

Abstract
The Von Hippel-Lindau gene (VHL) is frequently deleted or mutated in human renal cell carcinoma (RCC) at the early stage. According to the well-established theory, pVHL acts as a tumor suppressor through its E3 ligase activity, which targets hypoxia-inducing factor-1α (HIF-1α). However, the elevated expression of HIF-1α did not promote cell proliferation, indicating that there would be another target, which could promote cell proliferation at the early cancer stage of RCC. In this study, we show that estrogen receptor-α (ER-α) is a novel proteasomal degradation target of the pVHL E3 ligase. Indeed, the overexpression of VHL suppresses exo- and endogenous ER-α expression, whereas si-pVHL can increase ER-α expression. The negative regulation of pVHL on ER-α expression is achieved by its E3 ligase activity. Thus, pVHL can promote the ER-α ubiquitinylation. In addition, we revealed that ER-α and HIF-1α are competitive substrates of pVHL. Thus, under normal conditions, ER-α overexpression can increase the transcription factor activity of HIF-1α. Under the hypoxic condition, where HIF-1α is not a suitable target of pVHL, ER-α is more rapidly degraded by pVHL. However, in VHL-deficient cells, the expression of ER-α and HIF-1α is retained, so that the hypoxic condition did not suppress cell proliferation obviously compared with cells that are expressing pVHL. Thus, blocking of ER-α using its inhibitor could suppress the proliferation of VHL-deficient cells as effectively as hypoxia-induced growth suppression. Considering our results, blocking of ER-α signaling in VHL-deficient cancer cells would be beneficial for cancer suppression. Indeed, we showed the anti-proliferative effect of Faslodex in VHL-deficient cells.
Keywords: RCC, VHL, ER-alpha, hypoxia, cell proliferation
Introduction
VHL is a causal gene of a familial cancer syndrome1 called von Hippel-Lindau disease, and pVHL has been identified as an E3 ligase.2,3 Because the clinical features of VHL are adrenal gland tumors, hemangioblastomas and clear cell carcinomas of the kidney,4 the underlying molecular functions have been speculated to be related to endothelial cell regulation and cancer formation. Considering the fact that the VHL gene is frequently deleted or mutated in early clear cell carcinomas, its role as a tumor suppressor would be related to the initial step of tumor formation, such as cell proliferation or apoptosis inhibition.
Several decades ago, it was reported that pVHL is an E3 ligase, and that it promotes proteasome-dependent HIF-1α degradation. Because HIF-1α is a hypoxia-responding transcription factor and induces several types of genes, including VEGF-1, EPO and iNOS,5-8 the tumor-suppressive function of pVHL has been pinned down as angiogenesis suppression.
However, considering that angiogenesis is required at the late stage of cancer, and that VHL deletion or loss of function has been detected in the early cancer stage, it seems to be an insufficient explanation for the tumor-suppressive role of pVHL, and it is possible that there could be an additional target gene that is responsible for early cancer progression. In addition, the kidney is a highly vascularized tissue, so further angiogenesis seems to be dispensable for cancer progression.
Renal cell carcinoma has been revealed to be dependent on estrogen, although there is still a matter of debate.9 Indeed, in animal models, treatment with synthetic estrogen can induce RCC rather than other kinds of cancers.10,11 In addition, ER-α polymorphism seems to be related to various kinds of human cancers, including RCC.12 However, the relevance of ER-α and estrogen signaling in RCC progression has not been demonstrated until now. It was recently reported that the targeted deletion of VHL in the mammary gland induces the impairment of the mammary cell proliferation and differentiation, which is not rescued by HIF-1α co-deletion.13 In addition, the loss of VHL alone does not induce mammary tumors. These results indicate that pVHL may have an additional target in addition to HIF-1α that would be related to cell proliferation and differentiation.
In this study, we examined the relevance of pVHL and ER-α. In addition, we investigate the molecular functional linkage between ER-α and HIF-1α under the hypoxic condition. Thus, we revealed that ER-α is a novel target of pVHL that suspends cell proliferation under the hypoxic condition. Thus, blocking of ER-α using Faslodex, a specific blocker of ER-α, can suppress the proliferation of VHL-deficient RCC cells.
Results
pVHL suppresses ER-α expression
To investigate the relevance between pVHL and ER-α, we compared the expression of ER-α in UMRC2 (C2) cells and UMRC2-VHL (C2V) cells, an isogenic cell line stably transfected with VHL. In C2 cells, but not in C2V, we could observe the expression of ER-α (Fig. 1A). We then analyzed the effect of pVHL on exogenous ER-α expression. Ectopically expressed ER-α was clearly suppressed by pVHL co-transfection, whereas si-VHL, which could reduce pVHL expression at translation and transcription level (Fig. S1A and B), could induce exo- and endogenous ER-α expression (Fig. 1B). However, estrogen (Est) did not influence pVHL-mediated ER-α suppression (Fig. 1B). Furthermore, pVHL expression was also reduced when ER-α was co-transfected (Fig. 1B). To check the localization of ER-α and confirm that pVHL can regulate ER-α expression, we performed IF staining, and found that ER-α is mainly localized in the nucleus, while pVHL is distributed in the cytosol and the nucleus (Fig. 1C). This result is consistent with other reports that ER-α is a nuclear protein.14,15 However, the co-transfection of ER-α and pVHL could reduce the expression of both proteins (Fig. 1C), which is consistent with our WB analysis (Fig. 1B). To confirm this result, we measured the expression of ER-α in Caki-2, a RCC cell line in which pVHL is mutated,16 after transfection with VHL. Highly expressed ER-α was obviously suppressed by VHL transfection (Fig. 1D). In this case, Est did not block the pVHL-mediated ER-α reduction. In contrast, the increase of ectopically expressed ER-α by si-VHL could be observed in C2V cells (Fig. S1C). To avoid any artifacts caused by co-transfection, we next checked the expression of endogenous ER-α in MCF-7 cells and obtained the similar result that pVHL suppressed ER-α expression, whereas si-VHL could enhance it (Fig. 1E). In contrast, pVHL did not alter the expression of the androgen receptor in LNCaP (Fig. S1D). Next, we checked the effect of ER-α on endogenous pVHL expression using VHL-expressed C2V cells, and found that ER-α could suppress the pVHL expression. (Fig. S1E). We then examined the expression and localization of ER-α in pVHL-transfected MCF-7 cells and obtained the same result (Fig. 1F). However, the overexpression of HIF-1α, a well-known target of pVHL, did not alter the expression of ER-α (Fig. 1F). In addition, the elimination of pVHL but not HIF-1α could increase the nuclear expression of ER-α in MCF-7 cells (Fig. S1F). These results strongly suggest that ER-α is negatively regulated by pVHL in a ligand-independent manner.

Figure 1. pVHL suppresses ER-α. (A) Expression of ER-α in VHL-deficient C2 cells. MCF7 and MDA-MB.468 (MDA) cells were used as positive and negative controls of ER-α expression. Actin was used as a loading control. (B) pVHL suppresses ER-α. After transfection with the indicated vectors or si-RNAs for 24 h, 293 cells were treated with estrogen (Est; 600 μg/ml) for 6 h. The expression of ER-α was determined by an ER-α-specific Ab, and the expression of exogenous pVHL was determined by a FLAG-Ab. EV indicates an empty vector transfection. LE and SE indicate a long exposure and a short exposure, respectively. (C) The reduction of ER-α and pVHL. Two hundred and ninety-three cells were transfected with VHL and/or ER-α for 24 h. After fixation with Me-OH, 293 cells were stained with anti-ER-α (green), anti-pVHL (red) and DAPI (blue). (D) pVHL suppresses ER-α in pVHL-mutant Caki-2 cells. Caki-2 cells were transfected with the indicated vectors for 24 h and were treated with estrogen for 6 h. (E) pVHL suppresses endogenous ER-α. MCF7 cells were transfected with the indicated vectors or si-RNAs for 24 h and were treated with estrogen for 6 h. LE and SE indicate a long exposure and a short exposure, respectively. (F) The reduction of endogenous ER-α by VHL transfection. MCF-7 cells were transfected with VHL or HIF-1α for 24 h. After fixation with Me-OH, MCF7 cells were stained with anti-ER-α (green) and DAPI (blue). To check for any effects that could have been caused by the serum, cells were incubated in serum-free medium (SF) or complete medium (CM) for 6 h before harvest.
pVHL promotes the ubiquitin-mediated degradation of ER-α
To address how pVHL suppresses ER-α, we first checked the mRNA level of ER-α. However, we did not observe any obvious changes at the transcription level (data not shown). Then, we next checked the engagement with proteasomal degradation, because pVHL is a well-confirmed E3 ligase.2,3 To test this, we measured ER-α expression in the presence of proteasome inhibitors (MG132 and ALLN). These inhibitors could block the reduction of ER-α by pVHL (Fig. 2A). In addition, pVHL expression was also retained by the presence of the inhibitors (Fig. 2A). We could also observe the retention of ER-α in the nucleus in the presence of proteasome inhibitors by immunostaining (Fig. 2B). Moreover, pVHL was retained in the nucleus in the presence of proteasome inhibitors (Fig. 2B; Fig. S2A). In contrast, the cellular localization of ER-α was not altered by pVHL expression (Fig. 2B; Fig. S2B). To know the effect of proteasome inhibitor on reduction of endogenous pVHL by ER-α, we measured the expression of pVHL after transfection with ER-α. Consistent with our previous result, ER-α suppressed the pVHL expression. However, MG132 could block the reduction of both proteins (Fig. S2C). These results indicate that pVHL may recognize nuclear ER-α and target it for degradation. We next checked the half-life of ER-α. Since transfection with VHL strongly suppressed ER-α expression, we did not analyze the half-life of ER-α in VHL-transfected cells (Fig. 2C). Instead, we could observe an obvious extension of the ER-α half-life (from 6 h to over 24 h) after the addition of si-VHL (Fig. 2C). To confirm this result, we measured the half-life of endogenous ER-α in MCF-7 cells. Consistently, the ectopic expression of pVHL shortened the ER-α half-life from 12 h to less than 6 h, whereas si-VHL could extend it up to 12 h (Fig. 2D). Because pVHL is an E3 ligase, we used IP analysis to check the ubiquitinylation of ER-α by pVHL. HA-Ub-conjugated ER-α levels were increased in VHL-transfected cells (Fig. 2E). We next directly checked the level of Ub-conjugated ER-α through IP with an ER-α Ab. pVHL-mediated ER-α ubiquitinylation was not detected by si-VHL and mutant pVHL (R167W; E3 ligase activity-deficient mutant17; Fig. 2F). Thus, the pVHL mutant did not suppress ER-α expression (Fig. 2G). These results indicate that the E3 ligase activity of pVHL is required for ER-α suppression.

Figure 2. pVHL promotes ER-α degradation through an E3 ligase. (A) Blocking of proteasomal degradation induces ER-α expression. Two hundred and ninety-three cells were transfected with the indicated vectors or si-RNAs for 24 h and were treated with ALLN and MG132 (50 μg/ml) for 6 h. SE and LE indicate short exposure and long exposure. (B) Proteasome inhibitors block pVHL-induced reduction in nuclear ER-α. 293 cells were co-transfected with VHL and ER-α for 24 h and incubated with ALLN or MG132 for 6 h. After fixation with Me-OH, 293 cells were stained with anti-ER-α (red), anti-pVHL (green) and DAPI (blue). (C) si-VHL extends the half-life of ER-α. Two hundred and ninety-three cells were co-transfected with ER-α and si-VHL or pVHL for 24 h. To block de novo synthesis, cycloheximide (CHX; 5 μg/ml) was added for the indicated times. Although VHL transfection completely eliminated ER-α, si-VHL extended the ER-α half-life. LE and SE indicate a long exposure and a short exposure, respectively. (D) pVHL can reduce the half-life of ER-α. MCF7 cells were transfected with the indicated vectors or si-RNAs for 24 h. After washing with serum-free medium, de novo synthesis was blocked by cycloheximide treatment for indicated times, and the expression of ER-α was measured. (E and F) ER-α is ubiquitinylated through the E3 ligase activity of pVHL. Immunoprecipitation was performed using HA or ER-α antibodies, and the co-precipitated proteins were analyzed using the indicated antibodies. Whole-cell extracts were obtained from 293 cells transfected with the indicated vectors. (G) ER-α can be suppressed by wild-type but not mutant pVHL. Two hundred and ninety-three cells were transfected with the indicated vectors or si-RNAs for 24 h.
pVHL suppresses transcriptional activity of ER-α through a direct interaction
Since E3 ligase family can recognize their substrates by direct binding, we next examined the interaction between ER-α and pVHL. Through IP analysis, we could observe the interaction between ER-α and pVHL, with the similar affinity between HIF-1α and pVHL (Fig. 3A). The interaction between pVHL and HIF-1α or ER-α could be confirmed by GST pull-down analysis (Fig. 3B). We next examined the effect of pVHL on the activity of ER-α as a transcription factor. To address this, we monitored the luciferase activity using an ER-α response element-luciferase system (ERE-Luc). In 293 cells, the enhancement of luciferase activity by ER-α transfection was reduced by VHL transfection (Fig. 3C). We could also observe a reduction in ERE-Luc activity by VHL transfection and an increase in activity by si-VHL in MCF-7 cells (Fig. 3D), indicating that pVHL could regulate the transcription activity of endogenous ER-α. To confirm this result, we measured the expression of cyclin D1, a well-established target of ER-α,18 and found that ER-α-induced cyclin D1 expression could be suppressed by pVHL overexpression (Fig. 3E). In addition, the expression of Snail, which is also identified as a target of ER-α,19,20 was also reduced by pVHL (Fig. 3F). To confirm that reduction of Snail by VHL-transfection is achieved by ER-α suppression, we performed the similar experiment in ER-α negative MDA-MB468 cells. In this cell line, VHL did not suppress the Snail expression without ER-α transfection (Fig. 3G). Because Snail can suppress p53,21 GN25 and GN29, the specific inhibitors of Snail-p53 binding can induce p53 when Snail is overexpressed; we checked the effect of these inhibitors on p53 expression in VHL-deficient RCCs. Interestingly, GN25 and 29 could induce p53 expression in C2 cells (Fig. S2D). Moreover, Faslodex (an inhibitor of ER-α) and si-ER-α abolished chemically induced p53 expression, although they could increase the basal level of p53 expression (Fig. S2C). These results imply that a VHL deficiency leads to partial p53 suppression by ER-α-mediated Snail upregulation.

Figure 3. pVHL suppressed ER-α through a direct interaction. (A) Direct interaction between ER-α and pVHL. Immunoprecipitation was performed using a pVHL antibody, and the co-precipitated proteins were analyzed using the indicated antibodies. Whole-cell extracts were obtained from 293 cells transfected with the indicated vectors. (B) GST pull-down assay. Agarose-conjugated GST-pVHL was incubated with whole-cell extracts from 293 cells transfected with the indicated vectors in RIPA buffer for 4 h. PPT indicates proteins that co-precipitated with bead-conjugated pVHL, whereas Sup indicates the supernatant. (C) pVHL suppresses transcription activity of ER-α. Two hundred and ninety-three cells were transfected with ERE-Luc and pVHL or ER-α. The increase in ERE-Luc activity was clearly reduced by VHL transfection. (D) pVHL regulates endogenous ER-α transcription activity. MCF-7 cells were transfected with ERE-Luc and VHL or si-VHL for 24 h. ER-α transcription activity was determined by a luciferase activity. (E) pVHL can suppress cyclin D1 expression. After transfection with the indicated vectors for 24 h, estrogen was added for 6 h in A549 cells. Immunoblot analysis was performed using the indicated antibodies. LE and SE indicate a long exposure and a short exposure, respectively. (F and G) pVHL can suppress Snail expression. After transfection with indicated vectors for 24 h. RT-PCR was performed to determine the expression of Snail using matched specific primers.
ER-α inhibition suppresses cell proliferation in VHL-deficient cell lines
Since ER-α can promote cell proliferation by acting as a transcription factor (inducing cyclin D1, Myc or Snail),18-20,22 VHL-deficient cells might show ER-α-dependent cell proliferation. In contrast, VHL-intact cells would not respond to ER-α inhibition, because ER-α would already be suppressed by pVHL. To test this hypothesis, we monitored cell proliferation in RCC cell lines. First, we compared the proliferation of C2 and its isogenic cell line C2V and their responses to an ER-α inhibitor. In VHL-deficient C2 cells, Est could promote cell proliferation (23% increase), and Faslodex notably suppressed cell growth (approximately 40% reduction; Fig. 4A). In contrast, C2V cells did not respond to Est (6% increase) or Faslodex (less than 9% reduction; Fig. 4A). Interestingly, Faslodex-treated C2 cells showed a very similar growth potential to C2V control cells (Fig. S2E). These results suggest that the loss of pVHL would be sufficient for the activation of ER-α, and that the presence of pVHL can completely block ER-α activation. We could also observe a similar result in VHL-intact ACHN cells17 (RCC cell line; Fig. 4B) in which Est and Faslodex did not appear to alter cell growth. In contrast, pVHL-mutant A498 cells showed an obvious reduction in cell growth in response to Faslodex (Fig. 4C). Consistently, cyclin D1 expression was dramatically reduced by Faslodex in VHL-deficient A498 cells and C2 cells (Fig. 4D). Moreover, cells did not show a clear response to Est or Faslodex (Fig. 4D). To confirm that Faslodex-induced growth suppression is dependent on the presence of pVHL, we re-performed the cell counting in VHL-knockeddown ACHN cells and determined that si-VHL caused a reduction in the growth of these cells in response to Faslodex (Fig. 4E). These results indicate that the loss of pVHL can enhance cell proliferation, which is mediated by ER-α activation.

Figure 4. ER-α blocker suppresses cell proliferation in VHL-deficient cells. (A–C) Cell growth was estimated. Each cell line was treated with estrogen or Faslodex (5 μg/ml). Cells were maintained for maximum of 3 d, and cell numbers were calculated daily. (D) VHL-intact cell lines did not respond to estrogen or Faslodex. Each cell line was treated with estrogen or Faslodex for 6 h. After treatment, an immunoblot analysis was performed using the indicated antibodies. (E) The sensitivity to estrogen of Faslodex can be recovered by adding si-VHL to ACHN cells. Cell growth was estimated. After transfection with si-VHL, ACHN cells were treated with estrogen or Faslodex for 6 h. Cells were maintained for 4 d, and cell numbers were calculated daily.
pVHL-mediated ER-α suppression is enhanced by hypoxia
Our previous results showed that pVHL suppresses ER-α (Fig. 1). However, pVHL also suppresses HIF-1α.2,3 We thus investigated the molecular detail mechanism between ER-α and HIF-1α. We first checked the effect of HIF-1α or ER-α on transcription activity of ER-α or HIF-1α using ERE-Luc or iNOS-Luc assays in 293 cells. Differentially from the effect of HIF-1α on ERE-Luc, ER-α transfection could induce HIF-1α transcription activity (Fig. S3A and B). In addition, ER-α partially promoted resistance to pVHL-mediated suppression of HIF-1α (Fig. S3B). However, the increase in iNOS-Luc activity by ER-α transfection or in that of ERE-Luc by HIF-1α transfection was not detected in C2 cells (Fig. S3C and D). We also checked the binding between ER-α and HIF-1α and found that there was no interaction between them (data not shown). These results indicate that HIF-1α could escape from pVHL-mediated degradation in the presence of ER-α, and that ER-α would be a more suitable target of pVHL. To discern a more detailed mechanism, we measured the expression of HIF-1α and ER-α under the hypoxic condition, where HIF-1α is not a favorable target of pVHL.23 Under this condition, ER-α expression was reduced, whereas HIF-1α expression was increased (Fig. 5A). In contrast, we could not observe a reduction in ER-α or elevated expression of HIF-1α in response to CoCl2 in C2 cells (Fig. 5B). Although we observed an increase in HIF-1α following ER-α transfection in C2 cells under normoxia, considering our luciferase assay result, this finding is not related to pVHL or to the activation of HIF-1α. These results imply that under the hypoxic condition, ER-α is a primary target of pVHL. To confirm this result, we examined the localization of pVHL and ER-α under the hypoxic condition. Our hypothesis was that if ER-α is a primary target of pVHL, pVHL would be localized in the nucleus to target ER-α for destruction. From the subcellular fractionation analysis, we found that under the hypoxic conditions, pVHL is mainly localized in the nucleus, where ER-α expression was reduced to an undetectable level (Fig. 5C). Moreover, the elimination of ER-α could prevent the nuclear localization of pVHL (Fig. 5C). To confirm this result, we examined the localization of pVHL in the hypoxic condition through IF staining. Hypoxic stress promotes the partial translocation of pVHL from the cytosol to the nucleus (Fig. S4A). In addition, ER-α transfection could induce the partial nuclear translocation of pVHL (Fig. 5D; Fig. S4B). Indeed, hypoxic stress induced the nuclear localization of pVHL in ER-α-transfected cells (Fig. 5D; Fig. S4B). A more interesting feature is that the elimination of ER-α by si-RNA completely blocked the nuclear translocation of pVHL (Fig. 5D; Fig. S4C). These results are consistent with our cell fractionation data and support our hypothesis that under the hypoxic condition, pVHL mainly targets ER-α. However, HIF-1α is also located in the nucleus under hypoxic condition. We thus examined the effect of si-HIF-1α on pVHL localization. Nuclear pVHL did not return to the cytosol by si-HIF-1α (Fig. S5). These results suggest that the primary purpose of pVHL nuclear translocation under the hypoxic condition is the suppression of ER-α.

Figure 5. Hypoxia promotes pVHL-mediated ER-α degradation. (A and B) Hypoxia reduces the expression of ER-α in a pVHL-dependent manner. After transfection with the indicated vectors for 24 h, CoCl2 (400 μM) was added for 12 h in each cell line. Immunoblot analysis was performed using the indicated antibodies. (C) pVHL translocates from the cytoplasm to the nucleus under the hypoxic condition. Two hundred and ninety-three cells were transfected with the indicated vectors and were treated with CoCl2 for 12 h. After treatment, cells were harvested to isolate the cytoplasm, membrane/organelle, nucleus and insoluble fractions and these samples were analyzed by immunoblot. (D) Knockdown of ER-α blocks the nuclear translocation of pVHL. Two hundred and ninety-three cells were transfected with the indicated vectors or si-RNAs for 24 h and were treated with CoCl2 for 12 h in 293 cells. After fixation with Me-OH, cells were stained with anti-pVHL (green) and DAPI (blue). (E) ER-α promotes HIF-1α transcription activity. Two hundred and ninety-three cells were co-transfected with an iNOS-Luc reporter containing a HIF-1α response element and the indicated vectors for 24 h. In addition, cells were incubated with CoCl2 for 12 h to induce the hypoxic conditions. (F) Hypoxia-induced suppression of ER-α activity is achieved by pVHL. Two hundred and ninety-three cells transfected with ERE-Luc were incubated with CoCl2 for 12 h. A clear reduction in ERE-Luc activity in response to hypoxia was detected. (G) The resistance of hypoxia-induced ERE-Luc suppression in VHL-deficient cells. Under similar conditions to those described above, a reduction in ERE-Luc activity in response to CoCl2 in C2 cells was not observed. Instead, pVHL transfection could restore sensitivity to hypoxia.
To examine the functional relevance between pVHL localization and transcription activity of ER-α or HIF-1α, we measured the transcriptional activity of HIF-1α and ER-α. In 293 cells, CoCl2 dramatically increased the HIF-1α transcription activity, and co-transfection of ER-α could promote HIF-1α activity (Fig. 5E). Moreover, ER-α transfection could overcome the pVHL-mediated iNOS-Luc suppression (Fig. 5E). In contrast, ER-α did not show this beneficial effect on iNOS-Luc in the C2 and A498 RCC cell lines when pVHL was not transfected (Fig. S6A and B). These results also support that ER-α can facilitate HIF-1α activation by the repression of pVHL-mediated HIF-1α suppression. We next examined the effect of HIF-1α on ER-α activity. As shown previously, HIF-1α did not significantly affect ER-α transcription activity (Fig. S3A). In addition, hypoxic stress suppressed ER-α activity in 293 cells (Fig. 5F). Considering that the reduction of ER-α transcription activity is not significant in C2 cells treated with CoCl2 (Fig. 5G; Fig. S7A and B), the reduction of ER-α in the hypoxic condition (Fig. 5F) would be mediated by pVHL. This hypothesis could be supported by the fact that the transfection of pVHL could restore the CoCl2-induced ER-α suppression in VHL-deficient cell lines (Fig. 5G; Fig. S7A and B).
Although CoCl2 is popularly used as a hypoxia mimetic, it is not clear that low oxygen conditions also produce the same features. To address this, we measured the transcriptional activity of ER-α and HIF-1α under low oxygen conditions using hypoxic chamber. Consistently with CoCl2 experiments, hypoxia could suppress ERE-Luc activity in 293 cells (Fig. S8A). However, VHL-deficient C2 did not show the reduction of ERE-Luc activity in response to hypoxia (Fig. S8B). In addition, transcriptional activity of HIF-1α (iNOS-Luc) also showed the similar pattern in response to hypoxia with CoCl2 (Fig. S8C). Our results strongly suggest that hypoxia condition can promote pVHL-mediated ER-α suppression, whereas it can activate HIF-1α.
Hypoxia-induced cell growth arrest is achieved by pVHL-mediated ER-α suppression
In the previous result, we found that pVHL strongly suppressed ER-α activity under the hypoxic condition. Because the activation of ER-α can promote cell proliferation, we speculated that hypoxia would lead to cell growth arrest in VHL-intact cells. To address this, we assessed cell growth by cell counting. In VHL-mutant A498 cells, CoCl2 did not induce a significant growth suppression (Fig. 6A). In contrast, ACHN cells showed obvious growth suppression in response to CoCl2 (Fig. 6B). This result implies that CoCl2-induced growth suppression could be related to VHL status. We thus checked cell growth in C2 cells. Similarly to A498 cells, C2 cells also showed the resistance to CoCl2-induced growth suppression (Fig. 6C). In contrast, the growth of C2 cells was clearly suppressed by treatment with Faslodex (Fig. 6C). To test that the reduction in cell proliferation in response to hypoxia is mediated by pVHL-induced ER-α suppression, we checked cell growth by a colony-forming assay (Fig. 6D). In C2 cells, hypoxic stress did not suppress cell growth; however, Faslodex strongly suppressed cell growth (Fig. 6D). In contrast, C2V cells showed the obvious reduction of the cell growth in response to CoCl2 (Fig. 6D). Similarly, a MTT assay showed that the proliferative ability of C2 cells was reduced by inhibition of ER-α (Fig. 6E), whereas this was mainly accomplished by hypoxic stress in C2V cells (Fig. 6F). We could also obtain the same results from hypoxia (low oxygen-chamber) treatment (Fig. S9A and B). In fact, Faslodex did not enhance the CoCl2-induced cell proliferation reduction in MCF-7 cells (Fig. S9C and D). These results indicate that CoCl2-induced cell growth in pVHL-positive cells is achieved by the reduction of ER-α activity.

Figure 6. Blocking of ER-α suppresses cell proliferation in VHL-deficient tumors. (A and B) Hypoxia-induced growth arrest is dependent on pVHL status. The growth of cells that lacked VHL (A498; A) was not suppressed by hypoxia. In contrast, ACHN (VHL-positive) cells were sensitive to hypoxia-induced growth suppression. Each cell line was treated with CoCl2. Cells were maintained for 4 d, and cell numbers were calculated daily. (C) The inhibition of ER-α is more effective than hypoxia in cell growth suppression in VHL-deficient cells. C2 cells were incubated in CoCl2 or Faslodex-treated medium for 4 d. (D) Comparison of the growth of C2 and C2V cells. Differentially from C2 cells, where hypoxia did not notably suppress cell growth, C2V cells showed the sensitivity to hypoxia-induced growth suppression. In contrast, the growth of C2 cells was reduced by Faslodex. Cells were incubated with CoCl2 or Faslodex-containing medium for 3 d and fixed with PFA. Cells were visualized by staining with trypan blue. (E and F) pVHL is a critical factor for the determination of hypoxia- or Faslodex-induced growth suppression. Compared with C2V cells, in which Est or Faslodex did not alter cell viability, C2 cells were affected by Faslodex and Est. The opposite effect was observed with CoCl2 treatment. Cells were incubated with the indicated chemicals for 4 d. Cell viability was determined by a MTT assay.
Discussion
The loss of pVHL can induce von Hippel-Lindau disease, a well-known cancer syndrome. In addition, the genetic defects in VHL occur in 75% of human RCCs. Although it has been well-documented that pVHL suppresses HIF-1α and inhibits endothelial cell growth,5-8 its role in tumor suppression has not been fully elucidated until now. Moreover, why pVHL should be deleted at the early stage of RCC should be addressed to understand the carcinogenesis of RCC. In this study, we show that pVHL suppresses ER-α expression (Fig. 1). Since ER-α is known to be a powerful proliferation factor, ER-α should be suppressed in normal cells. In contrast, in tumor cells, the elevated expression or the activation of ER-α signaling would be beneficial for tumor formation. Indeed, the proliferation of VHL-deficient cells could be abolished by the inhibition of ER-α (Figs. 4A and D and 6E), whereas VHL-intact cells did not respond to treatment with the ER-α inhibitor Faslodex or Estrogen (Figs. 4A and B and 6F). These results strongly suggest that the proliferation of VHL-deficient RCC would be dependent on ER-α activity. In fact, it has been reported that the continuous injection of estrogen can particularly induce RCC in animal models.10,11
Interestingly, the estrogen-dependent proliferation of RCC seems to be related to a gender bias in the occurrence of RCC. In general, RCC frequently occurs in the aged male population.24 Considering that estrogen is elevated in aged males but is decreased in aged females, the deletion or inactivation of pVHL could cause RCC in the aged male population. However, it is unclear why females, who produce much more estrogen, have a lower incidence of RCC than males. It would be that an intrinsic protective mechanism against estrogen-induced proliferation may be present in the cells of females. However, until now, we did not know what could be responsible for this phenomenon.
In this study, we show that pVHL is preferentially localized in the nucleus when ER-α is highly expressed (Fig. 5C and D). In contrast, pVHL is predominantly expressed in the cytoplasm under normoxia (Fig. S2A) or si-ER-α-transfected conditions (Fig. 5C and D). In general, the interaction between pVHL and HIF-1α and the subsequent degradation of HIF-1α occurs in the cytoplasm.25 Considering these results and previous reports, the main role of pVHL in the nucleus seems to be to eliminate ER-α. Thus, under the hypoxic condition, where HIF-1α does not undergo hydroxylation by proline hydroxylase and is no longer a favorable substrate of pVHL, pVHL is translocated into the nucleus to block ER-α-mediated proliferation (Fig. 5C; Fig. S4A). In general, cell proliferation should be suppressed under the hypoxic condition, because the lack of nutrients and blood under these conditions is not suitable for cell growth. Under physiological conditions, pVHL suppresses HIF-1α and ER-α (Fig. 7). Under the hypoxic condition, HIF-1α is released from pVHL-mediated suppression and functions as a transcription factor, inducing the expression of pro-angiogenic factors such as VEGF or EPO.5-8 Meanwhile, pVHL blocks ER-α to suppress cell proliferation (Fig. 7). However, if the cell does not have functional pVHL because of a genetic deletion or epigenetic inactivation,26 ER-α functions as a transcription factor and induces the expression of cyclin D1, Myc and Snail (Fig. 7). Under these conditions, HIF-1α-mediated angiogenesis is accomplished, and the proliferation of VHL-deficient cells would be boosted, because the nutrient supply could be restored due to neo-vascularization.
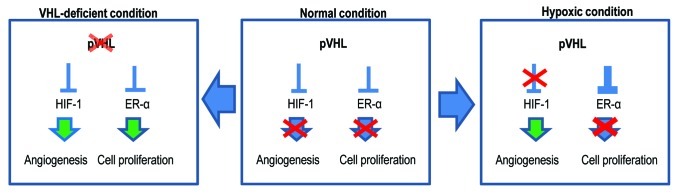
Figure 7. Diagram for the working mechanism. Under normal conditions (center), pVHL suppresses HIF-1α and ER-α through its E3 ligase activity. However, in the hypoxic condition (right panel), pVHL sequesters ER-α more strongly so that cells cannot proliferate. Instead, HIF-1α can be activated and induce angiogenesis. Because the hypoxic condition is generated by tissue damage, this mechanism may contribute to blocking an overgrowth of damaged cells. However, when pVHL is deleted or mutated (left panel), ER-1α-induced cell proliferation can be apparent. In addition, nutrients are then supplied by HIF-1α-mediated angiogenesis. These conditions would increase the probability of the occurrence of cancer.
Currently, we do not know how ER-α is activated in an estrogen-independent manner. Indeed, we checked the localization of ER-α in serum-free conditions, but it was localized to the nucleus in MCF-7, A498 and C2 cells. In addition, the overexpression of ER-α can increase ERE-Luc in these cell lines (Fig. 5F; Fig. S7A and B). Other reports also described that ER-α is present in the nucleus in the absence of estrogen.14,15 These results indicate that the elevated expression of ER-α due to the loss of pVHL can enhance transcription activity.
In this study, we also suggest that ER-α-mediated snail induction seems to be contributing to the suppression of p53 activity (Fig. S2D). Although we do not show the detailed mechanism, considering that snail can suppress p5321 and that blocking Snail-p53-binding through a small chemical can increase p53 expression,21 ER-α-mediated snail upregulation is another important target of RCC treatment. Indeed, we show a dramatic suppression of cell growth in pVHL-dependent RCC cells by the treatment with Faslodex (Figs. 4A and C and 6C–F).
Taken together, we revealed that pVHL acts as a tumor suppressor by suppressing HIF-1α as well as ER-α. Under normal conditions, pVHL strongly suppresses both, thereby inhibiting cell proliferation and neo-vascularization. However, HIF-1α is activated by hypoxia, and pVHL prefers to block ER-α activation more tightly to suppress improper cell proliferation. Thus, a loss of pVHL can promote cell proliferation, which is required for early tumor formation and angiogenesis, eventually leading to late cancer progression. Because of its dual roles, pVHL should be inactivated in RCC.
Materials and Methods
Cell culture and reagents
MCF7, MDA-MB.468, HEK293 and A549 cells were purchased from ATCC. ACHN and A498 cells were obtained from KCLB. UMRC2 (C2), UMRC2/VHL (C2V) and Caki-2 cells were kindly provided by Y.-S.J., Y.J.(Pusan National University). Cells were maintained in DMEM (Thermo), except for A549 (RPMI 1640; Thermo) cells. Estrogen, cycloheximide, Faslodex and CoCl2 were purchased from Sigma, and ALLN and MG132 were provided by Calbiochem. Antibodies against ER-α, actin, pVHL, HA, GFP, Cyclin D1, androgen receptor, GST and p53 (DO-1) were purchased from Santa Cruz, the antibody against HIF-1α was from BD Biosciences and the α-FLAG antibody was from Sigma.
Transfection of vectors and si-RNAs
ER-α and ERE-Luc expression vectors were gifts from Dr. J.H. Jung (Pusan National University). The α-FLAG-fused pVHL and iNOS-Luc vectors were kindly provided by Dr Y.J. Jung (Pusan National University). The HA-tagged HIF-1α expression vector was generously provided by Dr. Kim, Y.J. (Pusan National University). The HA-fused VHL R167W vector was purchased from Addgene. For the in vitro gene knockout, si-RNAs against target proteins were generated. The si-RNA target sequences are as follows: VHL: 5′ - ACA CAG GAG CGC ATT GCA CAT - 3′; HIF-1α: 5′ - TAC GTT GTG AGT GGT ATT ATT - 3′. For the mammalian expression of these vectors or si-RNAs, the transfection was performed using Jetpei (Polyplus) according to manufacturer’s protocol.
Immunoblot analysis and protein interaction studies
Proteins were extracted from cells with RIPA buffer. Samples were applied to SDS-PAGE, and immunoblot analysis was performed by means of a general protocol. Blotted membranes were incubated with primary antibodies for 1 h to overnight at 4°C. Secondary antibodies were HRP-linked goat anti-mouse, goat anti-rabbit and mouse anti-goat (Thermo). The whole-cell lysates were incubated first with the proper antibodies for 4 h at 4°C and then with protein A/G agarose beads (Invitrogen) for 2 h at 4°C. After centrifugation and washing with RIPA, the immunocomplexes were subjected to SDS-PAGE and immunoblot analysis. To determine the direct interaction between proteins, agarose bead-conjugated GST-pVHL was incubated with cell lysates for 4 h at 4°C. Using the same procedure with IP, precipitated proteins were analyzed by immunoblot analysis.
Immunofluorescence staining
Cells were seeded on a cover glass and transfected with the indicated vectors. After fixing with Me-OH for 30 min, the cells were incubated with blocking buffer [PBS + anti-human-Ab (1:500)] for 1 h. After washing with PBS, the cells were incubated with anti-pVHL, ER-α in blocking buffer (1: 100 ~200) for 4 h and subsequently with FITC-conjugated or Rhodamine-conjugated secondary antibodies in blocking buffer (1: 500) for 2 h. The nucleus was stained by DAPI. After washing with PBS, cover glasses were mounted with mounting solution (Vector Laboratories). The immunofluorescence signal was detected through fluorescence microscopy (Zeiss).
RNA isolation and RT-PCR
For RT-PCR, total cellular RNA was extracted using a Qiagen RNA extraction kit. After measurement of the RNA concentration, 1 μg of total RNA was reverse transcribed to cDNA using MMLV-RT (Invitrogen) and random hexamers. RT-PCR was performed with the primers specific to the Snail (5′-TATGCTGCCTTCCCAGGCTTG-3′ and 5′-ATGTGCATCTTGAGGGCACCC-3′) or GAPDH (5′-ATCTTCCAGGAGCGAGATCCC-3′ and 5′-AGTGAGCTTCCC GTTCAGCTC-3′) cDNA.
MTT assay and cell proliferation
To measure the cell viability, cells were treated with the indicated chemicals for 4 d. For the MTT assay, cells were incubated with 0.5 mg/ml of MTT solution (Calbiochem) for 4 h at 37°C. After removing the excess solution, the precipitated materials were dissolved in 200 μl DMSO and quantified by measuring the absorbance at 540 nm. For cell counting, we collected cells with media and stained them with Trypan blue (GIBCO) for 10 min at room temperature. Using a hemocytometer, we determined the number of viable cells.
Luciferase assay
To address ER-α or HIF-1α activity, ERE-Luc or iNOS-Luc vectors were transfected into cells for 24 h, and cells were treated with the indicated chemicals. After washing with wash buffer (Promega), the cells were lysed by lysis buffer (Promega). The luciferase activity was determined by a luminometer (MicroDigital).
Subcellular fractionation analysis
Indicated vectors or si-RNAs were transfected into 293 cells for 24 h. Cells were treated with CoCl2 for 12 h. After treatment, subcellular fractionation was performed using the cytoplasm, nucleus, membrane/organelle and insoluble isolation kit (Calbiochem) according to the manufacturer’s protocol.
Hypoxia using low-oxygen chamber
C2, C2V and 293 cells were transfected with each vector for 24 h. After transfection, cells were exposed to hypoxia (2% O2) treatment for 72 h (Trypan Blue staining and MTT) or 48 h (Luciferase) in a HEPA incubator (Thermo Forma).
Statistical analysis
To obtain the statistical significance, we performed the Student’s t-test.
Supplementary Material
Acknowledgment
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) and funded by the Ministry of Education, Science and Technology (2010–0022413). This work was supported by a 2-Year Research Grant of Pusan National University
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22794
References
- 1.Kaelin WG., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 2.Kaelin WG., Jr. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–73. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- 3.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 4.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–54. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoon D, Pastore YD, Divoky V, Liu E, Mlodnicka AE, Rainey K, et al. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J Biol Chem. 2006;281:25703–11. doi: 10.1074/jbc.M602329200. [DOI] [PubMed] [Google Scholar]
- 7.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 8.Boutin AT, Weidemann A, Fu Z, Mesropian L, Gradin K, Jamora C, et al. Epidermal sensing of oxygen is essential for systemic hypoxic response. Cell. 2008;133:223–34. doi: 10.1016/j.cell.2008.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Concolino G, Marocchi A, Conti C, Tenaglia R, Di Silverio F, Bracci U. Human renal cell carcinoma as a hormone-dependent tumor. Cancer Res. 1978;38:4340–4. [PubMed] [Google Scholar]
- 10.Wolf DC, Goldsworthy TL, Donner EM, Harden R, Fitzpatrick B, Everitt JI. Estrogen treatment enhances hereditary renal tumor development in Eker rats. Carcinogenesis. 1998;19:2043–7. doi: 10.1093/carcin/19.11.2043. [DOI] [PubMed] [Google Scholar]
- 11.Stefaniak T, Krajewski J, Kobiela J, Makarewicz W, Stanek A, Asano M, et al. Protein oxidation in male Syrian hamster kidney during estrogen-induced carcinogenesis. Pathophysiology. 2002;8:269–73. doi: 10.1016/S0928-4680(02)00019-6. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka Y, Sasaki M, Kaneuchi M, Fujimoto S, Dahiya R. Estrogen receptor alpha polymorphisms and renal cell carcinoma--a possible risk. Mol Cell Endocrinol. 2003;202:109–16. doi: 10.1016/S0303-7207(03)00071-6. [DOI] [PubMed] [Google Scholar]
- 13.Seagroves TN, Peacock DL, Liao D, Schwab LP, Krueger R, Handorf CR, et al. VHL deletion impairs mammary alveologenesis but is not sufficient for mammary tumorigenesis. Am J Pathol. 2010;176:2269–82. doi: 10.2353/ajpath.2010.090310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–51. doi: 10.1016/0092-8674(87)90581-2. [DOI] [PubMed] [Google Scholar]
- 15.Bukovsky A, Ayala ME, Dominguez R, Keenan JA, Wimalasena J, Elder RF, et al. Changes of ovarian interstitial cell hormone receptors and behavior of resident mesenchymal cells in developing and adult rats with steroid-induced sterility. Steroids. 2002;67:277–89. doi: 10.1016/S0039-128X(01)00159-3. [DOI] [PubMed] [Google Scholar]
- 16.Whaley JM, Naglich J, Gelbert L, Hsia YE, Lamiell JM, Green JS, et al. Germ-line mutations in the von Hippel-Lindau tumor-suppressor gene are similar to somatic von Hippel-Lindau aberrations in sporadic renal cell carcinoma. Am J Hum Genet. 1994;55:1092–102. [PMC free article] [PubMed] [Google Scholar]
- 17.Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science. 1995;269:1444–6. doi: 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]
- 18.Wilcken NR, Prall OW, Musgrove EA, Sutherland RL. Inducible overexpression of cyclin D1 in breast cancer cells reverses the growth-inhibitory effects of antiestrogens. Clin Cancer Res. 1997;3:849–54. [PubMed] [Google Scholar]
- 19.Park SH, Cheung LW, Wong AS, Leung PC. Estrogen regulates Snail and Slug in the down-regulation of E-cadherin and induces metastatic potential of ovarian cancer cells through estrogen receptor alpha. Mol Endocrinol. 2008;22:2085–98. doi: 10.1210/me.2007-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX, Shetuni B, et al. ERalpha signaling through slug regulates E-cadherin and EMT. Oncogene. 2010;29:1451–62. doi: 10.1038/onc.2009.433. [DOI] [PubMed] [Google Scholar]
- 21.Lee SH, Lee SJ, Jung YS, Xu Y, Kang HS, Ha NC, et al. Blocking of p53-Snail binding, promoted by oncogenic K-Ras, recovers p53 expression and function. Neoplasia. 2009;11:22–31, 6p, 31. doi: 10.1593/neo.81006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubik D, Dembinski TC, Shiu RP. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res. 1987;47:6517–21. [PubMed] [Google Scholar]
- 23.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 24.Stafford HS, Saltzstein SL, Shimasaki S, Sanders C, Downs TM, Sadler GR. Racial/ethnic and gender disparities in renal cell carcinoma incidence and survival. J Urol. 2008;179:1704–8. doi: 10.1016/j.juro.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mack FA, Rathmell WK, Arsham AM, Gnarra J, Keith B, Simon MC. Loss of pVHL is sufficient to cause HIF dysregulation in primary cells but does not promote tumor growth. Cancer Cell. 2003;3:75–88. doi: 10.1016/S1535-6108(02)00240-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stolle C, Glenn G, Zbar B, Humphrey JS, Choyke P, Walther M, et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417–23. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.