nM, respectively to KN-93. Unlike the results observed with the MCF-7 cell lines, a suboptimal dose of the CaM-K inhibitor did not reduce the doxorubicin IC50 for HT29/LXSN and HT29/NGAL cells (panel I).
Effects of enforced NGAL expression on sensitivity to the natural plant product berberine
The natural plant derivative berberine was also examined in our cell lines, and the results are presented in Figure 5. Berberine affects the regulation of many genes, including suppressing NFκB, ERK and MMP-9 expression while also inducing 5′ adenosine monophosphate-activated protein kinase (AMPK) expression.

Figure 5. Sensitivity of NGAL- and pLXSN-infected cells and doxorubicin-resistant MCF-7/DoxR cells to the natural product berberine, doxorubicin and the combination of doxorubicin and a constant dose of 250 nM berberine. Cells were collected and seeded (2,000 cells/well) in 96-well plates. The following day, serial 2-fold dilutions of berberine (A, D and G), doxorubicin (B, E and H) or serial 2-fold dilutions of doxorubicin and a constant dose of 250 nM berberine (C, F and I) were added to the wells. Four days later, MTT assays were performed. (A–C) MCF-10A/pLXSN (solid squares), MCF-10A/NGAL (solid upright triangles), (D–F) MCF-7/pLXSN (solid squares), MCF-7/NGAL (solid upright triangles) and 25 nM doxorubicin-selected MCF-7/DoxR cells (solid downward triangles), (G–I) HT-29/pLXSN (solid squares), HT-29/NGAL (solid upward triangles). A hatched horizontal line is present at the 50% relative growth mark from which the IC50 can be calculated. A vertical arrow indicates the IC50. The statistical significance was determined by the unpaired t-test (***, p < 0.001). All the experiments in this figure were performed at the same time (set up on the same day). These experiments were repeated multiple times and similar results were obtained.
Ectopic NGAL expression did not exhibit any effect on the sensitivity of MCF-10A cells to berberine over the concentrations examined (up to 2,000 nM), as the IC50s were not reached. Although the MCF-10A/NGAL cells did display more sensitivity to berberine at the highest concentrations used, they were not statistically different from the MCF-10A/pLXSN. Likewise, the addition of a constant dose (250 nM) of berberine did not appear to significantly lower the concentration of doxorubicin needed to reach the IC50 of doxorubicin alone in either MCF-10A/pLXSN or MCF-10A/NGAL cells (panel C).
MCF-7/pLXSN, MCF-7/NGAL and MCF-7/DoxR cells were also treated with berberine and are presented in Figure 5 (panels D, E and F). As seen in panel D, MCF-7/NGAL cells were very sensitive to berberine and had an IC50 of approximately 10 nM. In contrast, MCF-7/pLXSN and the doxorubicin-resistant MCF-7/DoxR were exceedingly more resistant to berberine and exhibited IC50s of approximately 2,000 nM and greater than 2,000 nM, respectively. The combinatorial effects of different concentrations of doxorubicin and a constant dose of berberine were determined and yielded some unexpected results. Although MCF-7/pLXSN and MCF-7/NGAL cells did not demonstrate a significant difference in their doxorubicin IC50s, the sensitivities of MCF-7/NGAL cells to a constant dose of berberine (250 nM) in the presence of different concentrations of doxorubicin were increased approximately 10-fold, while addition of a constant dose of berberine did not increase the sensitivity of MCF-7/pLXSN to doxorubicin (panel F). Moreover, berberine synergized with doxorubicin to dramatically lower the doxorubicin IC50 in the doxorubicin-resistant MCF-7/DoxR cells.
Similar to the results observed with the MCF-10A cell line, ectopic NGAL expression had a mild effect on the sensitivity of HT29 cells to berberine (Fig. 5, panels G, H and I). HT29/pLXSN cells were slightly more resistant to berberine than HT29/NGAL cells, with both having an IC50 of greater than 2,000 nM (panel G). A suboptimal dose of berberine did not produce a measurable reduction in the doxorubicin IC50s for HT29/LXSN and HT29/NGAL cells (panel I).
Inhibitor and doxorubicin IC50s for various NGAL and pLXSN retrovirally infected cell lines as well as the combination effects of doxorubicin with a constant dose of each inhibitor are presented in Table 1. The fold difference in sensitivities of the MCF-7/pLXSN, MCF-7/NGAL and MCF-7/DoxR cell lines to the various inhibitors are presented in Table 2. In this table, the values obtained with MCF-7/NGAL and MCF-7/DoxR are normalized to the empty vector-infected, doxorubicin-sensitive control line, MCF-7/pLXSN.
Table 2. Fold differences in sensitivity to small-molecule inhibitors, berberine and doxorubicin in MCF-7/pLXSN, MCF-7/NGAL and MCF-7/DoxR cells.1.
| Drug treatment | MCF-7/NGAL | MCF-7/DoxR |
|---|---|---|
| AG1478 (EGFR Inh) |
67 X↓ |
333 X↓ |
| ABT-737 (Bcl-2 Inh) |
1,000 X↓ |
667 X↓ |
| Rapamycin (mTORC1) |
1.3 X ↑ |
667 X↑ |
| KN-93 (CaM-K) |
9.2 X↓ |
23 X↑ |
| Berberine (AMPK, others) |
166 X↓ |
— |
| Doxorubicin (topoisomerase, others) |
1.3 X↓ |
25 X↑ |
| Dox + 250 nM AG1458 |
— |
6.7 X↑ |
| Dox + 50 nM ABT-737 |
2 X↑ |
1.1 X↑ |
| Dox + 5 nM Rapa |
47 X↑ |
1.7 X↑ |
| Dox + 250 nM KN-93 |
6 X↑ |
2.5 X↑ |
| Dox + 250 nM Berberine | 7 X↓ | 8.8 X↓ |
1Fold change in IC50s were normalized to the IC50s detected in MCF-7/pLXSN cells. ↓ indicates a decrease in IC50 in comparison to MCF-7/pLXSN control cells. ↑ indicates an increase in the IC50 compared with MCF-7/pLXSN control cells. Estimated data values obtained from drug titrations derived from graphs in Figures 1–5 and listed in Table 1.
Discussion
These studies were undertaken to determine whether increased NGAL expression altered the sensitivity of breast and CRC cells to certain small-molecule signal transduction inhibitors and the natural plant product berberine. The effects of the inhibitors and natural product were also examined in combination with the chemotherapeutic drug doxorubicin in cancer cell types that are normally sensitive to doxorubicin (breast) and cancer cell types which are normally resistant to doxorubicin (CRC) as well as immortalized breast epithelial cells (MCF-10A), which are not malignant. NGAL may have roles in iron transport, which may be associated with chemosensitivity in certain cancers. Some studies have shown that iron chelators will reduce chemotherapeutic drug resistance.99,100 NGAL expression has been associated with a poor prognosis in breast cancer. Importantly, our studies document that elevated NGAL expression altered the sensitivity to certain small-molecule inhibitors and the natural plant product berberine, especially in the MCF-7 breast cancer cell line. However, elevated NGAL expression had fewer effects on the immortalized breast epithelial MCF-10A cells and essentially no effects on the HT-29 CRC cell line.
In MCF-7/NGAL cells, it is likely that NGAL is altering either the uptake or efflux of some of the inhibitors and berberine into the cells. This is a novel, but logical function for NGAL. The expression of NGAL increased the toxicity of the EGFR, Bcl-2, CaM-K and berberine. In contrast, elevated NGAL expression antagonized the effects of the mTORC1 inhibitor rapamycin when combined with doxorubicin in MCF-7/NGAL cells. These are important results, as some cancer patients are treated with EGFR, Bcl-2 and mTORC1 inhibitors in combination with chemotherapeutic drugs. Interestingly, our results also identified that the sensitivity of the CRC line HT-29 to doxorubicin could be increased when co-treated with the Bcl-2 inhibitor ABT-737.
We included in our studies the doxorubicin-resistant derivative of the MCF-7 cell line, MCF-7/DoxR, to serve as a control for determining the effects of doxorubicin and the small-molecule inhibitors. Drug resistant breast cancer cells often express higher levels of proteins involved in drug transport. The doxorubicin-resistance of MCF-7/DoxR cells was eliminated by EGFR, Bcl-2, CaM-K and mTORC1 inhibitors and the natural plant product berberine.
The doxorubicin-resistant MCF-7/DoxR cell line was also infected with the retrovirus encoding NGAL;115 however, these cells were not more resistant or sensitive to doxorubicin than the pLXSN empty vector control MCF-7/DoxR cells (data not presented). NGAL expression was not detected at higher levels in drug-resistant MCF-7/DoxR/pLXSN cells than MCF-7/pLXSN.115 Moreover, the doxorubicin-resistant MCF-7/DoxR cells do not normally express NGAL; however, upon treatment with doxorubicin, increased NGAL protein has been detected.114 Elevated NGAL expression does not appear, by itself, to alter the sensitivity to doxorubicin in the cells examined, and the doxorubicin-resistance of MCF-7/DoxR cells could result from aberrant regulation of various signaling pathways or drug transporters in these cells.
The MCF-7/DoxR cells were very sensitive to the EGFR inhibitor AG1478 and the Bcl-2 inhibitor ABT-737. It is likely that the drug resistance present in MCF-7/DoxR is hypersensitive to EGFR pathway activation and Bcl-2 survival signaling. In contrast, MCF-7/DoxR cells were resistant to mTORC1 inhibition by itself, but the doxorubicin-resistance present in MCF-7/DoxR was eliminated when rapamycin was combined with doxorubicin in our assays. On the other hand, while MCF-7/NGAL cells were highly sensitive to rapamycin, the effects of rapamycin were eliminated when combined with doxorubicin. These results suggest that NGAL has different effects when certain small-molecule inhibitors are combined with chemotherapeutic drugs and could have clinical implications, as many cancer patients express elevated levels of NGAL.71-76,116
The doxorubicin-resistance of MCF-7/DoxR cells was eliminated when the cells were treated with the CaM-K inhibitor KN-93 and doxorubicin, as they exhibited a similar IC50 for doxorubicin as the drug-sensitive MCF-7/pLXSN cells. These results suggest a key role for the CaM-K pathway in the doxorubicin resistance of MCF-7/DoxR cells. The MCF-7/NGAL cells were also very sensitive to the CaM-K inhibitor. The CaM-K pathway is important in the regulation of the Ras/Raf/MEK/ERK pathway,117 the regulation of NFκB118 and AMPK.119 Also, the CaM-K pathway is activated after doxorubicin treatment and after other treatments which induce reactive oxygen species (ROS).120-122 A diagram illustrating where some of these drugs interact in signaling pathways is presented in Figure 6.
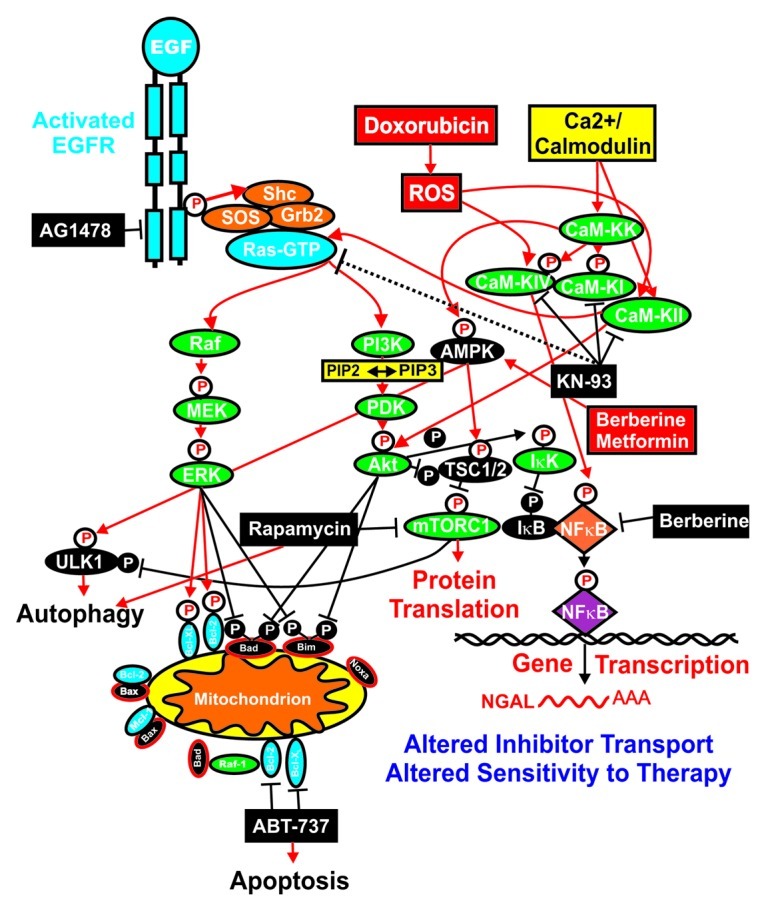
Figure 6. Overview of targeting of key pathways and effects on NGAL expression. Activation of many signaling cascades can occur after activation of the EGFR receptor or by treatment with doxorubicin, which induces reactive oxygen species (ROS) and the CaM-K cascade. The sites where certain signal transduction inhibitors (black rectangles) and the natural product berberine and the diabetes drug metformin are indicated. Berberine may activate AMPK (red rectangle) as well as inhibit NFκB (black rectangle). NGAL expression may alter the transport and efflux of certain small-molecule inhibitors, which, in some cases, may be deleterious to the cancer cell.
NGAL expression has been associated with a poor prognosis in breast and other cancers.80,116 While elevated expression of NGAL does not alter the IC50 for the chemotherapeutic drug doxorubicin in the cell lines examined, it did alter their sensitivity to certain small-molecule inhibitors in the breast cancer cell line MCF-7. Furthermore, elevated expression of NGAL did not appear to alter the responses of either the CRC line HT-29 or the immortalized epithelial line MCF-10A to doxorubicin.
Targeting the EGFR/Ras/PI3K/Akt/mTORC1 pathway is a key anticancer and anti-aging approach. There are many sites which are frequently mutated or aberrantly expressed and are being targeted in this pathway, from upstream receptors (e.g., EGFR, HER2)25,27,28,123,124 to downstream signaling proteins such as Ras,29,125,126 PI3K,26,30,127-138 PTEN,43,139,140 Akt,25-28 TSC1/TSC2,44-47,144 Rheb,25-28,145 mTOR25-28,146-165 and p70S6K.25-28,166 This pathway also plays important roles in cell growth and is often aberrantly regulated in diabetes and obesity.151 Many anti-diabetes drugs such as metformin and the traditional drug berebrine interfere with components that feed into this pathway or are regulated by this cascade,e and there is cross-talk between these pathways. It is interesting that rapamycin and berberine both had effects on NGAL-expressing cells. NGAL suppressed the effects that rapamycin had on lowering the doxorubicin IC50 in MCF-7/NGAL cells, while MCF-7/NGAL cells were very sensitive to the effects of berberine.
mTORC1 phosphorylates unc-51-like kinase 1 (ULK1), which results in the suppression of autophagy. The mTORC1 inhibitor rapamycin prevents phosphorylation of ULK1, and autophagy can occur.167-169 It appears that targeting key molecules which control energy may be approaches to control cancer and aging. Drugs such as rapamycin target mTORC1, and metformin is an indirect inhibitor of mTORC1. Metformin induces AMPK which activates TSC1 and subsequently suppresses mTORC1 activity.170-186 Berberine may elicit similar effects.187-189 Metformin may also induce the phosphorylation and inactivation of Raptor,190 a key regulatory component in the mTORC1 complex which is critical for the translation of many weak “oncogenic” mRNAs important in proliferation. Interestingly, diabetics treated with metformin have lower incidences of cancer and also do not exhibit as much aging191 and mice treated with metformin do not exhibit as much aging as untreated mice.183 Moreover, metformin may be able to prevent the survival of certain CICs.
Enhanced glycolysis (Warburg effect) is critical for CIC survival.192-197 Metformin disrupts the glycolytic metabotype and alters the ATM-mediated DNA damage response resulting in the acceleration of stress-induced senescence. Metformin in the presence of suppressed mTOR signaling slows down aging and alters the cellular senescence processes. Hence, metformin can alter the ability of cells to become immortalized into CICs and slows down cellular aging. By reducing the levels of DNA damage signaling, metformin has genoprotective effects198 and anticancer/tumor suppressor effects.199-202 Berberine may have similar effects on AMPK. Interestingly, MCF-7 cells that overexpressed NGAL were hypersensitive to berberine.
Materials and Methods
Cell lines and growth factors
Breast cancer cells lines (MCF-7) and the CRC line HT-29 were obtained from the ATCC. Cells were maintained in a humidified 5% CO2 incubator at 37°C with RPMI-1640 (RPMI) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals). This complete RPMI media is abbreviated cRPMI. The immortalized breast epithelial MCF-10A line was obtained from the ATCC and cultured in DMEM/F12 (Invitrogen) medium containing: 2.5 mM L-glutamine, supplemented with 5% heat-inactivated equine serum (Invitrogen), 500 ng/ml hydrocortisone (Sigma-Aldrich), 21.5 ng/ml epidermal growth factor (EGF) (Sigma-Aldrich), 10 μg/ml insulin (Sigma-Aldrich), 100 ng/ml cholera toxin (Sigma-Aldrich) and 15 mM HEPES (Sigma-Aldrich). AG1478 (EGFR inhibitor), KN-93 (CaM-K inhibitor), rapamycin (mTORC1 inhibitor), doxorubicin and berberine were purchased from Sigma-Aldrich. ABT-737 (BCL-2 inhibitor) was obtained from Dr. Michael Andreeff (MD Anderson Cancer Center).
Methylthiazol tetrazolium assay
Methylthiazol tetrazolium (MTT) assays were performed to determine a cell line’s sensitivity to chemotherapeutic drugs. Two thousand cells per well were plated in 96-well plates in 100 μL of cRPMI without phenol red (Invitrogen) and allowed to attach overnight under normal culture conditions. The next day, serial 2-fold dilutions of a chemotherapeutic drug were made, and 100 μL of each dilution were added to a corresponding well on the 96-well plate. Cells were incubated for 4 d under normal culture conditions. On the fourth day, 22.2 μL of a 5 mg/mL solution of thiazolyl blue tetrazolium bromide (Sigma-Aldrich) in 1X PBS was added to each well and incubated for 90 min at 37°C. The media was then removed, and 150 μL of dimethyl sulfoxide (DMSO) (Fisher Scientific) was added to resuspend formazin crystals to produce a purple color, which was subsequently read on a Multiskan EX Microplate photometer (Thermo Scientific) at a wavelength of 570 nm. Colormetric readings were normalized against plates of non-treated cells under identical culture conditions. Relative growth was calculated by dividing normalized cell growth values in the presence of drugs by normalized cell growth values in the absence of drugs, and the results were graphed. Drug concentrations that killed at least 50% of the cells (IC50) were determined from the calculated graphed values.114,115
NGAL retroviral transduced cells
The construction of the pLXSN/NGAL retroviral expression vector and the infection of cells with this retrovirus have been previously described.115
Acknowledgments
We thank Dr. Michael Andreeff for generously providing the Bcl-2 inhibitor, ABT-737. M.C. and G.M. were supported in part by grants from the Italian “Ministero dell’Istruzione, dell’Università e della Ricerca” (Ministry forEducation, Universities and Research) – MIUR PRIN 2008 and FIRB-MERIT (RBNE08YYBM). M.C. was also supported in part by a grant to the CNR from the Italian Ministry of Economy and Finance for the Project FaReBio di Qualità. M.L. was supported in part by a grant from the Italian Ministry of Health, Ricerca Finalizzata Stemness 2008 entitled “Molecular Determinants of Stemness and Mesenchymal Phenotype in Breast Cancer.” A.M.M. was supported in part by grants from: MIUR PRIN 2008 (2008THTNLC) and MIUR FIRB 2010 (RBAP10447J-003) and 2011 (RBAP11ZJFA_001). M.M. was supported in part from the Italian Association for Cancer Research (AIRC), the Cariplo Foundation and the Italian Ministry of Health. J.P. and R.T. were supported in part by a grant from Fondazione Umberto Veronesi entitled “Neutrophil gelatinase-associated lipocalin (NGAL) and matrix metalloproteinases (MMPS) as biomarkers of bladder cancer development and progression.” A.T. was supported in part by grants from the Italian “Ministero dell’Istruzione, dell’Università e della Ricerca” (Ministry for Education, University and Research) - MIUR - PRIN 2008 and grant from “Sapienza,” University of Rome 2009–11.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22786
References
- 1.Poulikakos PI, Rosen N. Mutant BRAF melanomas--dependence and resistance. Cancer Cell. 2011;19:11–5. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt P, Abken H. The beating heart of melanomas: a minor subset of cancer cells sustains tumor growth. Oncotarget. 2011;2:313–20. doi: 10.18632/oncotarget.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koomen JM, Smalley KS. Using quantitative proteomic analysis to understand genotype specific intrinsic drug resistance in melanoma. Oncotarget. 2011;2:329–35. doi: 10.18632/oncotarget.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N, et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. 2010;2:35ra41. doi: 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- 6.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smalley KS, Lioni M, Dalla Palma M, Xiao M, Desai B, Egyhazi S, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008;7:2876–83. doi: 10.1158/1535-7163.MCT-08-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–60. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shull AY, Latham-Schwark A, Ramasamy P, Leskoske K, Oroian D, Birtwistle MR, et al. Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One. 2012;7:e43369. doi: 10.1371/journal.pone.0043369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337–42. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chomel JC, Turhan AG. Chronic myeloid leukemia stem cells in the era of targeted therapies: resistance, persistence and long-term dormancy. Oncotarget. 2011;2:713–27. doi: 10.18632/oncotarget.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hochhaus A, La Rosée P, Müller MC, Ernst T, Cross NC. Impact of BCR-ABL mutations on patients with chronic myeloid leukemia. Cell Cycle. 2011;10:250–60. doi: 10.4161/cc.10.2.14537. [DOI] [PubMed] [Google Scholar]
- 20.Dienstmann R, Martinez P, Felip E. Personalizing therapy with targeted agents in non-small cell lung cancer. Oncotarget. 2011;2:165–77. doi: 10.18632/oncotarget.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steelman LS, Navolanic P, Chappell WH, Abrams SL, Wong EW, Martelli AM, et al. Involvement of Akt and mTOR in chemotherapeutic- and hormonal-based drug resistance and response to radiation in breast cancer cells. Cell Cycle. 2011;10:3003–15. doi: 10.4161/cc.10.17.17119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abrams SL, Steelman LS, Shelton JG, Wong EW, Chappell WH, Bäsecke J, et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle. 2010;9:1781–91. doi: 10.4161/cc.9.9.11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steelman LS, Abrams SL, Shelton JG, Chappell WH, Bäsecke J, Stivala F, et al. Dominant roles of the Raf/MEK/ERK pathway in cell cycle progression, prevention of apoptosis and sensitivity to chemotherapeutic drugs. Cell Cycle. 2010;9:1629–38. doi: 10.4161/cc.9.8.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCubrey JA, Steelman LS, Kempf CR, Chappell WH, Abrams SL, Stivala F, et al. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol. 2011;226:2762–81. doi: 10.1002/jcp.22647. [DOI] [PubMed] [Google Scholar]
- 26.Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, et al. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance (review) Int J Oncol. 2012;40:639–44. doi: 10.3892/ijo.2011.1312. [DOI] [PubMed] [Google Scholar]
- 27.Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 2011;3:192–222. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3:954–87. doi: 10.18632/oncotarget.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2:862–73. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011;2:435–47. doi: 10.18632/oncotarget.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson DH. p85 plays a critical role in controlling flux through the PI3K/PTEN signaling axis through dual regulation of both p110 (PI3K) and PTEN. Cell Cycle. 2010;9:2055–6. doi: 10.4161/cc.9.11.11926. [DOI] [PubMed] [Google Scholar]
- 32.Sokolosky ML, Stadelman KM, Chappell WH, Abrams SL, Martelli AM, Stivala F, et al. Involvement of Akt-1 and mTOR in sensitivity of breast cancer to targeted therapy. Oncotarget. 2011;2:538–50. doi: 10.18632/oncotarget.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kandouz M, Haidara K, Zhao J, Brisson ML, Batist G. The EphB2 tumor suppressor induces autophagic cell death via concomitant activation of the ERK1/2 and PI3K pathways. Cell Cycle. 2010;9:398–407. doi: 10.4161/cc.9.2.10505. [DOI] [PubMed] [Google Scholar]
- 34.Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle. 2011;10:1563–70. doi: 10.4161/cc.10.10.15703. [DOI] [PubMed] [Google Scholar]
- 35.Lehn S, Fernö M, Jirström K, Rydén L, Landberg G. A non-functional retinoblastoma tumor suppressor (RB) pathway in premenopausal breast cancer is associated with resistance to tamoxifen. Cell Cycle. 2011;10:956–62. doi: 10.4161/cc.10.6.15074. [DOI] [PubMed] [Google Scholar]
- 36.Musgrove EA, Sutherland RL. RB in breast cancer: differential effects in estrogen receptor-positive and estrogen receptor-negative disease. Cell Cycle. 2010;9:4607–15. doi: 10.4161/cc.9.23.13889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle. 2010;9:4153–63. doi: 10.4161/cc.9.20.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glazer RI. A new therapeutic basis for treating Li-Fraumeni Syndrome breast tumors expressing mutated TP53. Oncotarget. 2010;1:470–1. doi: 10.18632/oncotarget.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herbert BS, Chanoux RA, Liu Y, Baenziger PH, Goswami CP, McClintick JN, et al. A molecular signature of normal breast epithelial and stromal cells from Li-Fraumeni syndrome mutation carriers. Oncotarget. 2010;1:405–22. doi: 10.18632/oncotarget.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Napoli M, Girardini JE, Piazza S, Del Sal G. Wiring the oncogenic circuitry: Pin1 unleashes mutant p53. Oncotarget. 2011;2:654–6. doi: 10.18632/oncotarget.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris JL, Khanna KK. BRCA1 A‐complex fine tunes repair functions of BRCA1. Aging (Albany NY) 2011;3:461–3. doi: 10.18632/aging.100334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dever SM, Golding SE, Rosenberg E, Adams BR, Idowu MO, Quillin JM, et al. Mutations in the BRCT binding site of BRCA1 result in hyper-recombination. Aging (Albany NY) 2011;3:515–32. doi: 10.18632/aging.100325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poon JS, Eves R, Mak AS. Both lipid- and protein-phosphatase activities of PTEN contribute to the p53-PTEN anti-invasion pathway. Cell Cycle. 2010;9:4450–4. doi: 10.4161/cc.9.22.13936. [DOI] [PubMed] [Google Scholar]
- 44.Fidalgo da Silva E, Ansari SB, Maimaiti J, Barnes EA, Kong-Beltran M, Donoghue DJ, et al. The tumor suppressor tuberin regulates mitotic onset through the cellular localization of cyclin B1. Cell Cycle. 2011;10:3129–39. doi: 10.4161/cc.10.18.17296. [DOI] [PubMed] [Google Scholar]
- 45.Searle JS, Li B, Du W. Targeting Rb mutant cancers by inactivating TSC2. Oncotarget. 2010;1:228–32. doi: 10.18632/oncotarget.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gómez-Baldó L, Schmidt S, Maxwell CA, Bonifaci N, Gabaldón T, Vidalain PO, et al. TACC3-TSC2 maintains nuclear envelope structure and controls cell division. Cell Cycle. 2010;9:1143–55. doi: 10.4161/cc.9.6.11018. [DOI] [PubMed] [Google Scholar]
- 47.Bhatia B, Nahlé Z, Kenney AM. Double trouble: when sonic hedgehog signaling meets TSC inactivation. Cell Cycle. 2010;9:456–9. doi: 10.4161/cc.9.3.10532. [DOI] [PubMed] [Google Scholar]
- 48.Kolesnichenko M, Vogt PK. Understanding PLZF: two transcriptional targets, REDD1 and smooth muscle α-actin, define new questions in growth control, senescence, self-renewal and tumor suppression. Cell Cycle. 2011;10:771–5. doi: 10.4161/cc.10.5.14829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Campaner S, Doni M, Verrecchia A, Fagà G, Bianchi L, Amati B. Myc, Cdk2 and cellular senescence: Old players, new game. Cell Cycle. 2010;9:3655–61. doi: 10.4161/cc.9.18.13049. [DOI] [PubMed] [Google Scholar]
- 50.Bansal R, Nikiforov MA. Pathways of oncogene-induced senescence in human melanocytic cells. Cell Cycle. 2010;9:2782–8. doi: 10.4161/cc.9.14.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller KR, Kelley K, Tuttle R, Berberich SJ. HdmX overexpression inhibits oncogene induced cellular senescence. Cell Cycle. 2010;9:3376–82. doi: 10.4161/cc.9.16.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a) Cell Cycle. 2011;10:457–68. doi: 10.4161/cc.10.3.14707. [DOI] [PubMed] [Google Scholar]
- 53.Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Steelman LS, McCubrey JA. Cooperative effects of Akt-1 and Raf-1 on the induction of cellular senescence in doxorubicin or tamoxifen treated breast cancer cells. Oncotarget. 2011;2:610–26. doi: 10.18632/oncotarget.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Godlewski J, Bronisz A, Nowicki MO, Chiocca EA, Lawler S. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle. 2010;9:2742–8. doi: 10.4161/cc.9.14.12248. [DOI] [PubMed] [Google Scholar]
- 55.Mavrakis KJ, Leslie CS, Wendel HG. Cooperative control of tumor suppressor genes by a network of oncogenic microRNAs. Cell Cycle. 2011;10:2845–9. doi: 10.4161/cc.10.17.16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sayed D, Abdellatif M. AKT-ing via microRNA. Cell Cycle. 2010;9:3213–7. doi: 10.4161/cc.9.16.12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Radojicic J, Zaravinos A, Vrekoussis T, Kafousi M, Spandidos DA, Stathopoulos EN. MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell Cycle. 2011;10:507–17. doi: 10.4161/cc.10.3.14754. [DOI] [PubMed] [Google Scholar]
- 58.Ma S, Guan XY. MiRegulators in cancer stem cells of solid tumors. Cell Cycle. 2011;10:571–2. doi: 10.4161/cc.10.4.14772. [DOI] [PubMed] [Google Scholar]
- 59.Valastyan S, Weinberg RA. miR-31: a crucial overseer of tumor metastasis and other emerging roles. Cell Cycle. 2010;9:2124–9. doi: 10.4161/cc.9.11.11843. [DOI] [PubMed] [Google Scholar]
- 60.Oliveras-Ferraros C, Cufí S, Vazquez-Martin A, Torres-Garcia VZ, Del Barco S, Martin-Castillo B, et al. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: induction of the tumor suppressor miRNA let-7a and suppression of the TGFβ-induced oncomiR miRNA-181a. Cell Cycle. 2011;10:1144–51. doi: 10.4161/cc.10.7.15210. [DOI] [PubMed] [Google Scholar]
- 61.García JM, Silva J, Peña C, Garcia V, Rodríguez R, Cruz MA, et al. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer. 2004;41:117–24. doi: 10.1002/gcc.20062. [DOI] [PubMed] [Google Scholar]
- 62.Wang L, Wang WL, Zhang Y, Guo SP, Zhang J, Li QL. Epigenetic and genetic alterations of PTEN in hepatocellular carcinoma. Hepatol Res. 2007;37:389–96. doi: 10.1111/j.1872-034X.2007.00042.x. [DOI] [PubMed] [Google Scholar]
- 63.Chang S, Sharan SK. Epigenetic control of an oncogenic microRNA, miR-155, by BRCA1. Oncotarget. 2012;3:5–6. doi: 10.18632/oncotarget.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Faber AC, Wong KK, Engelman JA. Differences underlying EGFR and HER2 oncogene addiction. Cell Cycle. 2010;9:851–2. doi: 10.4161/cc.9.5.11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rudloff U, Samuels Y. A growing family: adding mutated Erbb4 as a novel cancer target. Cell Cycle. 2010;9:1487–503. doi: 10.4161/cc.9.8.11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raven JF, Williams V, Wang S, Tremblay ML, Muller WJ, Durbin JE, et al. Stat1 is a suppressor of ErbB2/Neu-mediated cellular transformation and mouse mammary gland tumor formation. Cell Cycle. 2011;10:794–804. doi: 10.4161/cc.10.5.14956. [DOI] [PubMed] [Google Scholar]
- 67.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010;2:185–99. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan L, Borregaard N, Kjeldsen L, Moses MA. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J Biol Chem. 2001;276:37258–65. doi: 10.1074/jbc.M106089200. [DOI] [PubMed] [Google Scholar]
- 71.Roy R, Louis G, Loughlin KR, Wiederschain D, Kilroy SM, Lamb CC, et al. Tumor-specific urinary matrix metalloproteinase fingerprinting: identification of high molecular weight urinary matrix metalloproteinase species. Clin Cancer Res. 2008;14:6610–7. doi: 10.1158/1078-0432.CCR-08-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang J, Bielenberg DR, Rodig SJ, Doiron R, Clifton MC, Kung AL, et al. Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci USA. 2009;106:3913–8. doi: 10.1073/pnas.0810617106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang J, Moses MA. Lipocalin 2: a multifaceted modulator of human cancer. Cell Cycle. 2009;8:2347–52. doi: 10.4161/cc.8.15.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bolignano D, Donato V, Lacquaniti A, Fazio MR, Bono C, Coppolino G, et al. Neutrophil gelatinase-associated lipocalin (NGAL) in human neoplasias: a new protein enters the scene. Cancer Lett. 2010;288:10–6. doi: 10.1016/j.canlet.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 75.Leng X, Wu Y, Arlinghaus RB. Relationships of lipocalin 2 with breast tumorigenesis and metastasis. J Cell Physiol. 2011;226:309–14. doi: 10.1002/jcp.22403. [DOI] [PubMed] [Google Scholar]
- 76.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–48. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 77.Matsuo S, Yamazaki S, Takeshige K, Muta T. Crucial roles of binding sites for NFκB and C/EBPs in IkappaB-zeta-mediated transcriptional activation. Biochem J. 2007;405:605–15. doi: 10.1042/BJ20061797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roudkenar MH, Kuwahara Y, Baba T, Roushandeh AM, Ebishima S, Abe S, et al. Oxidative stress induced lipocalin 2 gene expression: addressing its expression under the harmful conditions. J Radiat Res. 2007;48:39–44. doi: 10.1269/jrr.06057. [DOI] [PubMed] [Google Scholar]
- 79.Iannetti A, Pacifico F, Acquaviva R, Lavorgna A, Crescenzi E, Vascotto C, et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NFκB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc Natl Acad Sci USA. 2008;105:14058–63. doi: 10.1073/pnas.0710846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Franklin RA, Rodriguez-Mora OG, Lahair MM, McCubrey JA. Activation of the calcium/calmodulin-dependent protein kinases as a consequence of oxidative stress. Antioxid Redox Signal. 2006;8:1807–17. doi: 10.1089/ars.2006.8.1807. [DOI] [PubMed] [Google Scholar]
- 80.Martelli AM, Evangelisti C, Chappell W, Abrams SL, Bäsecke J, Stivala F, et al. Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia. 2011;25:1064–79. doi: 10.1038/leu.2011.46. [DOI] [PubMed] [Google Scholar]
- 81.Steelman LS, Franklin RA, Abrams SL, Chappell W, Kempf CR, Bäsecke J, et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia. 2011;25:1080–94. doi: 10.1038/leu.2011.66. [DOI] [PubMed] [Google Scholar]
- 82.Barré B, Coqueret O, Perkins ND. Regulation of activity and function of the p52 NF-κB subunit following DNA damage. Cell Cycle. 2010;9:4795–804. doi: 10.4161/cc.9.24.14245. [DOI] [PubMed] [Google Scholar]
- 83.Melvin A, Mudie S, Rocha S. Further insights into the mechanism of hypoxia-induced NFκB. [corrected] Cell Cycle. 2011;10:879–82. doi: 10.4161/cc.10.6.14910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bao G, Clifton M, Hoette TM, Mori K, Deng SX, Qiu A, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol. 2010;6:602–9. doi: 10.1038/nchembio.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle. 2007;6:1982–94. doi: 10.4161/cc.6.16.4603. [DOI] [PubMed] [Google Scholar]
- 86.Le NT, Richardson DR. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: a link between iron metabolism and proliferation. Blood. 2004;104:2967–75. doi: 10.1182/blood-2004-05-1866. [DOI] [PubMed] [Google Scholar]
- 87.Kovacevic Z, Richardson DR. The metastasis suppressor, Ndrg-1: a new ally in the fight against cancer. Carcinogenesis. 2006;27:2355–66. doi: 10.1093/carcin/bgl146. [DOI] [PubMed] [Google Scholar]
- 88.Kovacevic Z, Fu D, Richardson DR. The iron-regulated metastasis suppressor, Ndrg-1: identification of novel molecular targets. Biochim Biophys Acta. 2008;1783:1981–92. doi: 10.1016/j.bbamcr.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 89.Assinder SJ, Dong Q, Kovacevic Z, Richardson DR. The TGF-beta, PI3K/Akt and PTEN pathways: established and proposed biochemical integration in prostate cancer. Biochem J. 2009;417:411–21. doi: 10.1042/BJ20081610. [DOI] [PubMed] [Google Scholar]
- 90.Yu Y, Gutierrez E, Kovacevic Z, Saletta F, Obeidy P, Suryo Rahmanto Y, et al. Iron chelators for the treatment of cancer. Curr Med Chem. 2012;19:2689–702. doi: 10.2174/092986712800609706. [DOI] [PubMed] [Google Scholar]
- 91.Buss JL, Greene BT, Turner J, Torti FM, Torti SV. Iron chelators in cancer chemotherapy. Curr Top Med Chem. 2004;4:1623–35. doi: 10.2174/1568026043387269. [DOI] [PubMed] [Google Scholar]
- 92.Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics. 2009;2:2. doi: 10.1186/1755-8794-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sarafanov AG, Todorov TI, Centeno JA, Macias V, Gao W, Liang WM, et al. Prostate cancer outcome and tissue levels of metal ions. Prostate. 2011;71:1231–8. doi: 10.1002/pros.21339. [DOI] [PubMed] [Google Scholar]
- 94.Guo W, Schlicht M, Kucynda T, Zhou P, Valyi-Nagy K, Kajdacsy-Balla A. Iron increases the invasiveness of prostate cancer cells in vitro: Mechanisms and inhibition by the antioxidant ebselen. Cancer Res. 2012;72(Supplement 1) doi: 10.1158/1538-7445.AM2012-4322. [DOI] [Google Scholar]
- 95.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D’Angeli L, Bartoli A, et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2010;2:823–42. doi: 10.18632/aging.100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brazzolotto X, Andriollo M, Guiraud P, Favier A, Moulis JM. Interactions between doxorubicin and the human iron regulatory system. Biochim Biophys Acta. 2003;1593:209–18. doi: 10.1016/S0167-4889(02)00391-9. [DOI] [PubMed] [Google Scholar]
- 98.Davies NP, Suryo Rahmanto Y, Chitambar CR, Richardson DR. Resistance to the antineoplastic agent gallium nitrate results in marked alterations in intracellular iron and gallium trafficking: identification of novel intermediates. J Pharmacol Exp Ther. 2006;317:153–62. doi: 10.1124/jpet.105.099044. [DOI] [PubMed] [Google Scholar]
- 99.Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci USA. 2006;103:14901–6. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ganguly A, Chakraborty P, Banerjee K, Chatterjee S, Basu S, Sarkar A, et al. Iron N-(2-hydroxy acetophenone) glycinate (FeNG), a non-toxic glutathione depletor circumvents doxorubicin resistance in Ehrlich ascites carcinoma cells in vivo. Biometals. 2012;25:149–63. doi: 10.1007/s10534-011-9493-7. [DOI] [PubMed] [Google Scholar]
- 101.Monier F, Mollier S, Guillot M, Rambeaud JJ, Morel F, Zaoui P. Urinary release of 72 and 92 kDa gelatinases, TIMPs, N-GAL and conventional prognostic factors in urothelial carcinomas. Eur Urol. 2002;42:356–63. doi: 10.1016/S0302-2838(02)00350-0. [DOI] [PubMed] [Google Scholar]
- 102.Smith ER, Zurakowski D, Saad A, Scott RM, Moses MA. Urinary biomarkers predict brain tumor presence and response to therapy. Clin Cancer Res. 2008;14:2378–86. doi: 10.1158/1078-0432.CCR-07-1253. [DOI] [PubMed] [Google Scholar]
- 103.Roy R, Yang J, Moses MA. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol. 2009;27:5287–97. doi: 10.1200/JCO.2009.23.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schmidt C. Urine biomarkers may someday detect even distant tumors. J Natl Cancer Inst. 2009;101:8–10. doi: 10.1093/jnci/djn482. [DOI] [PubMed] [Google Scholar]
- 105.Leng X, Ding T, Lin H, Wang Y, Hu L, Hu J, et al. Inhibition of lipocalin 2 impairs breast tumorigenesis and metastasis. Cancer Res. 2009;69:8579–84. doi: 10.1158/0008-5472.CAN-09-1934. [DOI] [PubMed] [Google Scholar]
- 106.Hu L, Hittelman W, Lu T, Ji P, Arlinghaus R, Shmulevich I, et al. NGAL decreases E-cadherin-mediated cell-cell adhesion and increases cell motility and invasion through Rac1 in colon carcinoma cells. Lab Invest. 2009;89:531–48. doi: 10.1038/labinvest.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun Y, Yokoi K, Li H, Gao J, Hu L, Liu B, et al. NGAL expression is elevated in both colorectal adenoma-carcinoma sequence and cancer progression and enhances tumorigenesis in xenograft mouse models. Clin Cancer Res. 2011;17:4331–40. doi: 10.1158/1078-0432.CCR-11-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fernández CA, Yan L, Louis G, Yang J, Kutok JL, Moses MA. The matrix metalloproteinase-9/neutrophil gelatinase-associated lipocalin complex plays a role in breast tumor growth and is present in the urine of breast cancer patients. Clin Cancer Res. 2005;11:5390–5. doi: 10.1158/1078-0432.CCR-04-2391. [DOI] [PubMed] [Google Scholar]
- 109.Hu J, Van den Steen PE, Sang Q-X, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6:480–98. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 110.Bourguignon LY, Gunja-Smith Z, Iida N, Zhu HB, Young LJ, Muller WJ, et al. CD44v(3,8-10) is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. J Cell Physiol. 1998;176:206–15. doi: 10.1002/(SICI)1097-4652(199807)176:1<206::AID-JCP22>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 111.Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Abécassis I, Olofsson B, Schmid M, Zalcman G, Karniguian A. RhoA induces MMP-9 expression at CD44 lamellipodial focal complexes and promotes HMEC-1 cell invasion. Exp Cell Res. 2003;291:363–76. doi: 10.1016/j.yexcr.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 113.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 114.Chappell WH, Abrams SL, Stadelman KM, LaHair MM, Franklin RA, Cocco L, et al. Increased NGAL (Lnc2) expression after chemotherapeutic drug treatment. Adv Bio Reg. 2012;53 doi: 10.1016/j.jbior.2012.09.004. In press. [DOI] [PubMed] [Google Scholar]
- 115.Chappell WH, Abrams SL, Montano G, Cervello M, Martelli AM, Candido S, et al. Effects of ectopic expression of NGAL on doxorubicin sensitivity. Oncotarget. 2012 doi: 10.18632/oncotarget.691. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bauer M, Eickhoff JC, Gould MN, Mundhenke C, Maass N, Friedl A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res Treat. 2008;108:389–97. doi: 10.1007/s10549-007-9619-3. [DOI] [PubMed] [Google Scholar]
- 117.Salzano M, Rusciano MR, Russo E, Bifulco M, Postiglione L, Vitale M. Calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylates Raf-1 at serine 338 and mediates Ras-stimulated Raf-1 activation. Cell Cycle. 2012;11:2100–6. doi: 10.4161/cc.20543. [DOI] [PubMed] [Google Scholar]
- 118.Howe CJ, LaHair MM, Maxwell JA, Lee JT, Robinson PJ, Rodriguez-Mora O, et al. Participation of the calcium/calmodulin-dependent kinases in hydrogen peroxide-induced Ikappa B phosphorylation in human T lymphocytes. J Biol Chem. 2002;277:30469–76. doi: 10.1074/jbc.M205036200. [DOI] [PubMed] [Google Scholar]
- 119.Jensen TE, Rose AJ, Jørgensen SB, Brandt N, Schjerling P, Wojtaszewski JF, et al. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab. 2007;292:E1308–17. doi: 10.1152/ajpendo.00456.2006. [DOI] [PubMed] [Google Scholar]
- 120.Franklin RA, Rodriguez-Mora OG, Lahair MM, McCubrey JA. Activation of the calcium/calmodulin-dependent protein kinases as a consequence of oxidative stress. Antioxid Redox Signal. 2006;8:1807–17. doi: 10.1089/ars.2006.8.1807. [DOI] [PubMed] [Google Scholar]
- 121.Rodriguez-Mora OG, Lahair MM, Evans MJ, Kovacs CJ, Allison RR, Sibata CH, et al. Inhibition of the CaM-kinases augments cell death in response to oxygen radicals and oxygen radical inducing cancer therapies in MCF-7 human breast cancer cells. Cancer Biol Ther. 2006;5:1022–30. doi: 10.4161/cbt.5.8.2910. [DOI] [PubMed] [Google Scholar]
- 122.Rodriquez-Mora OG, LaHair MM, McCubrey JA, Franklin RA. CaM-KI and CAM-KK participate in the control of cell cycle progression in MCF-7 breast cancer cells. Cancer Res. 2005;65:5408–16. doi: 10.1158/0008-5472.CAN-05-0271. [DOI] [PubMed] [Google Scholar]
- 123.Rongo C. Epidermal growth factor and aging: a signaling molecule reveals a new eye opening function. Aging (Albany NY) 2011;3:896–905. doi: 10.18632/aging.100384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cervello M, McCubrey JA, Cusimano A, Lampiasi N, Azzolina A, Montalto G. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012;3:236–60. doi: 10.18632/oncotarget.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ratajczak MZ, Kucia M, Liu R, Shin DM, Bryndza E, Masternak MM, et al. RasGrf1: genomic imprinting, VSELs, and aging. Aging (Albany NY) 2011;3:692–7. doi: 10.18632/aging.100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.de Magalhães JP. A role for Ras signaling in modulating mammalian aging by the GH/IGF1 axis. Aging (Albany NY) 2011;3:336–7. doi: 10.18632/aging.100309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schmidt-Kittler O, Zhu J, Yang J, Liu G, Hendricks W, Lengauer C, et al. PI3Kα inhibitors that inhibit metastasis. Oncotarget. 2010;1:339–48. doi: 10.18632/oncotarget.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martelli AM, Tabellini G, Ricci F, Evangelisti C, Chiarini F, Bortul R, et al. PI3K/AKT/mTORC1 and MEK/ERK signaling in T-cell acute lymphoblastic leukemia: New options for targeted therapy. Adv Enzyme Regul. 2011 doi: 10.1016/j.advenzreg.2011.09.019. In press. [DOI] [PubMed] [Google Scholar]
- 129.Evangelisti C, Ricci F, Tazzari P, Tabellini G, Battistelli M, Falcieri E, et al. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia. 2011;25:781–91. doi: 10.1038/leu.2011.20. [DOI] [PubMed] [Google Scholar]
- 130.Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, et al. Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget. 2012;3:371–94. doi: 10.18632/oncotarget.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bressanin D, Evangelisti C, Ricci F, Tabellini G, Chiarini F, Tazzari PL, et al. Harnessing the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia: eliminating activity by targeting at different levels. Oncotarget. 2012;3:811–23. doi: 10.18632/oncotarget.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Garrett JT, Chakrabarty A, Arteaga CL. Will PI3K pathway inhibitors be effective as single agents in patients with cancer? Oncotarget. 2011;2:1314–21. doi: 10.18632/oncotarget.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010;1:530–43. doi: 10.18632/oncotarget.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zawel L. P3Kα: a driver of tumor metastasis? Oncotarget. 2010;1:315–6. doi: 10.18632/oncotarget.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weber GL, Parat MO, Binder ZA, Gallia GL, Riggins GJ. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget. 2011;2:833–49. doi: 10.18632/oncotarget.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM. INPP4B: the new kid on the PI3K block. Oncotarget. 2011;2:321–8. doi: 10.18632/oncotarget.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dbouk HA, Backer JM. A beta version of life: p110β takes center stage. Oncotarget. 2010;1:729–33. doi: 10.18632/oncotarget.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sacco A, Roccaro A, Ghobrial IM. Role of dual PI3/Akt and mTOR inhibition in Waldenstrom’s Macroglobulinemia. Oncotarget. 2010;1:578–82. doi: 10.18632/oncotarget.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Peng C, Chen Y, Li D, Li S. Role of Pten in leukemia stem cells. Oncotarget. 2010;1:156–60. doi: 10.18632/oncotarget.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Antico Arciuch VG, Russo MA, Dima M, Kang KS, Dasrath F, Liao XH, et al. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget. 2011;2:1109–26. doi: 10.18632/oncotarget.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia. 2012;26:2336–42. doi: 10.1038/leu.2012.136. [DOI] [PubMed] [Google Scholar]
- 142.Hou J, Lam F, Proud C, Wang S. Targeting Mnks for cancer therapy. Oncotarget. 2012;3:118–31. doi: 10.18632/oncotarget.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Alinari L, Christian B, Baiocchi RA. Novel targeted therapies for mantle cell lymphoma. Oncotarget. 2012;3:203–11. doi: 10.18632/oncotarget.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Major P. Potential of mTOR inhibitors for the treatment of subependymal giant cell astrocytomas in tuberous sclerosis complex. Aging (Albany NY) 2011;3:189–91. doi: 10.18632/aging.100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aizman E, Mor A, Levy A, George J, Kloog Y. Ras inhibition by FTS attenuates brain tumor growth in mice by direct antitumor activity and enhanced reactivity of cytotoxic lymphocytes. Oncotarget. 2012;3:144–57. doi: 10.18632/oncotarget.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Altman JK, Sassano A, Platanias LC. Targeting mTOR for the treatment of AML. New agents and new directions. Oncotarget. 2011;2:510–7. doi: 10.18632/oncotarget.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011;2:222–33. doi: 10.18632/oncotarget.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY) 2011;3:1130–41. doi: 10.18632/aging.100422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Finkel T. Breathing lessons: Tor tackles the mitochondria. Aging (Albany NY) 2009;1:9–11. doi: 10.18632/aging.100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Leontieva OV, Blagosklonny MV. Yeast-like chronological senescence in mammalian cells: phenomenon, mechanism and pharmacological suppression. Aging (Albany NY) 2011;3:1078–91. doi: 10.18632/aging.100402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Williamson DL. Normalizing a hyperactive mTOR initiates muscle growth during obesity. Aging (Albany NY) 2011;3:83–4. doi: 10.18632/aging.100290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Blagosklonny MV. Progeria, rapamycin and normal aging: recent breakthrough. Aging (Albany NY) 2011;3:685–91. doi: 10.18632/aging.100352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Panieri E, Toietta G, Mele M, Labate V, Ranieri SC, Fusco S, et al. Nutrient withdrawal rescues growth factor-deprived cells from mTOR-dependent damage. Aging (Albany NY) 2010;2:487–503. doi: 10.18632/aging.100183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY) 2010;2:924–35. doi: 10.18632/aging.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dulic V. Be quiet and you’ll keep young: does mTOR underlie p53 action in protecting against senescence by favoring quiescence? Aging (Albany NY) 2011;3:3–4. doi: 10.18632/aging.100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schug TT. mTOR favors senescence over quiescence in p53-arrested cells. Aging (Albany NY) 2010;2:327–8. doi: 10.18632/aging.100164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY) 2010;2:535–7. doi: 10.18632/aging.100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhao C, Vollrath D. mTOR pathway activation in age-related retinal disease. Aging (Albany NY) 2011;3:346–7. doi: 10.18632/aging.100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Blagosklonny MV. Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives. Aging (Albany NY) 2010;2:265–73. doi: 10.18632/aging.100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Blagosklonny MV. Why human lifespan is rapidly increasing: solving “longevity riddle” with “revealed-slow-aging” hypothesis. Aging (Albany NY) 2010;2:177–82. doi: 10.18632/aging.100139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Maki CG. Decision-making by p53 and mTOR. Aging (Albany NY) 2010;2:324–6. doi: 10.18632/aging.100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rosner M, Hengstschläger M. mTOR protein localization is cell cycle-regulated. Cell Cycle. 2011;10:3608–10. doi: 10.4161/cc.10.20.17855. [DOI] [PubMed] [Google Scholar]
- 163.Jiang Y. mTOR goes to the nucleus. Cell Cycle. 2010;9:868. doi: 10.4161/cc.9.5.11070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lisse TS, Hewison M. Vitamin D: a new player in the world of mTOR signaling. Cell Cycle. 2011;10:1888–9. doi: 10.4161/cc.10.12.15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Blagosklonny MV. Calorie restriction: decelerating mTOR-driven aging from cells to organisms (including humans) Cell Cycle. 2010;9:683–8. doi: 10.4161/cc.9.4.10766. [DOI] [PubMed] [Google Scholar]
- 166.Armour SM, Baur JA, Hsieh SN, Land-Bracha A, Thomas SM, Sinclair DA. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging (Albany NY) 2009;1:515–28. doi: 10.18632/aging.100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade Inhibitors: Targeting these pathways for cancer treatment and overcoming therapy resistance. Oncotarget. 2012 In press. [Google Scholar]
- 168.Grimaldi C, Chiarini F, Tabellini G, Ricci F, Tazzari PL, Battistelli M, et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia. 2012;26:91–100. doi: 10.1038/leu.2011.269. [DOI] [PubMed] [Google Scholar]
- 169.Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget. 2011;2:1302–6. doi: 10.18632/oncotarget.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010;9:3807–14. doi: 10.4161/cc.9.18.13131. [DOI] [PubMed] [Google Scholar]
- 171.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.van der Velden YU, Haramis AP. Insights from model organisms on the functions of the tumor suppressor protein LKB1: zebrafish chips in. Aging (Albany NY) 2011;3:363–7. doi: 10.18632/aging.100319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Martelli AM, Chiarini F, Evangelisti C, Ognibene A, Bressanin D, Billi AM, et al. Targeting the liver kinase B1/AMP-activated protein kinase pathway as a therapeutic strategy for hematological malignancies. Expert Opin Ther Targets. 2012;16:729–42. doi: 10.1517/14728222.2012.694869. [DOI] [PubMed] [Google Scholar]
- 174.Vakana E, Platanias LC. AMPK in BCR-ABL expressing leukemias. Regulatory effects and therapeutic implications. Oncotarget. 2011;2:1322–8. doi: 10.18632/oncotarget.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Godlewski J, Bronisz A, Nowicki MO, Chiocca EA, Lawler S. microRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle. 2010;9:2742–8. doi: 10.4161/cc.9.14.12248. [DOI] [PubMed] [Google Scholar]
- 176.Chen V, Shtivelman E. CC3/TIP30 regulates metabolic adaptation of tumor cells to glucose limitation. Cell Cycle. 2010;9:4941–53. doi: 10.4161/cc.9.24.14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chiacchiera F, Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle. 2010;9:1091–6. doi: 10.4161/cc.9.6.11035. [DOI] [PubMed] [Google Scholar]
- 178.Guo D, Cloughesy TF, Radu CG, Mischel PS. AMPK: A metabolic checkpoint that regulates the growth of EGFR activated glioblastomas. Cell Cycle. 2010;9:211–2. doi: 10.4161/cc.9.2.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Saha AK, Xu XJ, Balon TW, Brandon A, Kraegen EW, Ruderman NB. Insulin resistance due to nutrient excess: is it a consequence of AMPK downregulation? Cell Cycle. 2011;10:3447–51. doi: 10.4161/cc.10.20.17886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Amato S, Man HY. Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle. 2011;10:3452–60. doi: 10.4161/cc.10.20.17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mounier R, Lantier L, Leclerc J, Sotiropoulos A, Foretz M, Viollet B. Antagonistic control of muscle cell size by AMPK and mTORC1. Cell Cycle. 2011;10:2640–6. doi: 10.4161/cc.10.16.17102. [DOI] [PubMed] [Google Scholar]
- 182.Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Menendez JA. Polo-like kinase 1 regulates activation of AMP-activated protein kinase (AMPK) at the mitotic apparatus. Cell Cycle. 2011;10:1295–302. doi: 10.4161/cc.10.8.15342. [DOI] [PubMed] [Google Scholar]
- 183.Anisimov VN, Berstein LM, Popovich IG, Zabezhinski MA, Egormin PA, Piskunova TS, et al. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging (Albany NY) 2011;3:148–57. doi: 10.18632/aging.100273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, et al. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2:896–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Menendez JA, Cufí S, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Vellon L, et al. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging (Albany NY) 2011;3:1063–77. doi: 10.18632/aging.100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Halicka HD, Zhao H, Li J, Traganos F, Zhang S, Lee M, et al. Genome protective effect of metformin as revealed by reduced level of constitutive DNA damage signaling. Aging (Albany NY) 2011;3:1028–38. doi: 10.18632/aging.100397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gomes AP, Duarte FV, Nunes P, Hubbard BP, Teodoro JS, Varela AT, et al. Berberine protects against high fat diet-induced dysfunction in muscle mitochondria by inducing SIRT1-dependent mitochondrial biogenesis. Biochim Biophys Acta. 2012;1822:185–95. doi: 10.1016/j.bbadis.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhao HL, Sui Y, Qiao CF, Yip KY, Leung RK, Tsui SK, et al. Sustained antidiabetic effects of a berberine-containing Chinese herbal medicine through regulation of hepatic gene expression. Diabetes. 2012;61:933–43. doi: 10.2337/db11-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kim HS, Kim MJ, Kim EJ, Yang Y, Lee MS, Lim JS. Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression. Biochem Pharmacol. 2012;83:385–94. doi: 10.1016/j.bcp.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 190.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 2012;7:e33411. doi: 10.1371/journal.pone.0033411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, et al. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2:896–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Richardson AD, Scott DA. Reversing the Warburg effect through stromal autophagy. Cell Cycle. 2011;10:2830–1. doi: 10.4161/cc.10.17.16576. [DOI] [PubMed] [Google Scholar]
- 194.Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D’Angeli L, Bartoli A, et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2010;2:823–42. doi: 10.18632/aging.100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Darnell JE., Jr. STAT3, HIF-1, glucose addiction and Warburg effect. Aging (Albany NY) 2010;2:890–1. doi: 10.18632/aging.100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFβ-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle. 2010;9:4461–8. doi: 10.4161/cc.9.22.14048. [DOI] [PubMed] [Google Scholar]
- 197.Green AS, Chapuis N, Lacombe C, Mayeux P, Bouscary D, Tamburini J. LKB1/AMPK/mTOR signaling pathway in hematological malignancies: from metabolism to cancer cell biology. Cell Cycle. 2011;10:2115–20. doi: 10.4161/cc.10.13.16244. [DOI] [PubMed] [Google Scholar]
- 198.Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Martin-Castillo B, Menendez JA. Metformin activates an ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011;10:1499–501. doi: 10.4161/cc.10.9.15423. [DOI] [PubMed] [Google Scholar]
- 199.Mashhedi H, Blouin MJ, Zakikhani M, David S, Zhao Y, Bazile M, et al. Metformin abolishes increased tumor (18)F-2-fluoro-2-deoxy-D-glucose uptake associated with a high energy diet. Cell Cycle. 2011;10:2770–8. doi: 10.4161/cc.10.16.16219. [DOI] [PubMed] [Google Scholar]
- 200.Oliveras-Ferraros C, Cufí S, Vazquez-Martin A, Torres-Garcia VZ, Del Barco S, Martin-Castillo B, et al. Micro(mi)RNA expression profile of breast cancer epithelial cells treated with the anti-diabetic drug metformin: induction of the tumor suppressor miRNA let-7a and suppression of the TGFβ-induced oncomiR miRNA-181a. Cell Cycle. 2011;10:1144–51. doi: 10.4161/cc.10.7.15210. [DOI] [PubMed] [Google Scholar]
- 201.Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010;9:1057–64. doi: 10.4161/cc.9.6.10994. [DOI] [PubMed] [Google Scholar]
- 202.Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MN, et al. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010;9:188–97. doi: 10.4161/cc.9.1.10407. [DOI] [PubMed] [Google Scholar]