

Abstract
Antibiotics administered in low doses have been widely used as growth promoters in the agricultural industry since the 1950s, yet the mechanisms for this effect are unclear. Because antimicrobial agents of different classes and varying activity are effective across several vertebrate species, we hypothesized that such subtherapeutic administration alters the population structure of the gut microbiome as well as its metabolic capabilities. We generated a model of adiposity by giving subtherapeutic antibiotic therapy to young mice and evaluated changes in the composition and capabilities of the gut microbiome. Administration of subtherapeutic antibiotic therapy increased adiposity in young mice and increased hormones related to metabolism. We observed substantial taxonomic changes in the microbiome, changes in copies of key genes involved in the metabolism of carbohydrates to short-chain fatty acids, increases in colonic short-chain fatty acid levels, and alterations in the regulation of hepatic metabolism of lipids and cholesterol. In this model, we demonstrate the alteration of early-life murine metabolic homeostasis through antibiotic manipulation.
Antibiotics, discovered in the early twentieth century, came into widespread use after the Second World War, with substantial public health benefits. Antibiotic use has increased markedly, now approximating one antibiotic course per year in the average child in the United States1,2. However, there is increasing concern that antibiotic exposure may have long-term consequences3–5.
For more than 50 years we have known that the administration of low doses of antibacterial agents promotes the growth of farm animals, consequently, in the United States, the largest use of antibiotics and related antimicrobial substances is within farms, with low doses fed to large numbers of animals used for food production to increase weight gain by as much as 15%6,7. These effects are broad across vertebrate species, including mammals (cattle, swine, sheep) and birds (chickens, turkeys), and follow oral administration of the agents, either in feed or water, indicating that the microbiota of the gastrointestinal (GI) tract is a major target. That the effects are observed with many different classes of antibacterial agents (including macrolides, tetracyclines, penicillins and ionophores) indicates that the activity is not an agent-specific side effect, nor have the effects been observed with antifungals or antivirals.
The vertebrate GI tract contains an exceptionally complex and dense microbial environment, with bacterial constituents that affect the immune responses of populations of reactive host cells8 and stimulate a rich matrix of effecter mechanisms involved in innate and adaptive immune responses9. The GI tract also is a locus of hormone production, including those involved in energy homeostasis (such as insulin, glucagon, leptin and ghrelin) and growth (for example, glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 (GLP-1))10. Alterations in the populations of the GI microbiota may change the intra-community metabolic interactions11, modify caloric intake by using carbohydrates such as cellulose that are otherwise indigestible by the host12, and globally affect host metabolic, hormonal and immune homeostasis13. Full (therapeutic) dose antibiotic treatments alter both the composition of the gastrointestinal microbiota14 and host responses to specific microbial signals15. In combination with dietary changes, antibiotic administration has been associated with changes in the population structure of the microbiome. However, the effects of exposure to subtherapeutic antibiotic dosages have not been described.
Early studies of the effects of gut microbiota on metabolism were limited by the use of culture-based technologies that interrogated <5% of the extant GI tract microbes16. Culture-independent investigation of small subunit ribosomal RNA (ssrRNA) sequences allows the microbial population structure17 of the gut microbiota to be characterized with greater resolution. Despite inter-individual differences, substantial similarities exist18 among mammalian species in the GI microbiota at higher taxonomic levels and functional pathways, indicating a basis for the conserved responses to early-life subtherapeutic antibiotic treatment (STAT) within farms. Previous work has shown that obesity leads to variation in the GI microbiome12,19; we use the insights provided from modern agricultural practices to suggest an alternative approach, using a murine model of STAT to explore how antibiotic exposure modulates host metabolic phenotypes.
Early-life STAT increases adiposity
We exposed C57BL/6J mice at weaning to penicillin, vancomycin, penicillin plus vancomycin, chlortetracycline, or no antibiotic in their drinking water at levels in the mid-range of US Food and Drug Administration (FDA)-approved levels for subtherapeutic antibiotic use in agriculture6,7. After a 7 week exposure, the observed weights were within the expected range of growth for female C57BL/6J mice, and there was no significant difference in overall growth between the STAT and control mice (Fig. 1a). However, by dual energy X-ray absorptiometry (DEXA) scanning, (Fig. 1b) total fat mass was significantly higher in all four groups of STAT mice than in the control group (Fig. 1c). Per cent body fat also was increased in most STAT groups compared to controls (Fig. 1d). Lean weight was not significantly (P = 0.24) different in the STAT mice (15.0 ± 0.1 g) compared to controls (15.4 ± 0.3 g) (Fig. 1e). Thus, 7 weeks after starting the intervention, each of several STAT exposures changed body composition but not overall weight. Repeat STAT experiments showed similar results (Supplementary Fig. 2). There were no significant differences in calculated feed efficiency, expressed as weight gained per unit of feed consumed, between the STAT and control mice. In a larger, confirmatory experiment to assess when the morphometric changes appear, we began examining the mice immediately at weaning. Both the STAT males and females showed significantly increased early life growth rates (Supplementary Fig. 3A), and fat mass in the STAT animals began to diverge from controls by 16–20 weeks, in both males and females. Between 8 and 26 weeks, there were significantly increased rates of fat accumulation in both female and male STAT mice (0.042 and 0.045 g week−1, respectively) compared to controls; female mice also showed significantly increased total mass (Supplementary Fig. 3B). These studies confirm the increased adiposity associated with STAT in females, show parallel effects in males, and indicate that the morphometric changes begin at the earliest time (day 22) of measurement.
Figure 1. Weight and body composition of control and STAT mice.

a, Weight gain did not differ between control and STAT mice (n = 10 mice per group). b, Representative DEXA show per cent body fat in control (22.9%; top) and STAT (32.0%; bottom) mice. c, Total fat mass was significantly increased (P < 0.05) in all STAT groups compared to controls. d, Per cent body fat was significantly increased in all STAT groups (all P < 0.05) except vancomycin. e, Lean mass was lower in STAT mice, but not significantly different from controls. Data are presented as mean ± s.e.m. For all figures: all, all antibiotics; C, controls; Ct, chlortetracycline; P, penicillin; P+V, penicillin plus vancomycin; V, vancomycin.
Bone mineral density is increased in early-life growth
Bone mineral density (BMD) was evaluated by DEXA scanning in the control and STAT mice after 3 and 7 weeks of exposure. At 3 weeks, the BMDs in each of the five STAT groups (and overall) was significantly increased compared to controls (Fig. 2a). By 7 weeks, the BMDs in all mice increased, without significant differences between the STAT (0.045 ± 0.002 mg cm−2) and control mice (0.046 ± 0.003 mg cm−2) (Fig. 2a). Thus, an early bone developmental phenotype observed in each of the STAT groups normalized by 7 weeks. Parallel observations have been made in other STAT experiments (data not shown).
Figure 2. Bone development and serum GIP measurements.
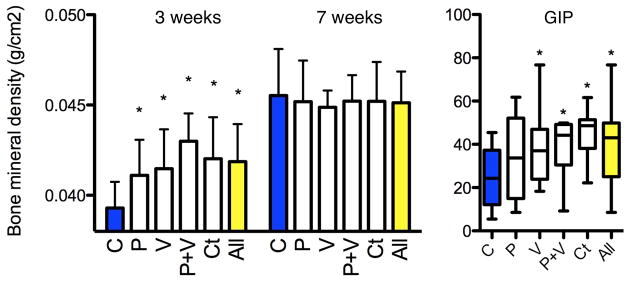
a, After 3 weeks of STAT, bone mineral density was significantly increased in each group (n = 10 mice per group) compared to controls (P < 0.05) but did not persist at 6 weeks. b, Serum GIP levels measured at death were significantly increased in the vancomycin, penicillin plus vancomycin, and chlortetracycline groups and in the aggregate antibiotic group compared to controls (P < 0.05). Data are presented as mean ± s.e.m. Box plots show median ± interquartile range (IQR) and 95% ranges (whiskers).
Increased GIP in STAT mice
To examine metabolic correlates to the changes in body composition, we assessed GIP, an incretin synthesized by small intestinal K cells20 with receptors located on adipocytes that stimulates lipoprotein lipase activity21. GIP was significantly elevated in the STAT mice (39.1 ± 2.5 pg ml−1) compared to controls (24.4 ± 4.2 pg ml−1), with the levels ranging from 34.3 ± 6.1 pg ml−1 in the penicillin group to 44.9 ± 3.7 pg ml−1 in the chlortetracycline group (Fig. 2b). Increases in GIP levels are consistent with existing mouse models of obesity, such as in IRS-1/GIPR and Kir6.2/GIPR double knockout mice that show increased levels of GIP, adiposity and serum glucose22,23. GIP elevation in STAT mice provides a mechanism for the observed adiposity increase24,25, but also could be secondary to the metabolic changes. There were no significant differences for fasting insulin-like growth factor (IGF)-I, insulin, peptide YY, leptin, or ghrelin levels between control and STAT mice (Supplementary Fig. 4). Glucose tolerance tests performed during week 6 of the experiment showed a trend towards hyperglycaemia in STAT mice (Supplementary Fig. 5).
STAT does not alter overall gut microbial census
To determine whether the STAT exposure leading to these metabolic changes affected the GI tract microbiome, microbial DNA extracted from faecal and caecal samples collected from the mice during the week of euthanasia or at necropsy, respectively, were studied. DNA concentrations measured from both caecal (77.62 ± 33.51 ng μl−1) and faecal (23.79 ± 14.41 ng μl−1) samples were not significantly different between control (n = 10) and STAT mice (n = 10 per group). The census in the STAT and control mice, determined through quantitative PCR using 338F/518R universal primers (Supplementary Table 1), showed no significant differences in bacterial counts or fungal census among the STAT and control groups (Fig. 3a). These data indicated that STAT exposure did not lead to substantial changes in the overall microbial census, and next led to us to conduct an assessment of the composition of the populations.
Figure 3. Changes in the faecal gut microbiome after 50 days of STAT.

a, There were no significant differences in microbial census between the STAT and control groups (n = 10 mice per group) evaluated by qPCR with universal primers for 16S rRNA and internal transcribed spacer (ITS). b, By 454-pyrosequencing, Firmicutes were shown to be increased in the STAT mice at multiple taxonomic levels. (Controls n = 10, penicillin n = 9, vancomycin n = 10, penicillin plus vancomycin n = 8, chlortetracycline n = 10; *P < 0.05.) Data are presented as mean ± s.e.m. c, Heat map of specimens showing relative abundance of bacteria present at >1% at the family taxonomic level. Hierarchical clustering based on Euclidean distance identified nonrandom branch distributions of control and STAT mice (P < 0.05). Lachno., Lachnospiraceae; Porphyr., Porphyromonadaceae.
STAT alters the composition of intestinal microbiota
To assess microbial populations in the STAT and control microbiomes, we analysed the relative distribution of taxonomic groups based on 16S rRNA v3 region sequence data. The extracted DNA was subjected to 454 pyrosequencing, yielding 555,233 readable sequences (5,784 ± 676 sequences per sample with mean length 188 ± 3 bp). The sequences were analysed at multiple (phylum to genus) taxonomic levels (Supplementary Fig. 6 and Supplementary Table 2). In both faecal and caecal samples, the ratio of Firmicutes to Bacteria was significantly elevated in the STAT mice compared to controls (Fig. 3b and Supplementary Fig. 7). Weighted Unifrac analysis of the dominant taxa (present in >1% of the total population) showed a nonrandom clustering of STAT and control mice (P < 0.05) (Fig. 3c). Importantly, deep branching was identified, with the mean weight of mice on the two major branch points on the heat map being significantly (P < 0.05) different (21.4 ± 3.1% (point A) versus 23.0 ± 2.8% (point B); Fig. 3c). A major contributor to the observed differences is increased Lachnospiraceae representation in the STAT mice. Minor taxonomic groups may have roles in the development of metabolic phenotypes, and STAT-associated increases in several minor taxa are consistent with this possibility (Supplementary Table 3). Additional rarefaction curves and Unifrac analyses, quantile plots, PCoA representations, and heat maps generated using non-Euclidean distance metrics (Supplementary Figs 8–11 and http://www.med.nyu.edu/medicine/labs/blaserlab/PDFs/Cho-et-al-Online-Figures.pdf) demonstrated consistent shifts within the microbial populations in the STAT-exposed groups. Although STAT did not change the overall bacterial census, even the minimal antibiotic doses caused shifts in taxonomic composition, such as the Lachnospiraceae bloom. We noted an increase in the relative concentrations of Firmicutes compared to Bacteroidetes in the STAT mice compared to controls, which accompanied the observed increases in adiposity. This observation extends previous findings26,27 of relative increases in the Firmicute population in ob/ob mice that are genetically prone to obesity. However, observations at such high taxonomic strata may not sufficiently describe the changes associated with obesity28,29; variations in DNA extraction efficiency and PCR-based sequencing of complex, heterogeneous microbial communities may bias census estimates of specific bacterial taxa. Furthermore, although the overall phenotypes (increased adiposity and hepatic lipogenesis) are consistent in the STAT groups, the intermediate steps may be more host- and treatment-specific. Our findings indicate that specific STAT exposures can be used as probes of microbiome structure and function.
STAT exposure alters gut microbiome SCFA metabolism
Because of the central role of short-chain fatty acid (SCFA) synthesis in colonic metabolism30,31, we examined the effect of STAT exposure on the gene counts of prokaryotic genes butyryl coA transferase (BCoAT) and formyltetrahydrofolate synthetase (FTHFS) that are involved in butyrate and acetate synthesis, respectively (Supplementary Fig. 12). Quantitative PCRs (qPCRs) for total bacteria, and degenerate qPCRs for BCoAT and FTHFS, were performed on caecal specimens in control and STAT mice. At 3 weeks, there were significant decreases in BCoAT gene copy numbers in the penicillin plus vancomycin, chlortetracycline, and aggregate groups. By 6 weeks, BCoAT copy numbers had increased in all the groups compared to the 3-week values, but there was no longer an aggregate difference. For FTHFS overall, there were no significant differences between control and STAT mice overall at 3 or 6 weeks, although there was variation within the antibiotic groups (Fig. 4a). Exploring the inter-antibiotic differences further, we noted that there were several patterns in the FTHFS qPCR with different melting curve peaks (Supplementary Fig. 13), indicating differences in the microbial population. In total, these results provide evidence that STAT treatment is dynamically affecting composition of genes related to SCFAs, probably in antibiotic-specific ways, but with overall conserved effects. Both BCoAT and FTHFS have a role in metabolism of carbohydrates into SCFAs32 and have been used to assess the functional characteristics of complex communities33; the observed changes in gene copy numbers in young STAT mice relative to controls as well as changes at different time points provide evidence that the STAT colonic microbiome alters SCFA metabolism. Our finding that the copy numbers of these two genes increase during growth (between 3 and 6 weeks) in both control and STAT mice (Fig. 4) suggests a greater dependence on SCFA synthesis pathways with maturation. Relative differences in the extent of these changes and in the melting curve peak populations may reflect changes in microbial populations. Furthermore, gene copy number (Fig. 4) in conjunction with melting curve peak patterns (Supplementary Fig. 13) may be useful for studying community genotypes of metabolic potential.
Figure 4. Caecal SCFA production after STAT exposure.

a, Quantitative PCR was performed for butyryl CoA transferase (BCoAT) and formyltetrahydrofolate synthetase (FTHFS) at experiment weeks 3 and 6 on STAT and control groups (n = 10 mice per group). At 3 weeks, BCoAT was diminished in two STAT groups and the aggregate group, a difference that persisted only in the chlortetracycline group. FTHFS copies do not show a consistent pattern. *P < 0.05, **P < 0.01, ***P < 0.001 comparing STAT to controls; hash symbol indicates significant difference between 3 and 6 weeks. b, SCFA concentration analysed by gas chromatography (GC) shows increases in SCFAs in each of the STAT groups compared to controls. c, The ratio of butyrate relative to acetate is significantly higher in the STAT mice than controls. Data are presented as mean ± s.e.m.
Direct measurements of SCFAs in the caecal contents of control and STAT mice demonstrate substantial increases in acetate, propionate and butyrate in all STAT groups (Fig. 4b); ratios of butyrate to acetate are also significantly altered by STAT exposure (Fig. 4c). These findings provide evidence that STAT exposure perturbs not only the composition of the GI microbiome but also the metabolic capabilities of the microbiome, specifically with respect to SCFAs. Increased SCFA concentrations and butyrate/acetate levels provide mechanisms for the STAT-induced adiposity phenotypes. SCFAs directly provide energy to colonocytes, and absorption into the portal circulation stimulates adipogenesis30,34. Metabolic cage experiments examining metabolic balance show no significant difference in caloric intake between control and STAT mice but lower caloric output in faecal pellets from STAT mice (Supplementary Fig. 14), providing evidence for selection for microbiota that can extract calories from otherwise indigestible constituents.
STAT alters hepatic metabolism of fatty acids and lipids
In confirmatory experiments using the identical STAT penicillin protocol, liver tissue was collected from both control and STAT mice. Standard histological analysis and triglyceride measurements showed no significant differences between control and STAT mice (Supplementary Fig. 15a, b). There were no significant differences in 16S rRNA gene counts, as detected by qPCR, from the liver specimens, providing evidence that bacterial translocation is not altered in the STAT mice (Supplementary Fig. 15c). Microarray analyses surveyed for differences in >45,000 genes, identifying 466 that by t-test or PTM were significantly up- or downregulated (397 significantly differed from controls by both tests) in the STAT compared to control mice. Focusing on pathways related to fatty acid metabolism and lipid metabolic processes, 22 and 47 genes, respectively, were differentially expressed between STAT and control mice (Fig. 5a). When specific genes were mapped to murine hepatic metabolic pathways, there were consistent changes in the same direction for several pathways, including upregulation in pathways related to lipogenesis and triglyceride synthesis (Fig. 5b). The changes in gene expression observed by microarray analyses were extended by qPCR assays of the same genes (Supplementary Fig. 16). These microarray and qPCR findings demonstrate substantial changes in the regulation of hepatic lipid, cholesterol and triglyceride metabolism that result from STAT-induced intraluminal intestinal changes.
Figure 5. Differentially regulated genes related to hepatic lipogenesis, identified through microarray and quantitative PCR analyses.

a, Microarray analysis of liver specimens surveyed for differences in >45,000 genes; 397 genes were significantly up- or downregulated. Heat maps generated by gene set enrichment analysis (GSEA)43 identify differences between the STAT and control mice (n = 6 mice per group), including pathways related to fatty acid metabolism and lipid metabolic processes. b, Mapping of metabolic genes detected by microarray onto specific pathways, including those related to lipogenesis and triglyceride synthesis, show consistent changes with STAT. Data are presented as mean ± s.e.m. White bars, controls; black bars, STAT; *P < 0.05, **P < 0.01. TG, triglycerides; VLDL, very low density lipoproteins.
Increased STAT adiposity is not metabolically altered
In the same confirmatory experiments (Supplementary Fig. 3), visceral adipose tissue dissected from control and STAT (penicillin) mice had no significant differences in adipocyte counts (Supplementary Fig. 17a, c) or in immunohistochemical staining for CD68+ macrophages (Supplementary Fig. 17b, d). These findings were extended by Gapdh-normalized (Supplementary Fig. 17e) metabolic gene qPCR analyses. There were no significant differences in leptin, adiponectin, resistin, sterol regulatory element binding transcription factor 1 (Srebf1, also called SREBP1c), peroxisome proliferator activated receptor γ (Pparg, also called PPARγ2), and fatty acid synthase (Fasn, also called FAS) levels between the control and STAT groups (Supplementary Fig. 17f). These findings provide evidence that STAT adipose tissue shows no substantial physiological difference compared to controls in cell density, local inflammation, or metabolic potential as demonstrated by quantitative PCR. Increased adiposity seems to be a downstream phenomenon primarily mediated by changes in the gut and liver.
Discussion
By developing a model to assess adiposity, we show that each of the several STAT approaches tested affects the adiposity of post-weaning C57BL/6J mice. Similarly, there was a consistent early change in bone development. Particularly in the dynamic phases of growth in young animals, STAT alterations of the microbiome may affect pluripotent cells that can become osteoblasts, adipocytes, or myocytes. Although the STAT model does not precisely replicate the weight gain observed in farm animals, possibly due to differences in the husbandry of laboratory mice in a clean environment versus animals raised on a farm, the broad effects of exposure demonstrated in this model provide evidence that altering the microbiome may have substantial consequences. Such changes in early-life body composition may be due to altered host responses35,36 and/or shifts in the metabolic characteristics of the gut microbiome. We postulate that the STAT exposures selected for microbiota with increased metabolic activity that were able to extract a higher proportion of calories from dietary complex carbohydrates that were relatively indigestible in the control mice. The increased SCFA concentrations are the metabolic products of this activity, which then may be delivered in increased quantities through the portal circulation to the liver, enabling enhanced lipogenesis (Supplementary Fig. 18). Enhanced caloric absorption has been implicated as a mechanism for increased weight gain in other murine obesity models12. The observed increase in adiposity is similar across several different STAT exposures, despite the different antibiotics used. This is consistent with the observation that a wide range of FDA-approved antibacterial agents are used for effective growth promotion in the agricultural industry6,7,37. Our studies of the intestine, liver and adipose tissue demonstrate that the active effect of the antibiotics is on the microbiota affecting the downstream liver; mechanistically, the adipose tissue is passively accepting the increased lipid load produced from more proximal activities.
Why might STAT exposures have such substantial effects on recipient animals? We propose that STAT represents a compounded perturbation, as described previously38, that has more serious consequences for long-term alterations of community state, possibly generating a different assemblage of species. The STAT-induced increase in SCFA production is a change in substrate availability that is a characteristic39 of ecological disturbance. Such overharvesting, at a developmentally sensitive time, may push the ecosystem beyond the normative recovery that usually follows infrequent disturbances38. In complex, co-evolved ecosystems, the intricate interactions among participants conform to equilibria40 that promote the robustness of a community41. However, when such equilibria are substantially perturbed, changes in host health and disease may result42. Other causative mechanisms for the changes in host phenotype could reflect that maturation of the gut microbiome is temporally altered by STAT exposure or that host responses are altered by microbiome changes. Ultimately, the interaction between the microbiome’s metabolic capabilities and numerous host reactive cells could account for the observed changes, including the increase in GIP. In our study, we confirm in a tractable experimental model the decades-long observations in farm animals that STAT exposure changes host development, and we demonstrate specific techniques useful for studying the metabolic effects of microbiome manipulation. Our study also indicates the possibility that modulation of the infant human gut microbiome by antibiotics could have long-term metabolic consequences affecting adiposity and bone development.
METHODS SUMMARY
Female C57BL/6J mice were given penicillin, vancomycin, penicillin plus vancomycin, or chlortetracycline (1 μg antibiotic per g body weight) via drinking water, or no antibiotics (control). Body weight was serially measured and body composition determined using dual energy X-ray absorptiometry (DEXA). At death, blood, caecal contents, liver and visceral adipose tissue were collected, and serum hormones measured. DNA was extracted from caecal contents and faecal pellets, and 16S rRNA gene v3 regions were barcoded and sequenced, using 454-FLX Titanium chemistry. Quality-filtered sequences were processed through the QIIME pipeline and analysed in the R statistical environment. Quantitative PCR assessed taxa and metabolic genes of interest, and expression profiling of hepatic RNA was performed by microarray.
Supplementary Material
Acknowledgments
This work was supported in part with grants from the NIH (T-RO1-DK090989, 1UL1-RR029893, UL1-TR000038), the Diane Belfer Program in Human Microbial Ecology, the Philip and Janice Levin Foundation, the Michael Saperstein Fellowship, and institutional funds provided by the J. Craig Venter Institute, and the NYU Genome Technology Center. We thank N. Javitt for advice and J. Chung for technical assistance.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions I.C. and M.J.B. designed the study; I.C., L.C., S.Y., Z.G., D.M., I.T. and K.R. performed experiments; B.A.M. and K.L. performed sequencing and sequencing analysis; J.Z. performed microarray analyses; I.C. and H.L. performed statistical interpretation and analyses; A.V.A. performed bioinformatics analyses and interpretation; I.C. and M.J.B. took primary responsibility for writing the manuscript.
Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.McCaig LF, Hughes JM. Trends in antimicrobial drug prescribing among office-based physicians in the United States. J Am Med Assoc. 1995;273:214–219. [PubMed] [Google Scholar]
- 2.Kozyrskyj AL, Ernst P, Becker AB. Increased risk of childhood asthma from antibiotic use in early life. Chest. 2007;131:1753–1759. doi: 10.1378/chest.06-3008. [DOI] [PubMed] [Google Scholar]
- 3.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nature Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dethlefsen L, Relman DA. Microbes and Health Sackler Colloquium: Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2010;108:4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manichanh C, et al. Reshaping the gut microbiome with bacterial transplantation and antibiotic intake. Genome Res. 2010;20:1411–1419. doi: 10.1101/gr.107987.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butaye P, Devriese LA, Haesebrouck F. Antimicrobial growth promoters used in animal feed: effects of less well known antibiotics on gram-positive bacteria. Clin Microbiol Rev. 2003;16:175–188. doi: 10.1128/CMR.16.2.175-188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jukes TH. Antibiotics in feeds. Science. 1979;204:8. doi: 10.1126/science.204.4388.8. [DOI] [PubMed] [Google Scholar]; Ozawa E. Studies on growth promotion by antibiotics. J Antibiot. 1955;8:205–214. [PubMed] [Google Scholar]
- 8.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 9.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansotia T, Drucker DJ. GIP and GLP-1 as incretin hormones: lessons from single and double incretin receptor knockout mice. Regul Pept. 2005;128:125–134. doi: 10.1016/j.regpep.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 11.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 13.Reikvam DH, et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS ONE. 2011;6:e17996. doi: 10.1371/journal.pone.0017996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson CJ, Young VB. Antibiotic administration alters the community structure of the gastrointestinal micobiota. Gut Microbes. 2010;1:279–284. doi: 10.4161/gmic.1.4.12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wlodarska M, et al. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect Immun. 2011;79:1536–1545. doi: 10.1128/IAI.01104-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCracken VJ, Simpson JM, Mackie RI, Gaskins HR. Molecular ecological analysis of dietary and antibiotic-induced alterations of the mouse intestinal microbiota. J Nutr. 2001;131:1862–1870. doi: 10.1093/jn/131.6.1862. [DOI] [PubMed] [Google Scholar]
- 17.Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 18.Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nature Rev Microbiol. 2011;9:279–290. doi: 10.1038/nrmicro2540. [DOI] [PubMed] [Google Scholar]
- 19.Ley RE, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buffa R, et al. Identification of the intestinal cell storing gastric inhibitory peptide. Histochemistry. 1975;43:249–255. doi: 10.1007/BF00499706. [DOI] [PubMed] [Google Scholar]
- 21.Miyawaki K, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nature Med. 2002;8:738–742. doi: 10.1038/nm727. [DOI] [PubMed] [Google Scholar]
- 22.Tsukiyama K, et al. Gastric inhibitory polypeptide is the major insulinotropic factor in KATP null mice. Eur J Endocrinol. 2004;151:407–412. doi: 10.1530/eje.0.1510407. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H, et al. Gastric inhibitory polypeptide modulates adiposity and fat oxidation under diminished insulin action. Biochem Biophys Res Commun. 2005;335:937–942. doi: 10.1016/j.bbrc.2005.07.164. [DOI] [PubMed] [Google Scholar]
- 24.Yip RG, Boylan MO, Kieffer TJ, Wolfe MM. Functional GIP receptors are present on adipocytes. Endocrinology. 1998;139:4004–4007. doi: 10.1210/endo.139.9.6288. [DOI] [PubMed] [Google Scholar]
- 25.Yamada Y, Seino Y. Physiology of GIP—a lesson from GIP receptor knockout mice. Horm Metab Res. 2004;36:771–774. doi: 10.1055/s-2004-826162. [DOI] [PubMed] [Google Scholar]
- 26.Ley RE, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy EF, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut. 2010;59:1635–1642. doi: 10.1136/gut.2010.215665. [DOI] [PubMed] [Google Scholar]
- 29.Fleissner CK, et al. Absence of intestinal microbiota does not protect mice from diet-induced obesity. Br J Nutr. 2010;104:919–929. doi: 10.1017/S0007114510001303. [DOI] [PubMed] [Google Scholar]
- 30.Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40:235–243. doi: 10.1097/00004836-200603000-00015. [DOI] [PubMed] [Google Scholar]
- 31.Hong YH, et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology. 2005;146:5092–5099. doi: 10.1210/en.2005-0545. [DOI] [PubMed] [Google Scholar]
- 32.Lovell CR, Leaphart AB. Community-level analysis: key genes of CO2-reductive acetogenesis. Methods Enzymol. 2005;397:454–469. doi: 10.1016/S0076-6879(05)97028-6. [DOI] [PubMed] [Google Scholar]
- 33.Henderson G, Naylor GE, Leahy SC, Janssen PH. Presence of novel, potentially homoacetogenic bacteria in the rumen as determined by analysis of formyltetrahydrofolate synthetase sequences from ruminants. Appl Environ Microbiol. 2010;76:2058–2066. doi: 10.1128/AEM.02580-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990;70:567–590. doi: 10.1152/physrev.1990.70.2.567. [DOI] [PubMed] [Google Scholar]
- 35.Backhed F, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 37.Jukes TH. Antibiotics in animal feeds. N Engl J Med. 1970;282:49–50. [PubMed] [Google Scholar]
- 38.Paine RT, Tegner MJ, Johnson EA. Compounded perturbations yield ecological surprises. Ecosystems. 1998;1:535–545. [Google Scholar]
- 39.Pickett ST, White PS. The Ecology of Natural Disturbance and Patch Dynamics. Academic; 1985. [Google Scholar]
- 40.Blaser MJ, Kirschner D. The equilibria that allow bacterial persistence in human hosts. Nature. 2007;449:843–849. doi: 10.1038/nature06198. [DOI] [PubMed] [Google Scholar]
- 41.Sole RV, Montoya JM. Complexity and fragility in ecological networks. Proc R Soc Lond B. 2001;268:2039–2045. doi: 10.1098/rspb.2001.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nature Rev Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.