

B cells engineer their genomes to generate the diversity of antibodies required for the immune response. Recent discoveries suggest promiscuous, genome-wide targeting of the B-cell mutator enzyme activation-induced deaminase (AID). This review discusses the mechanisms of AID substrate selection and action throughout the genome and the regulatory elements that prevent AID from inducing genomic instability and oncogenesis.
Keywords: activation-induced deaminase, DNA repair, transcription, somatic mutations
Abstract
The mechanisms by which B cells somatically engineer their genomes to generate the vast diversity of antibodies required to challenge the nearly infinite number of antigens that immune systems encounter are of tremendous clinical and academic interest. The DNA cytidine deaminase activation-induced deaminase (AID) catalyzes two of these mechanisms: class switch recombination (CSR) and somatic hypermutation (SHM). Recent discoveries indicate a significant promiscuous targeting of this B-cell mutator enzyme genome-wide. Here we discuss the various regulatory elements that control AID activity and prevent AID from inducing genomic instability and thereby initiating oncogenesis.
The adaptive immune system
Cellular origins
Detection of antigens by the mammalian immune system triggers a two-pronged response, originating in both the adaptive and the innate system wings. The adaptive immune response, which we consider in greater detail in this review, consists of two types of antigen-responsive cells: B and T cells. Secreting antibodies and cytokines, respectively, these antigen-responsive cells recruit macrophages, neutrophils, mast cells, basophils, eosinophils, and natural killer cells to participate in the immune response. These latter six cell types all derive from the pluripotent hematopoietic stem cell (HSC), a common precursor found in the bone marrow (for a flowchart of lymphocyte development, see Fig. 1A) that gives rise to both common lymphoid progenitor (CLP) and common myeloid progenitor (CMP) cells. CLP cells, the precursors of B and T lymphocytes, are the key antigen-responsive cells of the adaptive immune response. B cells recognize an antigen both when it is in soluble form and also when it has been processed; polypeptide epitopes are displayed on the surface of follicular dendritic cells (Chen et al. 1978; Suzuki et al. 2009), conventional dendritic cells (Huang et al. 2005), and macrophages (Koppel et al. 2005), the latter two of which derive from CMP cells and are produced during the sentinel innate immune response. Dendritic cells and macrophages travel to the site of an infection and function as phagocytic cells (macrophages) or as cells specialized in taking up antigens and presenting peptide fragments to lymphocytes for recognition (dendritic cells). Briefly, then, the HSC-derived myeloid progenitor macrophages and dendritic cells present antigens to the HSC-derived lymphoid progenitor B and T cells, allowing the immune response to proceed. CMP cells are not restricted to just phagocytic or antigen presentation roles, however, as they also encompass members of the granulocyte and mast cell lineages. Mast cells, when activated, play a role in protection from pathogens by releasing histamine and other inflammatory mediators from granular structures they possess; granulocytes, including neurophils, eosinophils, and basophils, travel to the site of infection and release antimicrobials to neutralize infectious agents. To summarize, the immune system has two responses to antigens: the innate response, which immediately secretes factors such as histamines to eliminate the perceived foreign invading species and digests the invading species into small peptides to be displayed on the surface of antigen-presenting cells, which then allow the members of the adaptive immune response (B cells and T cells) to be summoned to continue the attack on the invading species.
Figure 1.
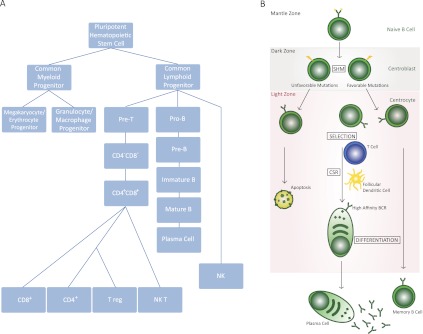
Development of B cells. (A) Development of various components of the immune system from HSCs. B cells emerge from the common lymphoid progenitor, a cell type that can differentiate to precursors of B or T cells. Pro-B cells continue their development to pre-B cells, which can then convert to mature B cells, often being converted to plasma cells in a GC-driven reaction. (B) A schematic of GC-based pathways that control B-cell development. The schematic highlights the various compartments of the GC where B cells undergo CSR and SHM following an encounter with an antigen. In addition, this schematic demonstrates that B cells are activated for CSR following interaction with T cells and follicular dendritic cells. The maturation of B cells due to SHM and CSR results from migration between the LZ and DZ of the GC, as discussed in the text.
Although B cells recognize both membrane-bound and soluble antigens, membrane-bound antigens seem to be more effective at inducing B-cell activation than soluble antigens (Batista and Neuberger 1998). Antigens are recognized through the B-cell receptor (BCR) located on the membrane surface of B cells. The BCR consists of an antigen-binding membrane immunoglobulin domain (mIg) in complex with immunoreceptor tyrosine kinase motifs (ITAMs), which mediate intracellular signal transmission (Reth 1989). When bound by antigen, the BCR cross-links and induces phosphorylation of the associated ITAMs by the Src family kinase Lyn, resulting in the formation of the signalosome complex. The signalosome initiates downstream events that lead to both B-cell activation and internalization and processing of the bound antigen. The processed antigen is then loaded onto the major histocompatibility complex (MHC) and presented by the B cell to T cells (Rock et al. 1984; DeFranco 1997). Antigen presentation in the context of MHC class II molecules generates interaction between B cells and CD4+ helper T cells that then activate B cells to produce antigen-specific antibodies. The opportunity for interaction of the antigen with an antibody is increased by B-cell localization to follicles within peripheral lymphoid tissues, including the spleen, lymph nodes, Peyer's patches, and tonsils, which filter antigens from blood, lymph, and alimentary tract contents.
B cells activated by antigens establish an immune cell interaction site within the B-cell follicle called the germinal center (GC). Lymph nodes are divided into T-cell zones and B follicles; within the B follicles, GCs exist that are divided into dark zones (DZs) and light zones (LZs) (Victora and Nussenzweig 2012). It is in the GC that B cells undergo the processes of somatic hypermutation (SHM) and antibody affinity maturation and terminally differentiate into antibody-secreting plasma cells and memory B cells.
The GC reaction
Following antigen-mediated activation, the GC response is initiated. In the GC, the B cell differentiates into either a plasma cell that secretes antibodies or a memory cell with the long-lived memory of the antigen (Harwood and Batista 2010). The GCs are the main site where SHM and class switch recombination (CSR) occur, two processes that are initiated by the incorporation of somatic mutations in the Ig variable region genes and the constant region genes, respectively (Weigert et al. 1970; Jacob et al. 1991). The activated B cells either immediately begin secreting low-affinity antibodies at extrafollicular sites of plasmablast growth and differentiation or move into the primary follicle. The primary follicle is a structure composed of recirculating IgM+IgD+ B cells (markers on B cells that have not undergone CSR) within a network of follicular dendritic cells carrying antigens on their surface, and these structures are critical for optimal selection of B cells (Klein and Dalla-Favera 2008; Victora and Nussenzweig 2012). These proliferating B cells form a mantle zone around the GC to generate the secondary follicle, and thereafter, the complete structure of the GC is observed. The various zones of the GC and the developmental stages of B cells that are present in these zones are illustrated in Figure 1B.
Live-cell imaging studies performed by several groups have demonstrated that B cells move bidirectionally between the anatomic zones of the GC: the LZ and DZ (Allen et al. 2007a,b; Hauser et al. 2007; Schwickert et al. 2007). However, none of these studies addressed the question of B-cell selection as a result of GC polarization. To address this question and also to understand how antigen-driven selection is mediated, Victora et al. (2010) developed a method of labeling and imaging GC B cells. This labeling method uses a transgenic mouse that expresses photoactivatable green fluorescent protein (PA-GFP) that can be traced with multiphoton laser-scanning microscopy as well as flow cytometry. Within these transgenic mice, all hematopoietic cells express PA-GFP, which can be photoactivated within intact lymph nodes and with microanatomical precision up to 10 μm. Using these cell populations for flow cytometry and cell sorting, Victora et al. (2010) found that B-cell division is restricted to the DZ with net B-cell migration from the DZ to the LZ. Selection for the B cells to return to the DZ for clonal expansion is controlled by T helper cells located in the GC LZ (Victora et al. 2010). These findings argue for the cyclic re-entry model of affinity maturation whereby GC B cells undergo several cycles of proliferation and hypermutation in the DZ followed by selection in the LZ of cells with improved antigen binding.
For the purpose of affinity maturation and generation of high-affinity immunoglobulin molecules during an immune response, the cells that are undergoing SHM exist in the DZ. These cells display high proliferation rates; the rates for cell cycle completion of DZ B cells range from 6 to 12 h (MacLennan 1994). Correspondingly, increased transcription of cell cycle regulators and proapoptotic factors have been reported in the DZ, while negative regulators of cell cycle and anti-apoptotic factors are down-regulated (Hu et al. 1997; Victora and Nussenzweig 2012). Changes in expression of cell surface receptors are also observed (Klein et al. 2003). Within the DZ of the GC, B cells initiate the processes of SHM and CSR. Both processes require the activity of the B-cell-expressed mutator enzyme activation-induced cytidine deaminase (AID), which is expressed by DZ B cells and deaminates deoxycytidines (dCs) to deoxyuridines (dUs) in the IgH locus to catalyze SHM and CSR (AID enzymatic activity is discussed in detail later) (Petersen-Mahrt et al. 2002; Rada et al. 2002).
CSR
There are five Ig classes in the CH region: IgM, IgD, IgG, IgE, and IgA. These classes determine the effector function and antigen response of the antibody. IgM is the first Ig class produced during B-cell development, and subsequent antigen exposure induces CSR to downstream classes. Figure 2B illustrates the various classes of constant region genes; Figure 2C details the various cytokines that can cause transcription from these alternative constant region genes. CSR is an IgH locus-specific DNA deletion and recombination event that occurs between the most upstream of the switch sequences (Sμ) and one of the downstream switch sequences (Sx) to allow the selection of a constant region gene (CH) that will then be expressed along with the rearranged VDJ exon to code for the heavy chain of the antibody expressed by that particular B cell. (The recombination and deletion reactions leading to CSR are illustrated in Fig. 3 [Honjo and Kataoka 1978].) CH gene deletion profiling studies using murine myeloma cells designated the CH gene order on the chromosome, later confirmed using chromosome walking studies, as follows: 5′-JH-(6.5 kb)-Cμ-(4.5 kb)-Cδ-(55 kb)-Cγ3-(34kb)-Cγl-(21 kb)-Cγ2b-(15 kb)-Cγ2a-(14 kb)-Cɛ-(12kb)-Cα-3′ (Fig. 2B; Shimizu et al. 1982).
Figure 2.

Essential components of CSR machinery. (A) Schematic representation of AID subdomains and various identified phosphorylation sites (in red asterisks). Note especially that (1) mutagenic analysis of AID protein has lead to identification of a nuclear localization signal (NLS) and a nuclear export signal (NES), (2) AID has a conserved cytidine deaminase motif, and (3) the phosphorylation sites S3, T27, S38, T140, and Y184 are characterized to a certain extent but require further work for complete functional characterization. (B) Structure of the Ig locus following VDJ recombination. Each constant region exon (Cx) is preceded by a cognate C sequence (Sx). The S sequences have their own transcriptional control elements that are activated by specific signaling pathways. (C) The combinations of various known cytokines that can activate S sequence transcription are shown. These cytokines often can be used to stimulate CSR in B cells grown in ex vivo cultures.
Figure 3.

Schematic of CSR intermediate steps in B cells. Transcription of various S sequences, in this case IgSμ and IgSɛ, leads to generation of secondary DNA structures that are substrates of the ssDNA cytidine deaminase AID. AID-mediated DNA deamination leads to generation of DNA DSBs that are intermediates in a DNA deletion–recombination reaction, leading to completion of CSR. Recombination between S sequences leads to deletion of intermediate DNA sequences that loop out as a circle. In this case, the IgSμ recombines with the downstream IgSɛ, with the intermediate sequence shown to be looped out, leading to IgE CSR. B cells with this configuration of their IgH locus will be able to synthesize IgE. The details of each step in this illustration are discussed in the text.
Recombination occurs at stretches of tandem repeats of short, G-rich sequences located 5′ of each CH gene (with the exception of Cδ), termed switch (S) regions (Kataoka et al. 1980; Dunnick et al. 1993). Each CH region has unique repeats; however, these regions frequently contain short motifs, such as GAGCT or TGGGG (Dunnick et al. 1980; Kataoka et al. 1981). Recombination sites can be located throughout the tandem repeats or at the 5′ end, middle, or 3′ end of the switch region (Dunnick et al. 1993). Switch recombination initiates at the Sμ region, which can join with any of the other downstream S regions using a mechanism that results in looping-out of the intervening DNA between the S regions (Iwasato et al. 1990). Figure 3 schematically illustrates the various steps of the CSR process (specifically for an isotype switch from IgM to IgE) and demonstrates the formation of an excised DNA circle.
CSR requires proliferation of B cells. Switching to IgG and IgA is not detected until the third cell cycle after stimulation, and additional divisions are necessary for IgE switching (Hodgkin et al. 1996; Hasbold et al. 1998; Deenick et al. 1999). Down-regulation of IgM and IgD occurs following cytokine stimulation (Hasbold et al. 1998). Additionally, the mode of activation (LPS or anti-CD40) can alter the relationship with division (Deenick et al. 1999). Division-linked switching falls in a Gaussian probability distribution, centered around a mean division number. Furthermore, divisions exist at which switching to IgG2b and IgG3 overlap, supporting a model of stochastic and independent switching in which no intermediate acceptor sites are used (Deenick et al. 1999). The link between division and CSR may be attributed to the requirements of the first step in the CSR mechanism, transcriptional activation through the S region (Kinoshita et al. 1998), which is known to be essential for CSR (Gu et al. 1993).
SHM
While CSR induces antibody diversity by permitting antigen recognition by different classes of antibodies, SHM incorporates point mutations in the recombined V(D)J exon of the heavy and light chain-encoding genes to enhance the affinity of the antibody to specific antigens. If the mutated V genes result in high specificity for antigens, these B cells are selectively enriched and clonally expanded. In analyses of SHM mutations in mice and humans, mutations begin to appear 100–200 base pairs (bp) downstream from the transcription start site (TSS) and gradually decline as the distance from the promoter increases. The peak of mutations is observed at 400–500 bp downstream from the TSS, restricting mutation to the variable region and not the constant region (Lebecque and Gearhart 1990; Rada and Milstein 2001). However, synthetic duplication of the promoter upstream of both the variable region and the constant region results in mutations in both the variable and constant regions at a similar frequency, linking SHM to transcription initiation (Peters and Storb 1996). Additionally, deletion of the endogenous germline promoter upstream of the Sμ region results in extremely low levels of CSR, indicating that both the presence of an intact promoter region and transcription are required for CSR to occur (Jung et al. 1993; Zhang et al. 1993; Bottaro et al. 1994). To investigate the role of the promoter in SHM and CSR, several studies were performed in which the endogenous promoter was replaced with a transgene or a heterologous promoter (Bottaro et al. 1994; Tumas-Brundage and Manser 1997). In all of these promoter replacement studies, CSR and SHM occurred, thereby indicating that specificity does not lie within the promoter. It is possible, therefore, that the primary role of the promoter actually is to drive transcription, which is necessary for the AID-containing “mutator complex” to target the S regions. However, not all heterologous promoters can support CSR and SHM. Replacement of the endogenous promoter with chicken β-actin or human EF-1 resulted in higher levels of transcription than were observed with the endogenous IgL promoter. Interestingly, unlike the chicken β-actin promoter, the EF-1 promoter did not support efficient SHM (Yang et al. 2006). These experiments suggest that additional levels of regulation may exist to control transcription and AID binding, including transcription factors that bind at the promoter. A schematic representation of how AID mutations spread during SHM is shown in Figure 4 and discussed in detail later.
Figure 4.

The DNA deamination model for AID activity in B cells. AID deaminates cytidine residues to uracils that are identified by the cellular BER pathway (UNG) or the MMR pathway (MSH2/MSH6) for repair. In this process of DNA lesion repair that depends on the DNA polymerase η, neighboring residues (A/T-based pairs) may be mutated. There are multiple possibilities how a lesion could be repaired. Based on the activity of error-prone DNA polymerases (DNA pol η), change of the neighboring A/T-based pair could be to T/A, G/C, or C/G base pairs. The nascently formed uracil residue can also be excised by the APEs (APE1/2) that lead to creation of ssDNA nicks. ssDNA nicks on both strands of S sequences can generate DNA DSBs that are intermediates during CSR.
AID
Diversification of the antibody repertoire by SHM and CSR absolutely requires the activity of the AID protein (Muramatsu et al. 2000). AID belongs to the APOBEC family of cytidine deaminases and is capable of deaminating dCs to dUs in vitro on both ssDNA substrates and ssDNA generated by the formation of RNA–DNA hybrids (Chaudhuri et al. 2003; Ramiro et al. 2003). As this protein is capable of introducing mutations into the DNA at a relatively high rate (up to 10−4 to 10−3 per base per division) (Liu and Schatz 2009), a network of transcriptional and post-transcriptional regulatory mechanisms are in place to control each step of its DNA mutator activity. The activity of AID is regulated transcriptionally (e.g., HoxC4 and NFκB) (Xu et al. 2012), post-transcriptionally (miR-155 and miR-181) (de Yebenes et al. 2008; Dorsett et al. 2008; Teng et al. 2008), and post-translationally (e.g., AID phosphorylation [Basu et al. 2005; McBride et al. 2006, 2008; Pasqualucci et al. 2006; Chatterji et al. 2007; Aoufouchi et al. 2008; Basu et al. 2008] and ubiquitination [Aoufouchi et al. 2008]) by various factors, as outlined in Figure 5 and discussed in other recently published reviews (Orthwein and Di Noia 2012; Vuong and Chaudhuri 2012). Biochemical assays demonstrate that AID is a single-strand deaminase, requiring transcription to create access to ssDNA at sites of transcription bubbles and R loops (Chaudhuri et al. 2003; Yu et al. 2003). In vitro cytidine deamination on T7 transcribed DNA templates reveals that a significant level of deamination occurs on the physiological nontemplate strand (G-rich strand). A very modest amount of deamination occurs when the inverted G-rich strand (now acting as the template strand) is transcribed (Chaudhuri et al. 2003). Strand-specific analysis of reaction products from transcription-coupled deamination reactions reveals transcription targets AID deamination in an orientation-dependent fashion, with deamination preferentially targeted to the displaced single strand of the R loop (nontemplate strand) (Chaudhuri et al. 2003). However, analysis of ung−/− msh−/− mice (where ung is the DNA base excision repair [BER] enzyme uracil DNA glycosylase [UNG], and msh are mismatch repair [MMR] factors that function in the spreading of AID-initiated mutagenesis) indicates that in the absence of UNG and MMR pathway components, there is no preference for AID targeting preferentially to the nontemplate over the template strand, suggesting that AID can deaminate both strands of DNA with equal frequency (Xue et al. 2006). The proposed mechanism by which Ung and Msh2/6 propagate AID mutations during SHM and CSR is shown in Figure 4 and discussed in much greater detail in the next section.
Figure 5.

Various steps of AID regulation. AID expression is regulated by the transcription activation of the AID gene via various pathways. Activation of the B-cell receptor, CD40 receptor, or Toll-like receptor pathways stimulates AID locus transcription. Once AID is expressed, its activity is further regulated post-translationally by various identified AID cofactors listed in the table at the bottom of the figure. The proposed function of each identified cofactor is described, although further research will reveal the mechanism of AID cofactor complex function with additional detail. These factors can control AID's stability in the cytoplasm, regulate its nucleocytoplasmic distribution, stabilize its binding to target DNA sequences, and/or activate its DNA deamination activity.
Discovery of AID as ‘B-cell mutator factor’
AID was identified by subtractive hybridization cloning of cDNA expressed in mouse intestinal CH12F3 lymphoma cells before and after stimulation for CSR (Muramatsu et al. 1999). These experiments revealed increased expression of a novel gene in the stimulated sets of CH12F3 cells, and this gene was named activation-induced cytidine deaminase (AID). The ORF of the AID cDNA encodes a protein of 198 amino acids (or 24 kDa), with the catalytic domain's amino acid sequence homologous to that of the messenger RNA (mRNA)-editing enzyme APOBEC-1, which suggests that AID may function as an RNA-editing deaminase rather than as a DNA cytidine deaminase (see Fig. 2A for AID domain structure; Muramatsu et al. 1999, 2000). AID and APOBEC-1 are located in close proximity on the same chromosomes (chromosomes 6 and 12 in mice and humans, respectively), suggesting a gene duplication event may have occurred to facilitate the evolution of AID (Conticello et al. 2005). However, recent studies have clearly established that AID functions on DNA substrates to catalyze CSR and SHM; we discuss some of these studies in later sections of this review.
To establish the requirement of AID in CSR and SHM, a germline AID-deficient mouse strain was generated in which the endogenous AID exon 2 was replaced by a neomycin resistance cassette (Muramatsu et al. 2000). AID deficiency completely blocked CSR and SHM in primary splenic B cells, although LPS-induced transcriptional activation of the relevant Ig region germline transcripts occurred at normal levels (Muramatsu et al. 2000). Detailed transcriptional analysis of AID target S sequences upstream of constant region-coding exons (switch sequences are shown as Sx in Fig. 3) revealed similar expression of all isotype S sequence transcripts in response to LPS with and without cytokines in AID−/−, AID+/+, and AID+/− splenic B cells (Muramatsu et al. 2000). In a simultaneously published and very relevant study, Revy et al. (2000) reported that patients harboring various loss-of-function mutations of the AID gene suffered from severe immune deficiency and were classified as hyper-IgM syndrome 2 (HIGM2) patients. Subsequently, while introduction of human wild-type AID-expressing constructs in AID-deficient mouse B cells rescued CSR, introduction of human HIGM2 mutations failed to rescue CSR, firmly establishing that integrity of AID function in B cells is absolutely required for CSR (Ta et al. 2003).
The mechanism by which AID initiates CSR and SHM has been intensely debated. During the last decade, several elegant biochemical and genetic experiments have led to the proposal that AID mutates DNA sequences in the B-cell genome to initiate CSR and SHM (DNA deamination model). Expression of AID in bacteria reveals a DNA cytidine residue mutator phenotype acting directly on dC/dG pairs, suggesting that AID has the ability to function as a DNA cytidine deaminase (Petersen-Mahrt et al. 2002). The observation that deficiency of the DNA BER enzyme UNG, now a broadly accepted downstream factor of AID-mutated DNA lesions, reduced AID-generated mutation rates further supported AID activity in DNA mutagenesis in bacteria (Petersen-Mahrt et al. 2002). Additional biochemical evidence that AID can mutate DNA was obtained when it was shown that either recombinant AID or AID purified from murine B cells could directly deaminate ssDNA structures generated in transcription-coupled DNA deamination reactions in in vitro conditions (Bransteitter et al. 2003, 2004; Chaudhuri et al. 2003, 2004; Dickerson et al. 2003). Based on these and other observations, Di Noia and Neuberger (2002) predicted that BER and MMR pathways are two machineries that are used to mutagenically repair AID-generated DNA lesions. Further evidence for the hypothesis that UNG identified AID-deaminated cytidine residues was the demonstration that inhibition of UNG by a bacteriophage-encoded protein shifts the pattern of IgV mutations in chicken DT40 B cells from transversion to transition dominance (Di Noia and Neuberger 2002). Mice deficient in UNG (ung−/−) display a similar phenotype and a substantial decrease in CSR efficiency (to ∼10% of ung+/+ mice), supporting the formation of a possible DNA abasic site during CSR (Rada et al. 2002). When UNG deficiency is combined with that of MMR factors (ung−/−msh2−/− or ung−/−msh6−/−), CSR is completely blocked, providing compelling evidence that AID-generated DNA lesions are targeted by components of cellular DNA repair pathways, leading to the catalysis of CSR (Rada et al. 2002; Shen et al. 2006). Other biochemical studies that substantiate AID's role as a DNA deamination enzyme include the observations that AID is found to be associated with transcriptionally active switch sequences in CSR-stimulated primary B cells via chromatin immunoprecipitation (ChIP) assays (Chaudhuri and Alt 2004) and a recent study that revealed the presence of deoxyuracil residues at regions of AID-generated mutations, thereby contributing to the already impressive volume of evidence that points toward AID as a DNA cytidine deaminase in B cells (Maul et al. 2011).
Mechanism of AID-initiated DNA mutagenesis
During SHM, AID-initiated DNA mutagenesis is proposed to occur by a two-phase DNA deamination mechanism, initiated by the action of AID. In phase 1, AID initiates mutations by specific deamination of dC to dU (Neuberger et al. 2005; Xue et al. 2006). Sequence hot spots for mutation in the V regions occur primarily within DNA sequence motifs RGYW/WRCY (where R = dA/dG, Y = dC/dT, and W = dA/dT, and the hot spot dG:dC is bold). Further analysis examining the mutability of the RGYW motif reveals that CGYW/WRCG is mutable in vitro but not in vivo. This discrepancy may be due to the presence of a dCdG dinucleotide repeat characteristic of CpG islands, which, when mutated in vivo, may be repaired by DNA repair enzymes (Rogozin and Diaz 2004). Therefore, a modification of an AID-driven SHM hot spot motif is proposed to be DGYW/WRCH (D = dA,dG,dT and H = dT,dC,dA) (Rogozin and Diaz 2004). Studies of AID-initiated DNA mutagenesis during CSR indicate that similar RGYW motifs are mutagenized by AID in switch sequences. Similar to SHM, CSR uses transcription to generate AID substrates, and the highest density of mutagenesis in S sequences is located with respect to TSSs of each isotype's S sequence. For example, at Sμ and most Sγ sequences, the switch sequence-associated mutation domains commence ∼150 nucleotides downstream from the exon 1 TSS and extend over several kilobases through the S region (Xue et al. 2006). Studies using cell line-based CSR assays have proposed that it is not the sequence of the S region that is critical for CSR, but the overlapping AID hot spot motif on the two DNA strands that is important for CSR catalysis. Indeed, the densities of these AID hot spot motifs correlate with CSR efficiency and thus the quality of the switch region (Han et al. 2011).
Spreading of AID-initiated mutations during SHM
Faithful replication of the dU–dG mispair generated in phase 1 results in a transition mutation at dC–dG pairs, where the dU residue is processed as a dT. Therefore, after replication, the resulting daughter cells contain a dU–dA pair that, after replication, is propagated as a dT–dA pair. A transition mutation results because later generations will have a dC-to-dT or dG-to-dA transition due to the dT–dA pair in place of the original dC–dG pair (Neuberger et al. 2005). The second possibility for repair leads to transversion and transition mutations in which the aberrant dU residues are detected by UNG and processed by the BER pathway (Rada et al. 2002). Transversion mutations are created when the original dC–dG pair is replaced by a dA–dT or dG–dC pair. This replacement could occur, since an abasic site is a type of noninstructional lesion where the polymerase can incorporate any base. Replication past the site requires a normal replication polymerase or a trans-lesion polymerase (Neuberger et al. 2005). A schematic representation of these pathways, which presents different possibilities for AID-initiated mutagenesis, is shown in Figure 4.
Phase 2 mutations occur when the dU/dG lesion is recognized by the Msh2/Msh6 heterodimer and subsequently processed by the MMR pathway, leading to mismatches mainly restricted to neighboring dA–dT pairs (Phung et al. 1998; Wiesendanger et al. 2000; Neuberger et al. 2005). Aberrant repair due to low-fidelity polymerase η (Rogozin et al. 2001) action at these sites creates a characteristic repair pattern in which dA residues are targeted approximately twice as often as dT residues on the top, nontranscribed (nontemplate) strand (Neuberger et al. 2005). Strand-biased spread of mutations in SHM appears to arise from the preferential targeting of the error-prone MMR pathway to top-strand Us (Unniraman and Schatz 2007). Although AID can access both strands equally, differential repair arises due to the targeting of the MMR machinery, but not the base excision machinery, to lesions on the top but not the bottom strand (Unniraman and Schatz 2007). As a result of SHM, if the mutated V genes result in high specificity for antigens, these B cells are selectively enriched and clonally expanded.
Conversion of AID lesions to DNA double-strand breaks (DSBs) during CSR
The presence of the AID-induced dU triggers BER, with dU being removed by UNG. An absence of UNG results in repair of the dU:dG lesions by replication, resulting in a dC → dT transition (and dG → dA) (Petersen-Mahrt et al. 2002; Xue et al. 2006). Consequently, AID-initiated mutations are enhanced by a deficiency in UNG. Following UNG action, an abasic site is left at the site of dU removal. This site must be repaired by the action of the BER enzyme apurinic/apyrimidinic endonuclease (APE), which cuts the phosphodiester bond of the DNA at abasic sites, resulting in single-stranded breaks (SSBs) (Christmann et al. 2003). There are three homologs of APE in mammals: APE1, APE2, and PALF/APLF/XIP1 (Hadi et al. 2002; Iles et al. 2007). APE1 is considered to be the main APE, as it is required for embryonic development (Xanthoudakis et al. 1996); however, both APE1 and APE2 are required for optimal CSR (Guikema et al. 2007). Reduced switching to all isotypes is observed from splenic B cells isolated from ape1+/−, ape2Y/−, and ape1+/−/ape2Y/− (where Ape2Y/- is null, since it is located on the X chromosome) mice in one study but was not completely observed in another study (Sabouri et al. 2009). Switch region microhomologies are not altered in these cells, suggesting that APE1/APE2 does not function in the processing of S–S junctions. However, upon analysis of Sμ, it is observed that DSBs in this region are reduced, suggesting that APE1/APE2 functions at abasic sites to initiate DSBs (Guikema et al. 2007). The deoxyribose phosphate group (dRP) is still attached to the 5′ end at the break after APE action. The lyase activity of DNA polymerase β excises this group to process the SSBs, which act as precursors to DSBs (Stavnezer and Schrader 2006) that are generated possibly due to high-density breaks on both strands of the AID substrate switch sequence (Faili et al. 2004; Wilson et al. 2005). After DSB formation, 5′ and 3′ single-strand overhangs must be processed to create appropriate ends for end-joining pathways to function. The donor and acceptor S regions frequently lack homology, suggesting homologous recombination is not the ideal mechanism of switch sequence joining (Dunnick et al. 1993). S regions are recombined using proteins of the nonhomologous end-joining pathway (NHEJ) (Ma et al. 2005) or the microhomology-dependent alternative end-joining pathway (Pan-Hammarstrom et al. 2005; Yan et al. 2007; Boboila et al. 2010). For in-depth recent literature of the NHEJ and alternative end-joining pathways during lymphocyte development, please see Boboila et al. (2012). Figure 4 illustrates possible mechanisms of conversion of AID lesions to DNA DSBs.
Cotranscriptional regulation of AID activity by RNA processing and RNA polymerase II (polII) stalling machineries
Transcripts generated at AID's DNA target sequences
In the IgH locus, there are germline promoters upstream of S sequences that drive transcription in response to specific activators and cytokines. The transcripts produced from these promoters span through a noncoding I exon and intronic S sequence and are polyadenylated downstream from the C exons. These transcripts are later spliced to fuse the I exon to the C region to produce the germline or sterile noncoding transcript. Gene targeting experiments in which I promoters or 3′ IgH enhancer elements are deleted result in the absence or reduction of germline transcription and, correspondingly, CSR efficiency, illustrating the necessity of transcription orientation for this process (Chaudhuri et al. 2007).
Bidirectional transcription occurs at S regions, with transcripts produced in both the sense and antisense orientations having similar expression patterns (Perlot et al. 2008). Interestingly, Cμ exons, located between the VHDHJH and downstream switch (Cx) exons and not targeted by AID, are transcribed only in the sense direction, suggesting that antisense transcripts may contribute to AID activity targeting (Perlot et al. 2008). However, using a transcriptional terminator cassette to provide poly(A) sites in both sense and antisense orientations (pAp cassette), it was shown that only the sense transcript through the switch regions is essential and sufficient for CSR, whereas the antisense transcript is dispensable (Haddad et al. 2011). An insertion of the cassette 168 bp downstream from the Iμ exon in mice (pApμ) has no effect on V(D)J recombination but leads to decreased germline transcription through Sμ, with a mild defect in CSR to downstream isotypes. An insertion 251 bp downstream from the Iγ3 exon (pApγ) leads to a complete and specific abrogation of CSR to IgG3 in pApγ mice due to a lack of sense transcription from Sγ3. However, an insertion 500 bp downstream from Sγ3 core repeats (Sγ3-pAp), which transcribes in the sense direction only, results in normal CSR to IgG3 despite the absence of Sγ3 antisense transcription, indicating the dispensability of antisense transcription for CSR (Haddad et al. 2011). Unlike the endogenous antisense transcription in the Ig switch sequences, the transcriptional orientation of the sense transcript is important for CSR. CSR studies of mouse B cells with inverted S region sequences without alteration of switch region promoter function illustrate the dependence of S region function on transcript orientation. Inversion of Sγ1 results in a decrease of IgG1 switching to just 25% of that observed with the Sγ1 in its wild-type configuration, indicating a clear orientation dependence, likely due to the fact that the regions where DNA breaks occur during CSR with respect to the promoter are now altered in the inverted Ig Sγ1 alleles (Shinkura et al. 2003). These results provide evidence that even if antisense RNA transcription occurs in the IgG1 switch sequences, the transcripts are not sufficient for generation of secondary DNA structures that can be identified by AID. Rather, the distance of secondary DNA structure elements formed from the sense transcript relative to the TSS of switch sequences is critical for catalysis of CSR.
During the process of transcription of S regions, stable RNA:DNA structures are formed that are resistant to RNase A, phenol extraction, SDS–proteinase K, and heating to 65°C (Daniels and Lieber 1995). Several secondary elements are formed, such as stable long R loops (>1 kb) in which a purine-rich RNA strand pairs with a pyrimidine-rich DNA strand (Yu et al. 2003; Roy et al. 2008; Ruiz et al. 2011). Other RNA secondary structures have also been shown to form at these locations, such as stem loops (Tashiro et al. 2001). These structural features are proposed to provide ssDNA substrates for AID. Consistent with this expectation, ectopically expressed AID in a yeast THO mutant strain, which is defective in mRNP biogenesis and accumulates R loops, deaminates cytosine residues on a marker DNA sequence predominantly on the nontemplate DNA strand. A high level of chromosome translocations was also observed in this yeast strain. In contrast, a yeast strain defective for R loop formation, hpr1-101, did not show increased frequency of chromosome translocations (Ruiz et al. 2011). These experiments provide evidence that cotranscriptionally generated secondary DNA structures attract AID to transcribed DNA substrates and contribute toward DNA deamination.
RNA processing pathways that regulate CSR
It has long been hypothesized that IgH locus transcription has functions beyond that of generating proper DNA substrates for RAG (a VDJ recombination-specific endonuclease) or AID activity. Even prior to the discovery of AID, it was proposed that proper RNA processing of the Ig switch region transcripts is required for optimal CSR. More recently, multiple laboratories have been investigating the influence of various RNA processing enzymes and noncoding germline transcript intermediates on CSR. These studies have relied on earlier publications that demonstrate that the integrity and processing of Ig switch transcripts at the IgG1 locus are required for targeting of CSR. Evidence includes the observation that deletion of the 5′ end of the Igγ1 switch region leads to a strong reduction in antibody production (Jung et al. 1993) and that targeted deletion of the Iγ1 switch sequence splice acceptor and donor sequences does not permit CSR to proceed (Lorenz et al. 1995). Given that the Iγ1 exon is a relatively short sequence, its function during CSR has been investigated. In a report by Hein et al. (1998), replacement of both the endogenous promoter and the noncoding I exon of the IgG1 locus with a heterologous promoter led to a loss of CSR. This CSR deficit could be rescued when a splice donor site was incorporated along with a heterologous promoter in lieu of the endogenous Igγ1 promoter and I exon. These experiments lead to the proposal that processing of the Iγ1 transcript, potentially via splicing machinery, is required for robust CSR (Stavnezer et al. 1985; Esser and Radbruch 1989; Lorenz et al. 1995; Hein et al. 1998). These studies provided compelling reasons to investigate the role of RNA processing during CSR. Recently published studies have implicated components of the RNA splicing machinery complex (CTNNBL1 and PTBP2) as being required for efficient CSR, thereby providing further cause for determining the exact role of RNA splicing in AID activity (Conticello et al. 2008; Nowak et al. 2011). The role of these factors and RNA splicing in general during SHM has not been thoroughly determined due to the lack of existing RNA splicing-deficient mouse models that can be a source of proliferating GC B cells. The generation of mouse models is crucial because deletion of CTNNBL1 in mouse cell lines does not drastically alter CSR to IgA (Han et al. 2010). Thus, with the advent of new technologies for conditionally and rapidly inactivating developmentally relevant genes, the role of RNA splicing during SHM undoubtedly will be investigated in the near future.
RNA polII stalling in the IgH locus during SHM and CSR
Although SHM prefers hot spot motifs that occur frequently in different DNA sequences at various positions in the genome, in the variable region (IgV) sequences of the IgH locus, the 5′ end-located mutations start occurring from a leader intron and continue into downstream sequences relative to the IgV TSS. Mutations upstream of the 5′ TSS are rare, and no definite 3′ end to the mutations is known, although the mutations normally range 1.5–2.0 kb from the TSS. These observations create an intriguing set of rules that require better understanding to explain the biological mechanisms that govern SHM. Prior to the discovery of AID, Storb et al. (1998) proposed “a model in which a mutator factor associates with the RNA polymerase at the promoter, travels with the polymerase during elongation, and causes mutations during polymerase pausing,” which turned out to be stunningly accurate. A study by Peters and Storb (1996) demonstrated that introduction of an Igκ transgene allele sequentially linked with a VLJL promoter and with a Cκ promoter displayed robust transcription when expressed in B cells. Furthermore, the transcripts were bimodal in distribution and distinctly initiated from the two neighboring promoters. The downstream Cκ promoter was not occluded by the upstream VLJL promoter activity, indicating that separate transcripts originate from these promoters and promote mutations. However, in the absence of Cκ promoter activation, although transcripts from the VκJκ promoter still overlapped with the Cκ region, mutations were not observed. These studies indicate that a mutator factor may join the transcription complex during its initiation phase and induce promoter-proximal mutations. Once the RNA polII enters the elongation or termination phase, the frequency of mutations rapidly declines. To characterize the promoter-proximal nature of mutagenesis in Ig genes, Storb et al. (1998) inserted a sequence containing a readily mappable restriction enzyme site in the Vκ sequence of the transgene. The experiments produced increased SHM depending on the sequence and nucleotide content of the targeted sequence. These studies point toward an increase in mutator activity and SHM at sequences that support stalling of the RNA polII complex.
These earlier studies have been supported and extended by the recent advent of deep sequencing technologies. A genomic screen (Pavri et al. 2010) of activated B cells revealed that AID is associated with sites of Spt5 occupancy, a marker of stalled RNA polII. This analysis found that Iμ showed the strongest AID and Spt5 occupancy, followed by the IgH enhancer region in the IgH locus. In addition, Spt5 and AID have high occupancy in the promoter-proximal regions of many genes outside the immunoglobulin loci (Pavri et al. 2010). Another biochemistry-based study of AID-interacting proteins that stimulate AID DNA deamination activity led to the proposal that an Spt5-interacting RNA processing complex, RNA exosome, binds to the RNApolII/Spt5-bound AID and promotes its access to both strands of the DNA. A proposed model is based on the assumption that the AID-bound RNA exosome displaces the nascent RNA bound to the RNA/DNA hybrid of the transcribed switch sequence DNA. To this end, stable knockdown of RNA exosome subunits in CH12F3 B-cell lines decreased CSR efficiency, but transient knockdowns of the RNA exosome did not affect CSR (Basu et al. 2011; Begum et al. 2012). Thus, it is likely that efficient removal of the RNA exosome complex from cells requires generation of stable knockdown of RNA exosome subunits. Furthermore, it was observed that in B cells, AID interacts with the RNA exosome complex and facilitates the RNA exosome's binding to the transcribed switch sequences in the immunoglobulin locus. Taken together, these studies provide evidence that transcription-coupled RNA processing of germline transcripts in the IgH locus may be a key regulator of AID-mediated mutagenesis in B cells. All of the studies for AID activity regulation by Spt5 or RNA exosome complex were performed with CSR-competent B cells or B-cell lines. The mechanism of interaction of the RNA polII with the RNA exosome complex and other factors such as Spt5 in regulating SHM in the IgH locus and genome-wide AID target sequences is an exciting field of investigation. Figure 6 provides a schematic representation of the proposed mechanism by which stalled RNA polII is recognized by AID and other factors and induces mutations.
Figure 6.

Step-by-step mechanism of cotranscriptional AID regulation. RNA polII, while transcribing AID's substrate sequences, stalls at various regions due to the formation of secondary DNA structures like R loops. Following stalling, AID (red) interacts with its cofactor, Spt5 (yellow). The ssDNA is stabilized by the ssDNA-binding protein complex RPA (orange). RNA exosome (multisubunit complex; red) binds the nascent transcript and excludes it from the DNA/RNA hybrid to present AID with both the template and nontemplate strand as DNA deamination substrates. In vivo, AID has been observed to mutate both template and nontemplate strands of transcribed S sequences. Details of this proposed AID deamination mechanism are discussed in the text. Other AID cofactors have been observed to regulate AID's functions during DNA deamination by various means via mechanisms that are related and unrelated to that shown above. Further experimentation is required to completely understand AID activity in vivo.
AID-generated DNA lesions can lead to chromosomal instability
Deep sequencing studies have been used to investigate Ig and non-Ig targets of AID action. Chromosome translocations between the IgH switch region and the c-myc oncogene, characteristic of the human B-cell malignancy Burkitt's lymphoma, are now identified as markers of AID aberrant activity (Adams et al. 1982; Taub et al. 1982; Leder et al. 1983). Previous studies by Ramiro et al. (2004) using mice deficient in AID on an IL6 transgenic background (AID-IL6tg) evaluated mature B-cell genomic stability. Multiple translocations of IgH/c-myc were detected in AID+/−Il6tg mice; however, none was detected in the AID−/−Il6tg mice, indicating that AID contributes to the genomic instability in B cells by mediating IL6-induced c-myc/IgH translocations in vivo (Ramiro et al. 2004). Large-scale DNA sequence analysis (Liu et al. 2008) of Peyer's patch B cells from wild-type and AID-deficient mice revealed that AID acts broadly on the genome, with significant action on genes outside of the immunoglobulin loci. Numerous genes linked to B-cell tumorigenesis are deaminated by AID, including Myc, Pim1, Pax5, Ocab, H2afx, Rhoh, and Ebf1. However, a system of checks and balances put in place by the BER machinery, MMR machinery, and error-prone DNA polymerases active in B cells determines the distribution of AID-induced mutations throughout these loci (Liu et al. 2008). In addition, the homologous recombination repair pathway may be important in maintaining genome integrity. A deficiency in the homologous repair factor XRCC2 leads to AID-dependent, widespread, highly cytotoxic breaks throughout the genome (Hasham et al. 2010). These experiments suggest that both high-fidelity (homologous recombination) and error-prone (MMR pathway) DNA repair act as two separate but interrelated levels of protection for the B-cell genome.
To uncover guidelines that control translocation mechanisms in B cells, Alt and coworkers (Chiarle et al. 2011) developed the method of high-throughput genome-wide translocation sequencing (HTGTS) in mouse B cells. This method can be used to isolate chromosomal translocations that occur between a fixed site and other locations throughout the genome. The unique endonuclease I-SceI was used by Chiarle et al. (2011) to generate DSBs within fixed sites in the c-myc or IgH loci, both frequent targets of recurrent translocations leading to B-cell oncogenesis. These fixed DSBs were used as bait to clone translocations occurring with other DSB sites induced by AID throughout the B-cell genome in cells stimulated for CSR. Both AID-dependent and AID-independent hot spots were identified. Most c-myc translocation hot spots were AID-dependent, with the Sμ, Sγ1, and Sɛ regions being the strongest hot spots. This observation is consistent with previous in vivo studies demonstrating AID's role in IgH/c-myc translocations (Ramiro et al. 2004; Pasqualucci et al. 2008). Additionally, many of the translocations identified were within the same chromosome, chromosome 15, in which the I-SceI site for c-myc was located, indicating the existence of chromosomal territories in the nucleus that restrict the range of chromosomal translocations. Although DSBs were translocated across the genome in these HTGTS studies, transcribed chromosomal regions were the preferred targets for DSBs. A global run-on sequencing (GRO-seq) technique was applied to I-SceI-expressing AIDWT and AID−/− B cells following cytokine-mediated CSR activation. These studies revealed a distinct peak in both wild-type and AID−/− cells that rested ∼300–600 bp on the sense side of active TSSs but spanned from 600 bp on the antisense side to 1 kb on the sense side. Genes containing translocation hot spots were found to have a large number of translocations within this area, indicating a relationship between active TSSs and translocations genome-wide (Chiarle et al. 2011).
In order to identify chromosomal rearrangements within primary cells, Nussenzweig and coworkers (Klein et al. 2011) developed the method of translocation capture sequencing (TC-seq). In this method, primary B cells engineered to express the I-SceI endonuclease recognition site within either the c-myc or IgH locus were infected with a retrovirus containing the I-SceI enzyme in the presence or absence of AID. Following infection, to study possible chromosomal rearrangements, genomic DNA was isolated and processed for paired-end deep sequencing. Sequencing revealed a distinct enrichment of intrachromosomal rearrangements on chromosome 15. The localization of these rearrangements around the I-SceI site supports a mechanism of DNA end resection and rejoining. In addition, Klein et al. (2011) also observed a bias for translocations to transcribed genes, with fewer translocations than expected at silent and tracely transcribed genes, while observing distinctly more rearrangements than predicted at low, medium, and highly transcribed genes. Although translocations between IgH and c-myc displayed a dependence on AID, the overall profile of rearrangements did not depend on AID. Genes enriched with polII and epigenetic markers of active transcription, including histone markers H3K4 trimethylation, H3 acetylation, and H3K36 trimethylation, were also enriched in regions harboring more frequent rearrangements. Overall, these studies by the Alt (Chiarle et al. 2011) and Nussenzweig (Klein et al. 2011) laboratories lend support to a relationship between active TSSs and genome-wide translocations. Also, these studies found that most rearrangements occurred within breaks on the same chromosome—up to 80%–90% of the breaks observed.
Chromosome conformation also plays a role in rearrangements. Chromosomes have been shown to be organized into nuclear compartments that are delineated by an open or closed chromatin conformation (Gilbert et al. 2004; Lieberman-Aiden et al. 2009; Cremer and Cremer 2010). Therefore, within open chromatin, transcriptionally active genes may be localized to a particular compartment within the cell that then facilitates genomic rearrangements (Zhang et al. 2012). In support of this idea, numerous cytogenetic studies have found the nuclear location of several translocated genes to be in close proximity to one another (Goncalves et al. 1999; Roix et al. 2003; Osborne et al. 2007). A recent study by Zhang et al. (2012) combined spatial three-dimensional (3D) nuclear organization mapping with HTGTS to investigate the relationship between genomic spatial organization and translocation frequency. Zhang et al. (2012) created HTGTS libraries in which the I-SceI recognition site had been integrated in chromosomes 2, 7, and 18 in pro-B cells that were deficient in ATM (atm−/−) to promote genomic instability. Sequencing revealed frequent translocations occurring in eight loci within five antigen receptor loci, thus indicating a strong bias for RAG1/2-mediated DSBs in translocation formation. In order to overcome this bias determined by the high frequency of RAG-mediated DSBs, cells were γ-irradiated to create DSBs throughout the genome. Hi-C analysis (chromosome capture followed by deep sequencing) was then performed on irradiated cells to map the spatial organization of the pro-B-cell genome. This analysis revealed a greater frequency of intrachromosomal interactions as compared with interchromosomal interactions, and the frequency of interaction decreased as genomic distance increased. The combined Hi-C analyses and HTGTS analyses from this study demonstrated that both the 3D organization of the genome and spatial proximity of chromosomes do influence translocation frequency.
Along similar experimental lines, Hakim et al. (2012) used a chromosome capture technique from selected loci (4C technique) to map IgH and c-myc long-range interactions in peripheral B cells. These 4C profiles revealed correlations between interacting loci and RNA polII, mRNA transcripts, or activating histone markers. Additionally, the interactome of a transcriptionally silent control gene, mycn, appeared random and did not correlate with any features associated with actively transcribed genes. Analysis of translocations in the absence of AID using AID−/− B cells revealed a nonrandom distribution of translocations, which was consistent with a preference for chromosome interaction between epigenetically accessible interacting genomic loci. As an analog to measuring AID-mediated breaks, replication protein A (RPA) ChIP-seq was employed in DNA repair protein 53bp1−/− mice that overexpressed AID. This method of sequencing found that RPA recruitment was AID-dependent and also was enhanced in the absence of 53BP1. Combining results of the various sequencing methods used in this study, Hakim et al. (2012) proposed that the frequency of DNA breaks directly accounts for the rate of translocation. Another recent study using 3D fluorescence in situ hybridization techniques measured the distance between non-Ig genes and Ig genes in the GC. In these analyses, no significant correlation was observed between the proximity of Ig genes and the level of mutations in non-Ig genes. The myc oncogene was not found to be any closer to IgH than other known AID mutated genes in these studies. Based on these observations, the investigators concluded that the proximity of the Ig locus to other genes is not a major determinant in causing AID mutations at non-Ig genes (Gramlich et al. 2012).
However, in a report published around the same time, Skok and colleagues (Rocha et al. 2012), using a TC-seq approach similar to that of Hakim et al. (2012), arrived at a new set of conclusions. In their study, Skok and colleagues (Rocha et al. 2012) reported that AID-mediated IgH translocation partners are in fact found in chromosomal domains that are in contact with the IgH locus during isotype switching. In contrast to Hakim et al. (2012), who used nonoverlapping 200-kb fixed windows, Rocha et al. (2012) partitioned the mouse genome into 200-kb-long windows centered around TSSs and also performed a domain-centered analysis. The resulting domainograms allow for an analysis at different length scales. This study is consistent with the view that had prevailed prior to the recent TC genomics strategy discussed above. In summary, these are early days in the field of translocation genomics, and fine-tuning of techniques as well as concepts will be required to completely grasp the fluid nature of chromosome dynamics and genome stability. In this fast-paced field of B-cell translocation mapping research, the next obvious step is to resolve how the density of breaks as well as chromosomal proximity are identified and regulated by the cellular machinery to prevent genomic catastrophe due to various chromosomal translocations.
Concluding remarks: AID targeting mechanisms and B-cell genome instability
Increased IgH translocations have been found in B cells deficient in the tumor suppressor proteins ATM, Nbs1, p19Arf, and p53, which have been activated for class switching (Franco et al. 2006; Ramiro et al. 2006). As IgH translocations are inhibited by the above-mentioned tumor suppressor proteins, this observation suggests that the translocations proceed through activation of DNA damage and genotoxic or oncogenic stress-induced pathways (Ramiro et al. 2006). AID has also been shown to be involved in solid tumor models by introducing chromosomal translocations at oncogenes involved in prostate cancer (TMPRSS2:ERG and TMPRSS2:ETV1) as a result of genotoxic stress (Lin et al. 2009). Deep sequencing studies, including that of Lin et al. (2009), have now been expanded by several groups that have considered the implications and mechanisms of both AID-dependent and AID-independent translocations. These studies have confirmed the dependence on AID for c-myc and IgH translocations, with the likely method of recombination occurring through resection and end-joining. Taken together, it seems likely that AID is responsible for various oncogenic chromosomal aberrations that should now be robustly identified, and these observations should be extended to clinically relevant studies.
One important question to investigate is how AID assembles its cofactors on substrates in the Ig locus and genome-wide to deaminate DNA. The answer to this question may provide a distinction between AID binding to chromosomes genome-wide and actually mutating them. When RNA polII encounters a nucleosome, it pauses with a distinctive 10-bp periodicity and then backtracks by 10–15 bp. Backtracking of the RNA polymerase following stalling can reveal a nascent RNA transcript (Adelman et al. 2005) that can be a substrate for the AID cofactor 3′–5′ exonucleolytic RNA exosome. Previous studies have found that RNA polII stalls during transcription of S regions (Rajagopal et al. 2009), and AID has been demonstrated to be associated with the paused RNA polII complex via association with pausing factor Spt5 at both Ig and non-Ig targets (Pavri et al. 2010). These observations support the model that transcription-facilitated AID accesses target sites throughout the genome and provides a mechanism for recruitment of its cofactor, RNA exosome, at certain sequences. The nature of the various transcripts due to the fate of RNA polII at AID target sequences that will generate RNA exosome substrates may provide an exciting link between noncoding RNA biogenesis and genome stability.
Taken together, the studies examined throughout this review underscore the importance of identification of the mechanisms of AID substrate selection and action genome-wide. Overexpression of AID has been shown to lead to organ-specific cancers in a transgenic mouse model; however, it is not known whether such a wide occurrence of tumorigenesis is due to AID's ability to create DNA breaks and translocations or more subtle epigenetic alterations via DNA demethylation and alteration of histone modification patterns. Notably, these studies have also revealed the link between genomic and spatial orientation of DNA DSBs and both external and internal factors of the cell that can contribute to the generation of translocations, and these are obviously areas of investigation that will be pursued in the future.
Acknowledgments
We thank Dr. Roberto Chiarle (Harvard Medical School) and Dr. Gabriel Victora (Massachusetts Institute of Technology) for critical input regarding some sections of the manuscript. We apologize to many authors whose work may not have been directly referenced in this review due to space constraints. This work was supported by grants from NIH (1DP2OD008651-01) and National Institute of Allergy and Infectious Diseases (NIAID) (1R01AI099195-01A1), Trustees of Columbia University faculty startup funds, and Irma Hirschl Foundation (to U.B.). C.K. was supported by Columbia University, Pathology Educational Fund. D.K. is supported by a grant from NIAID (F31AI098411-01A1).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.200014.112.
References
- Adams JM, Gerondakis S, Webb E, Mitchell J, Bernard O, Cory S 1982. Transcriptionally active DNA region that rearranges frequently in murine lymphoid tumors. Proc Natl Acad Sci 79: 6966–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman K, Marr MT, Werner J, Saunders A, Ni Z, Andrulis ED, Lis JT 2005. Efficient release from promoter-proximal stall sites requires transcript cleavage factor TFIIS. Mol Cell 17: 103–112 [DOI] [PubMed] [Google Scholar]
- Allen CD, Okada T, Cyster JG 2007a. Germinal-center organization and cellular dynamics. Immunity 27: 190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CD, Okada T, Tang HL, Cyster JG 2007b. Imaging of germinal center selection events during affinity maturation. Science 315: 528–531 [DOI] [PubMed] [Google Scholar]
- Aoufouchi S, Faili A, Zober C, D'Orlando O, Weller S, Weill JC, Reynaud CA 2008. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med 205: 1357–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, Alt FW 2005. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature 438: 508–511 [DOI] [PubMed] [Google Scholar]
- Basu U, Wang Y, Alt FW 2008. Evolution of phosphorylation-dependent regulation of activation-induced cytidine deaminase. Mol Cell 32: 285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, et al. 2011. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 144: 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista FD, Neuberger MS 1998. Affinity dependence of the B cell response to antigen: A threshold, a ceiling, and the importance of off-rate. Immunity 8: 751–759 [DOI] [PubMed] [Google Scholar]
- Begum NA, Stanlie A, Nakata M, Akiyama H, Honjo T 2012. The histone chaperone SPT6 is required for AID target determination through H3K4me3 regulation. J Biol Chem 287: 32415–32429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Yan C, Wesemann DR, Jankovic M, Wang JH, Manis J, Nussenzweig A, Nussenzweig M, Alt FW 2010. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med 207: 417–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Alt FW, Schwer B 2012. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol 116: 1–49 [DOI] [PubMed] [Google Scholar]
- Bottaro A, Lansford R, Xu L, Zhang J, Rothman P, Alt FW 1994. S region transcription per se promotes basal IgE class switch recombination but additional factors regulate the efficiency of the process. EMBO J 13: 665–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Scharff MD, Goodman MF 2003. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci 100: 4102–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Calabrese P, Goodman MF 2004. Biochemical analysis of hypermutational targeting by wild type and mutant activation-induced cytidine deaminase. J Biol Chem 279: 51612–51621 [DOI] [PubMed] [Google Scholar]
- Chatterji M, Unniraman S, McBride KM, Schatz DG 2007. Role of activation-induced deaminase protein kinase A phosphorylation sites in Ig gene conversion and somatic hypermutation. J Immunol 179: 5274–5280 [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Alt FW 2004. Class-switch recombination: Interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol 4: 541–552 [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 422: 726–730 [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, Alt FW 2004. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature 430: 992–998 [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, et al. 2007. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol 94: 157–214 [DOI] [PubMed] [Google Scholar]
- Chen LL, Frank AM, Adams JC, Steinman RM 1978. Distribution of horseradish peroxidase (HRP)-anti-HRP immune complexes in mouse spleen with special reference to follicular dendritic cells. J Cell Biol 79: 184–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. 2011. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147: 107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christmann M, Tomicic MT, Roos WP, Kaina B 2003. Mechanisms of human DNA repair: An update. Toxicology 193: 3–34 [DOI] [PubMed] [Google Scholar]
- Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS 2005. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol 22: 367–377 [DOI] [PubMed] [Google Scholar]
- Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS 2008. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell 31: 474–484 [DOI] [PubMed] [Google Scholar]
- Cremer T, Cremer M 2010. Chromosome territories. Cold Spring Harb Perspect Biol 2: a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GA, Lieber MR 1995. RNA:DNA complex formation upon transcription of immunoglobulin switch regions: Implications for the mechanism and regulation of class switch recombination. Nucleic Acids Res 23: 5006–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deenick EK, Hasbold J, Hodgkin PD 1999. Switching to IgG3, IgG2b, and IgA is division linked and independent, revealing a stochastic framework for describing differentiation. J Immunol 163: 4707–4714 [PubMed] [Google Scholar]
- DeFranco AL 1997. The complexity of signaling pathways activated by the BCR. Curr Opin Immunol 9: 296–308 [DOI] [PubMed] [Google Scholar]
- de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, He L, Ramiro AR 2008. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med 205: 2199–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia J, Neuberger MS 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 419: 43–48 [DOI] [PubMed] [Google Scholar]
- Dickerson SK, Market E, Besmer E, Papavasiliou FN 2003. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med 197: 1291–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, Di Virgilio M, Reina San-Martin B, Heidkamp G, Schwickert TA, et al. 2008. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 28: 630–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnick W, Rabbitts TH, Milstein C 1980. An immunoglobulin deletion mutant with implications for the heavy-chain switch and RNA splicing. Nature 286: 669–675 [DOI] [PubMed] [Google Scholar]
- Dunnick W, Hertz GZ, Scappino L, Gritzmacher C 1993. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res 21: 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser C, Radbruch A 1989. Rapid induction of transcription of unrearranged Sγ1 switch regions in activated murine B cells by interleukin 4. EMBO J 8: 483–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faili A, Aoufouchi S, Weller S, Vuillier F, Stary A, Sarasin A, Reynaud CA, Weill JC 2004. DNA polymerase η is involved in hypermutation occurring during immunoglobulin class switch recombination. J Exp Med 199: 265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, et al. 2006. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell 21: 201–214 [DOI] [PubMed] [Google Scholar]
- Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA 2004. Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell 118: 555–566 [DOI] [PubMed] [Google Scholar]
- Goncalves PP, Meireles SM, Neves P, Vale MG 1999. Ionic selectivity of the Ca2+/H+ antiport in synaptic vesicles of sheep brain cortex. Brain Res Mol Brain Res 67: 283–291 [DOI] [PubMed] [Google Scholar]
- Gramlich HS, Reisbig T, Schatz DG 2012. AID-targeting and hypermutation of non-immunoglobulin genes does not correlate with proximity to immunoglobulin genes in germinal center B cells. PLoS ONE 7: e39601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Zou YR, Rajewsky K 1993. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 73: 1155–1164 [DOI] [PubMed] [Google Scholar]
- Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, Schrader CE 2007. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med 204: 3017–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad D, Oruc Z, Puget N, Laviolette-Malirat N, Philippe M, Carrion C, Le Bert M, Khamlichi AA 2011. Sense transcription through the S region is essential for immunoglobulin class switch recombination. EMBO J 30: 1608–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadi MZ, Ginalski K, Nguyen LH, Wilson DM 3rd 2002. Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J Mol Biol 316: 853–866 [DOI] [PubMed] [Google Scholar]
- Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, et al. 2012. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484: 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Masani S, Yu K 2010. Cutting edge: CTNNBL1 is dispensable for Ig class switch recombination. J Immunol 185: 1379–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Masani S, Yu K 2011. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proc Natl Acad Sci 108: 11584–11589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood NE, Batista FD 2010. Early events in B cell activation. Annu Rev Immunol 28: 185–210 [DOI] [PubMed] [Google Scholar]
- Hasbold J, Lyons AB, Kehry MR, Hodgkin PD 1998. Cell division number regulates IgG1 and IgE switching of B cells following stimulation by CD40 ligand and IL-4. Eur J Immunol 28: 1040–1051 [DOI] [PubMed] [Google Scholar]
- Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, Wilpan RY, He Y, King BL, Mills KD 2010. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat Immunol 11: 820–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, von Andrian UH, Shlomchik MJ, Haberman AM 2007. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity 26: 655–667 [DOI] [PubMed] [Google Scholar]
- Hein K, Lorenz MG, Siebenkotten G, Petry K, Christine R, Radbruch A 1998. Processing of switch transcripts is required for targeting of antibody class switch recombination. J Exp Med 188: 2369–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin PD, Lee JH, Lyons AB 1996. B cell differentiation and isotype switching is related to division cycle number. J Exp Med 184: 277–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo T, Kataoka T 1978. Organization of immunoglobulin heavy chain genes and allelic deletion model. Proc Natl Acad Sci 75: 2140–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BT, Lee SC, Marin E, Ryan DH, Insel RA 1997. Telomerase is up-regulated in human germinal center B cells in vivo and can be re-expressed in memory B cells activated in vitro. J Immunol 159: 1068–1071 [PubMed] [Google Scholar]
- Huang NN, Han SB, Hwang IY, Kehrl JH 2005. B cells productively engage soluble antigen-pulsed dendritic cells: Visualization of live-cell dynamics of B cell-dendritic cell interactions. J Immunol 175: 7125–7134 [DOI] [PubMed] [Google Scholar]
- Iles N, Rulten S, El-Khamisy SF, Caldecott KW 2007. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol Cell Biol 27: 3793–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Shimizu A, Honjo T, Yamagishi H 1990. Circular DNA is excised by immunoglobulin class switch recombination. Cell 62: 143–149 [DOI] [PubMed] [Google Scholar]
- Jacob J, Kelsoe G, Rajewsky K, Weiss U 1991. Intraclonal generation of antibody mutants in germinal centres. Nature 354: 389–392 [DOI] [PubMed] [Google Scholar]
- Jung S, Rajewsky K, Radbruch A 1993. Shutdown of class switch recombination by deletion of a switch region control element. Science 259: 984–987 [DOI] [PubMed] [Google Scholar]
- Kataoka T, Kawakami T, Takahashi N, Honjo T 1980. Rearrangement of immunoglobulin γ 1-chain gene and mechanism for heavy-chain class switch. Proc Natl Acad Sci 77: 919–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka T, Miyata T, Honjo T 1981. Repetitive sequences in class-switch recombination regions of immunoglobulin heavy chain genes. Cell 23: 357–368 [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Tashiro J, Tomita S, Lee CG, Honjo T 1998. Target specificity of immunoglobulin class switch recombination is not determined by nucleotide sequences of S regions. Immunity 9: 849–858 [DOI] [PubMed] [Google Scholar]
- Klein U, Dalla-Favera R 2008. Germinal centres: Role in B-cell physiology and malignancy. Nat Rev Immunol 8: 22–33 [DOI] [PubMed] [Google Scholar]
- Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J Jr, Miljkovic V, Cattoretti G, Califano A, Dalla-Favera R 2003. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci 100: 2639–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. 2011. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147: 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel EA, Wieland CW, van den Berg VC, Litjens M, Florquin S, van Kooyk Y, van der Poll T, Geijtenbeek TB 2005. Specific ICAM-3 grabbing nonintegrin-related 1 (SIGNR1) expressed by marginal zone macrophages is essential for defense against pulmonary Streptococcus pneumoniae infection. Eur J Immunol 35: 2962–2969 [DOI] [PubMed] [Google Scholar]
- Lebecque SG, Gearhart PJ 1990. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5′ boundary is near the promoter, and 3′ boundary is approximately 1 kb from V(D)J gene. J Exp Med 172: 1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder P, Battey J, Lenoir G, Moulding C, Murphy W, Potter H, Stewart T, Taub R 1983. Translocations among antibody genes in human cancer. Science 222: 765–771 [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, et al. 2009. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 139: 1069–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Schatz DG 2009. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol 30: 173–181 [DOI] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG 2008. Two levels of protection for the B cell genome during somatic hypermutation. Nature 451: 841–845 [DOI] [PubMed] [Google Scholar]
- Lorenz M, Jung S, Radbruch A 1995. Switch transcripts in immunoglobulin class switching. Science 267: 1825–1828 [DOI] [PubMed] [Google Scholar]
- Ma Y, Lu H, Schwarz K, Lieber MR 2005. Repair of double-strand DNA breaks by the human nonhomologous DNA end joining pathway: The iterative processing model. Cell Cycle 4: 1193–1200 [DOI] [PubMed] [Google Scholar]
- MacLennan IC 1994. Germinal centers. Annu Rev Immunol 12: 117–139 [DOI] [PubMed] [Google Scholar]
- Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, Gramlich HS, Schatz DG, Woodgate R, Wilson DM 3rd, et al. 2011. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol 12: 70–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, Nussenzweig MC 2006. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci 103: 8798–8803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC 2008. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med 205: 2585–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem 274: 18470–18476 [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563 [DOI] [PubMed] [Google Scholar]
- Neuberger MS, Di Noia JM, Beale RC, Williams GT, Yang Z, Rada C 2005. Somatic hypermutation at A.T pairs: Polymerase error versus dUTP incorporation. Nat Rev Immunol 5: 171–178 [DOI] [PubMed] [Google Scholar]
- Nowak U, Matthews AJ, Zheng S, Chaudhuri J 2011. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol 12: 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Di Noia JM 2012. Activation induced deaminase: How much and where? Semin Immunol 24: 246–254 [DOI] [PubMed] [Google Scholar]
- Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P 2007. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol 5: e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan-Hammarstrom Q, Jones AM, Lahdesmaki A, Zhou W, Gatti RA, Hammarstrom L, Gennery AR, Ehrenstein MR 2005. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J Exp Med 201: 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R 2006. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci 103: 395–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC 3rd, Nussenzweig MC, Dalla-Favera R 2008. AID is required for germinal center-derived lymphomagenesis. Nat Genet 40: 108–112 [DOI] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, et al. 2010. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143: 122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlot T, Li G, Alt FW 2008. Antisense transcripts from immunoglobulin heavy-chain locus V(D)J and switch regions. Proc Natl Acad Sci 105: 3843–3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Storb U 1996. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity 4: 57–65 [DOI] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Harris RS, Neuberger MS 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418: 99–103 [DOI] [PubMed] [Google Scholar]
- Phung QH, Winter DB, Cranston A, Tarone RE, Bohr VA, Fishel R, Gearhart PJ 1998. Increased hypermutation at G and C nucleotides in immunoglobulin variable genes from mice deficient in the MSH2 mismatch repair protein. J Exp Med 187: 1745–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Milstein C 2001. The intrinsic hypermutability of antibody heavy and light chain genes decays exponentially. EMBO J 20: 4570–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol 12: 1748–1755 [DOI] [PubMed] [Google Scholar]
- Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R, Gearhart PJ 2009. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med 206: 1237–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC 2003. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol 4: 452–456 [DOI] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC 2004. AID is required for c-myc/IgH chromosome translocations in vivo. Cell 118: 431–438 [DOI] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, Eisenreich TR, Chen J, Dickins RA, Lowe SW, et al. 2006. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature 440: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reth M 1989. Antigen receptor tail clue. Nature 338: 383–384 [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell 102: 565–575 [DOI] [PubMed] [Google Scholar]
- Rocha PP, Micsinai M, Kim JR, Hewitt SL, Souza PP, Trimarchi T, Strino F, Parisi F, Kluger Y, Skok JA 2012. Close proximity to Igh is a contributing factor to AID-mediated translocations. Mol Cell 47: 873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Benacerraf B, Abbas AK 1984. Antigen presentation by hapten-specific B lymphocytes. I. Role of surface immunoglobulin receptors. J Exp Med 160: 1102–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, Diaz M 2004. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J Immunol 172: 3382–3384 [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA 2001. Somatic mutation hotspots correlate with DNA polymerase η error spectrum. Nat Immunol 2: 530–536 [DOI] [PubMed] [Google Scholar]
- Roix JJ, McQueen PG, Munson PJ, Parada LA, Misteli T 2003. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet 34: 287–291 [DOI] [PubMed] [Google Scholar]
- Roy D, Yu K, Lieber MR 2008. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol Cell Biol 28: 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz JF, Gomez-Gonzalez B, Aguilera A 2011. AID induces double-strand breaks at immunoglobulin switch regions and c-MYC causing chromosomal translocations in yeast THO mutants. PLoS Genet 7: e1002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabouri Z, Okazaki IM, Shinkura R, Begum N, Nagaoka H, Tsuchimoto D, Nakabeppu Y, Honjo T 2009. Apex2 is required for efficient somatic hypermutation but not for class switch recombination of immunoglobulin genes. Int Immunol 21: 947–955 [DOI] [PubMed] [Google Scholar]
- Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, Nussenzweig MC 2007. In vivo imaging of germinal centres reveals a dynamic open structure. Nature 446: 83–87 [DOI] [PubMed] [Google Scholar]
- Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U 2006. Somatic hypermutation and class switch recombination in Msh6−/−Ung−/− double-knockout mice. J Immunol 177: 5386–5392 [DOI] [PubMed] [Google Scholar]
- Shimizu A, Takahashi N, Yaoita Y, Honjo T 1982. Organization of the constant-region gene family of the mouse immunoglobulin heavy chain. Cell 28: 499–506 [DOI] [PubMed] [Google Scholar]
- Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW 2003. The influence of transcriptional orientation on endogenous switch region function. Nat Immunol 4: 435–441 [DOI] [PubMed] [Google Scholar]
- Stavnezer J, Schrader CE 2006. Mismatch repair converts AID-instigated nicks to double-strand breaks for antibody class-switch recombination. Trends Genet 22: 23–28 [DOI] [PubMed] [Google Scholar]
- Stavnezer J, Sirlin S, Abbott J 1985. Induction of immunoglobulin isotype switching in cultured I.29 B lymphoma cells. Characterization of the accompanying rearrangements of heavy chain genes. J Exp Med 161: 577–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storb U, Peters A, Klotz E, Kim N, Shen HM, Kage K, Rogerson B, Martin TE 1998. Somatic hypermutation of immunoglobulin genes is linked to transcription. Curr Top Microbiol Immunol 229: 11–19 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG 2009. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med 206: 1485–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, Nonoyama S, Tashiro J, Ikegawa M, Ito S, et al. 2003. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol 4: 843–848 [DOI] [PubMed] [Google Scholar]
- Tashiro J, Kinoshita K, Honjo T 2001. Palindromic but not G-rich sequences are targets of class switch recombination. Int Immunol 13: 495–505 [DOI] [PubMed] [Google Scholar]
- Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P 1982. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci 79: 7837–7841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN 2008. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 28: 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumas-Brundage K, Manser T 1997. The transcriptional promoter regulates hypermutation of the antibody heavy chain locus. J Exp Med 185: 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unniraman S, Schatz DG 2007. Strand-biased spreading of mutations during somatic hypermutation. Science 317: 1227–1230 [DOI] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC 2012. Germinal centers. Annu Rev Immunol 30: 429–457 [DOI] [PubMed] [Google Scholar]
- Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC 2010. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143: 592–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong BQ, Chaudhuri J 2012. Combinatorial mechanisms regulating AID-dependent DNA deamination: Interacting proteins and post-translational modifications. Semin Immunol 24: 264–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert MG, Cesari IM, Yonkovich SJ, Cohn M 1970. Variability in the λ light chain sequences of mouse antibody. Nature 228: 1045–1047 [DOI] [PubMed] [Google Scholar]
- Wiesendanger M, Kneitz B, Edelmann W, Scharff MD 2000. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J Exp Med 191: 579–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, Yasui A, Woodgate R, Gearhart PJ 2005. MSH2-MSH6 stimulates DNA polymerase η, suggesting a role for A:T mutations in antibody genes. J Exp Med 201: 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T 1996. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci 93: 8919–8923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zan H, Pone EJ, Mai T, Casali P 2012. Immunoglobulin class-switch DNA recombination: Induction, targeting and beyond. Nat Rev Immunol 12: 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue K, Rada C, Neuberger MS 2006. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2−/− ung−/− mice. J Exp Med 203: 2085–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, et al. 2007. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449: 478–482 [DOI] [PubMed] [Google Scholar]
- Yang SY, Fugmann SD, Schatz DG 2006. Control of gene conversion and somatic hypermutation by immunoglobulin promoter and enhancer sequences. J Exp Med 203: 2919–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR 2003. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4: 442–451 [DOI] [PubMed] [Google Scholar]
- Zhang J, Bottaro A, Li S, Stewart V, Alt FW 1993. A selective defect in IgG2b switching as a result of targeted mutation of the Iγ 2b promoter and exon. EMBO J 12: 3529–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J 2012. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148: 908–921 [DOI] [PMC free article] [PubMed] [Google Scholar]