

The Sir2–Sir4 sirtuin complex is required for epigenetic silencing of mating type and telomeric loci in budding yeast. However the mechanisms of allosteric regulation of sirtuin activities are unknown. Hsu et al. now report the structure of a Sir2–Sir4 complex. They show that Sir4 binding stabilizes Sir2 domains and stimulates Sir2 deacetylase activity. Their findings pinpoint specific Sir2 and Sir4 residues important for their interaction and uncover an allosteric mechanism of Sir2 activation.
Keywords: chromatin, crystallography, epigenetics, histone deacetylation, structure, transcriptional silencing, yeast
Abstract
The budding yeast Sir2 (silent information regulator 2) protein is the founding member of the sirtuin family of NAD-dependent histone/protein deacetylases. Its function in transcriptional silencing requires both the highly conserved catalytic domain and a poorly understood N-terminal regulatory domain (Sir2N). We determined the structure of Sir2 in complex with a fragment of Sir4, a component of the transcriptional silencing complex in Saccharomyces cerevisiae. The structure shows that Sir4 is anchored to Sir2N and contacts the interface between the Sir2N and the catalytic domains through a long loop. We discovered that the interaction between the Sir4 loop and the interdomain interface in Sir2 is critical for allosteric stimulation of the deacetylase activity of Sir2. These results bring to light the structure and function of the regulatory domain of Sir2, and the knowledge should be useful for understanding allosteric regulation of sirtuins in general.
Sir2 is a silent information regulator (Sir) protein universally required for transcriptional silencing in the budding yeast Saccharomyces cerevisiae. It forms a complex with Sir4 at the mating type and telomeric loci (Rusche et al. 2003) and interacts with Net1 for silencing of the rDNA locus (Shou et al. 1999; Straight et al. 1999). Biochemically, Sir2 deacetylates Lys16 of histone H4 (H4K16) in a NAD-dependent manner (Imai et al. 2000; Landry et al. 2000; Smith et al. 2000). Sir2 homologs, known as sirtuins, are found from bacteria to mammals. There are five sirtuins (Sir2 and HST1–4) in budding yeast and seven (SIRT1–7) in humans (Brachmann et al. 1995; Frye 2000). In all eukaryotes studied to date, at least some sirtuins are involved in histone deacetylation. For example, in Schizosaccharomyces pombe, Sir2 deacetylates H3K9, and human SIRT1 deacetylates H3K9, H4K16, and K26 of linker histone H1 (Shankaranarayana et al. 2003; Vaquero et al. 2004). The catalytic activities of Sir2 proteins are essential for their function in epigenetic silencing, as a hypoacetylated environment is a prerequisite for the formation of a repressive higher-order chromatin structure. Nonhistone proteins have also emerged as important substrates of sirtuins. Notably, substrates for mammalian SIRT1 include peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) (Rodgers et al. 2005), FOXO transcription factors (Brunet et al. 2004; Motta et al. 2004), and the tumor suppressor protein p53 (Luo et al. 2001; Vaziri et al. 2001). Consequently, SIRT1 has been implicated in aging, metabolism, tumorigenesis, and neurodegenerative and cardiovascular diseases (Guarente 2011).
The molecular mechanism of NAD-dependent histone/protein deacetylation by sirtuins has been studied in some depth. Deacetylation of the substrate acetyl-lysine is coupled with the cleavage of the nicotinamide group from NAD, and the acetyl group becomes covalently attached to the NAD cleavage product to form a novel compound, O-acetyl-ADP-ribose (Denu 2005; Sauve et al. 2006). Sirtuins share a conserved catalytic core, which comprises a Rossmann fold large domain and a zinc-containing small domain (Finnin et al. 2001; Min et al. 2001; Avalos et al. 2002; Zhao et al. 2003a; Sanders et al. 2010). The NAD molecule is bound in a cleft between the two domains, with the nicotinamide moiety facing toward the inside of the protein (Min et al. 2001; Zhao et al. 2003b; Avalos et al. 2004). Two possible sites for binding the nicotinamide group, termed the B and C sites, were predicted and confirmed to accommodate the nicotinamide moiety of NAD in the absence and presence of substrate acetyl-lysine, respectively (Min et al. 2001; Zhao et al. 2003b; Avalos et al. 2004). The structures of sirtuins with substrate peptides reveal that the substrate peptides are bound at the entry of the cleft primarily via hydrogen bonds between main chain atoms of the enzyme and the substrate peptides (Avalos et al. 2002; Zhao et al. 2003b). The acetyl-lysine of the substrate peptides is inserted into a hydrophobic channel, and, as predicted, the nicotinamide moiety of NAD is forced into the C pocket, where the space is more restricted (Avalos et al. 2004; Zhao et al. 2004). Based on this observation, a ground-state destabilization model of NAD-dependent protein lysine deacetylation has been proposed (Avalos et al. 2004).
An important unresolved matter concerns the regulation of sirtuin activities. Some sirtuins, including yeast Sir2, HST1, and human SIRT1, contain extra domains important for interaction with other proteins, and the protein–protein interactions significantly impact the activities of sirtuins (Frye 2000; Tanny et al. 2004; Kim et al. 2007). Here we focus our study on the interaction between Sir2 and Sir4. Sir2 is a protein of 562 residues containing a highly conserved catalytic domain at the C-terminal end and an ∼240-residue N-terminal region without recognizable sequence motifs. Genetic studies showed that the first 93 amino acids of Sir2 are dispensable for silencing function (Cockell et al. 2000), and the regions flanking the catalytic domain are important for directing Sir2's silencing function to specific genomic loci, most likely by interacting with Sir4 for the mating type and telomeric loci and with Net1 for the rRNA region (Cuperus et al. 2000). Sir4 is a large scaffold protein harboring binding sites for multiple factors important for transcriptional silencing, including Sir1, Sir2, Sir3, Rap1, and N-terminal tails of histones H3 and H4 (Moretti et al. 1994; Hecht et al. 1995, 1996; Triolo and Sternglanz 1996; Moazed et al. 1997; Strahl-Bolsinger et al. 1997). The Sir2-binding site is located at the C-terminal half of Sir4, within a region encompassing residues 745–1172 (Moazed et al. 1997; Tanny et al. 2004). Interestingly, Sir4 binding significantly stimulated the deacetylase activity of Sir2 in vitro, whereas no such effect was observed with the binding of Net1 (Tanny et al. 2004). The stimulation of Sir2 activity appears to be through an allosteric mechanism, as interaction with Sir4 involves regions outside of the conserved catalytic domain of Sir2 (Cockell et al. 2000). An analogous phenomenon is also observed with mammalian SIRT1—its interaction with AROS enhances its NAD-dependent deacetylase activity (Kim et al. 2007). Modulation of sirtuin activities can have profound biological effects, yet the molecular mechanisms of allosteric regulation of sirtuin activities remain unknown.
To understand the structural basis for Sir2–Sir4 interaction and the molecular mechanism of stimulation of the histone deacetylase activity of Sir2, we determined the structure of a Sir2–Sir4 complex. The cocrystal structure pinpointed specific Sir2 and Sir4 residues important for their interaction and uncovered an allosteric mechanism of Sir2 activation.
Results
Overall structure
By systematic deletion and pull-down experiments, we narrowed down the Sir2 interaction domain (SID) in Sir4 and cocrystallized a fragment encompassing residues 737–893 (Sir4SID) in the presence of NAD, with a large fragment of Sir2 lacking only 86 nonfunctional residues at the N terminus (Supplemental Fig. S1). The structure was solved by the selenomethionyl (SeMet) multiwavelength anomalous dispersion (MAD) method, and the final 2.9-Å model was refined against a data set collected from a C543S mutant of the Sir2 fragment (Table 1). The structure shows that Sir2 consists of a C-terminal catalytic domain (Sir2C, amino acids 237–555) and a helical N-terminal domain (Sir2N, amino acids 101–236), arranged in a horseshoe shape (Fig. 1A,B). The two domains are independently folded. The catalytic domain structure is highly conserved among sirtuins from archaea to mammals (Sanders et al. 2010); it consists of a Rossmann fold domain and a smaller zinc-containing regulatory domain (Fig. 1A). The NAD-binding site is located in the cleft between the two domains. An ADP ribose (ADPr) molecule is bound in this cleft, even though NAD was used for cocrystallization. This is a common phenomenon in sirtuin–NAD complexes, probably due to hydrolysis of the nicotinamide group of NAD during crystallization. Five α helices form the compact core of Sir2N. A three-residue segment N-terminal to α1 (amino acids 119–121) and a 21-residue loop connecting α4 and α5 (amino acids 189–209) are disordered and located next to each other on the Sir2N surface distal from the catalytic domain (Fig. 1A). Apart from covalent joining, the two Sir2 domains, separated between residues 236 and 237, only have limited contact (Fig. 1B). A key residue involved in interdomain interactions is Arg235, which makes a salt bridge with Asp506, a hydrogen bond with the side chain of Gln492, and two hydrogen bonds with the carbonyl groups of Val490 and Glu504 (Fig. 1B). The two domains have a calculated total buried surface area of 622 Å2 (definition: twice the buried surface area on an individual component), which is small for typical protein recognition and indicates dynamic relative positioning of the two Sir2 domains.
Table 1.
Statistics of crystallographic analysis
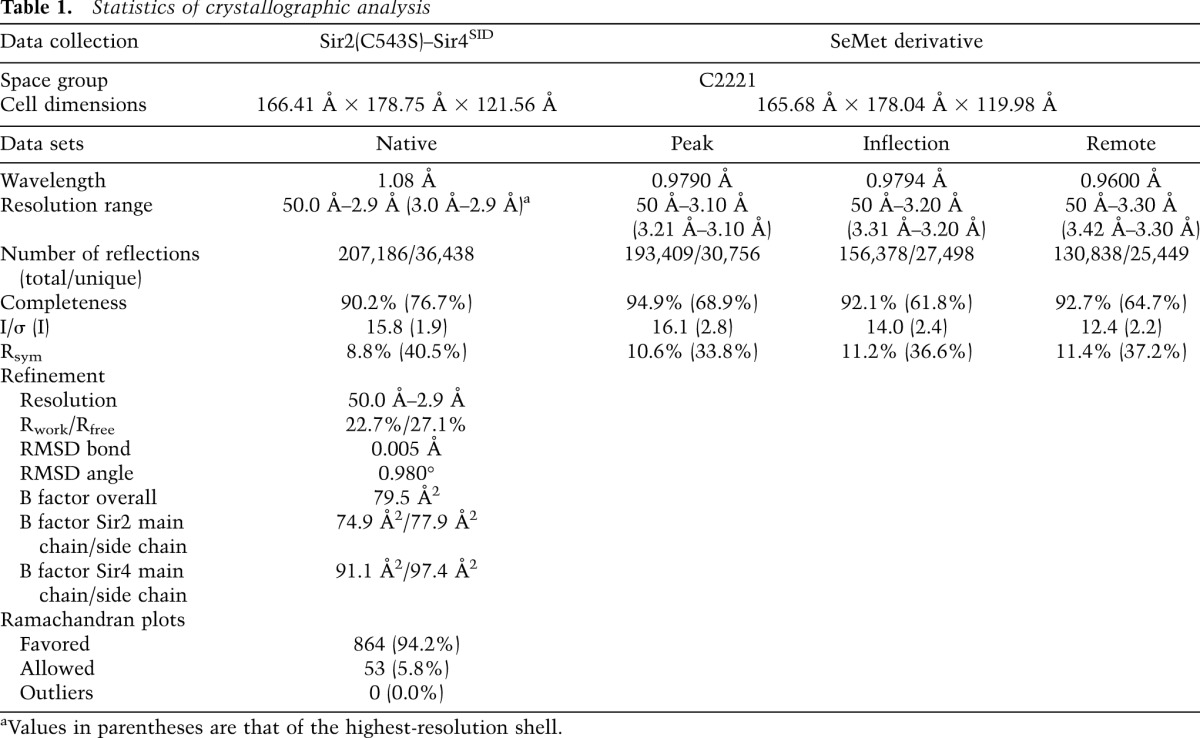
Figure 1.

Overall structure. (A) Sir2 has a helical N-terminal domain (Sir2N; cyan) and a conserved C-terminal catalytic domain (Sir2C; green). The Sir4SID is shown in magenta. An ADPr molecule bound to Sir2C is shown in a stick model, and a zinc ion is shown as a sphere. (B) Limited interface between Sir2N (cyan) and Sir2C (green) domains. Involved residues are shown in stick models, with the carbon bonds colored the same as in the ribbon model. Sir2N residues are labeled in brown, and Sir2C residues are labeled in black. (C) The structure of Sir4SID viewed from a direction opposite to that in A.
In isolation, Sir4SID does not appear to have a stable structure, as it lacks a hydrophobic core (Fig. 1C). The main structural feature is a pair of anti-parallel α helices, αA and αB, which are held together by a hydrogen bond between Asn816 of αA and Gln838 of αB, hydrophobic interactions between Leu835 of αB and Ile817 and Ile821 of αA, and weaker van der Waals contacts. Helix αA is connected to a short N-terminal segment (amino acids 743–752) via a long disordered loop (amino acids 753–804), and the C-terminal end of αB is connected to a long irregular loop, followed by a short helix (αC) and another long loop. The loose Sir4SID structure indicates that it may not have a defined structure on its own, and the observed Sir4SID conformation is stabilized by interaction with Sir2.
Structural conservation and differences
All sirtuins with known structures contain only the catalytic core, whereas yeast Sir2 and Hst1 and metazoan SIRT1s have identifiable extra domains. Comparing the structure of the catalytic core of Sir2 with that of yeast Hst2, a representative structure of eukaryotic sirtuins, shows a conserved overall arrangement of the Rossmann fold and the zinc-containing regulatory domains as well as the architecture of the active/NAD-binding site (Fig. 2A). Notably, the NAD-binding site is composed of several connected pockets. In the Sir2 structure, the ribose moiety of the ADPr molecule is situated in a pocket called the B site (Min et al. 2001; Zhao et al. 2003b), formed by conserved residues, including Phe274, Arg275, and Tyr281 located on the loop (termed the cofactor-binding loop hereafter) connecting the two domains of Sir2C and catalytically important Gln344 and His364 from the Rossmann fold domain (Fig. 2B,C). The B site is connected to the substrate acetyl-lysine-binding channel, which is formed by hydrophobic residues Phe445, Leu449, and Val476. Curiously, a methionine residue of Sir4, Met747, occupies the substrate-binding channel. When an acetyl-lysine is bound in the channel, the nicotinamide moiety of NAD is pushed into a pocket deep inside the protein called the C site, which is formed by three invariant residues, Ser266, Asn345, and Asp347. The nicotinamide group is cleaved off in the C site (Min et al. 2001; Avalos et al. 2004; Zhao et al. 2004). A back channel, suggested to be the nicotinamide escape/exchange tunnel, is also found in Sir2C (Fig. 2C; Avalos et al. 2004).
Figure 2.

The catalytic domain of Sir2. (A) Superposition of the structures of Sir2C (green) and Hst2 (PDB ID: 1SZD) (yellow). The cofactor-binding loop (magenta) from a different Hst2 structure (PDB ID: 1Q1A) is also superimposed for comparison. Three regions of major differences are circled with red dashed lines: the cofactor-binding loop (I), a Sir2-specific insertion in the zinc-binding region (II), and the very C terminus of Sir2, which has two extra β strands (III). (B) A close-up view of the substrate- and NAD-binding pocket. A methionine of Sir4SID (magenta) is sticking into the hydrophobic substrate acetyl-lysine-binding channel. Selected Sir2 residues are shown in a stick model and labeled in black. (C) A sliced view of the active site showing the substrate-binding channel, the catalytic B and C sites, and a back channel connected to the C site.
There are three main structural differences between the catalytic cores of Sir2 and Hst2 (Fig. 2A; Zhao et al. 2004). First, the cofactor-binding loop has a significantly different conformation. The loop region immediately C-terminal to β1 contains a stretch of highly conserved amino acids, forming a GAGΩSX3GIPXFRX3-4GΩΦ sequence motif, where X represents any residue, and Ω and Φ represent hydrophobic and aromatic residues, respectively (Min et al. 2001). Several residues of this motif, such as Phe274, Arg275, and Tyr281, directly interact with ADPr, linking cofactor binding with the conformation of this loop. A variety of conformations of this loop have been observed in sirtuin structures with different molecules bound in the active site (Sanders et al. 2010). Thus, the conformational variability of this cofactor-binding loop reflects different modes of NAD/ADPr binding in the active site. Second, an insertion of ∼30 residues in Sir2 following the four-cysteine zinc-coordinating motif introduces an extra helix and a long coil, changing the zinc ribbon structure found in other sirtuins into a structural motif resembling the four-cysteine coordinated zinc substructure of a PHD finger, as indicated by a search of structure similarity using the Dali server (Fig. 3A). Sir2 is crystallized as a homodimer in one asymmetric unit, and the zinc-binding region of Sir2 is involved in self-interaction. However, the physiological significance of this interaction is unclear, as similar interactions were not observed in the Sir2 structure lacking most of the N-terminal domain (Protein Data Bank [PDB] ID: 2HJH). Finally, a β hairpin at the C terminus of Sir2 forms an extended β sheet with the six parallel strands of the Rossmann fold domain (β10 and β11 in Fig. 2A).
Figure 3.

Sir2-specific zinc-binding region and Sir2 structures with or without the N-terminal domain. (A) Superposition of the zinc-binding regions of Sir2 (green for the conserved region, and red for the Sir2-specific insertion) and Hst2 (yellow). Four cysteines coordinating the binding of the zinc ion are shown as stick models (Sir2: carbon bonds are in green; Hst2: carbon bonds are in yellow), the zinc ion is shown as a gray sphere, and the coordination bonds are indicated with magenta dashed lines. (B) Superposition of Sir2 structures from the Sir2–Sir4 complex (colored as in Fig. 1A) and one lacking the N-terminal domain (PDB ID: 2HJH) (colored in salmon). ADPr and a lysine (K222) are shown in stick models. (C) Superposition of the N-terminal loop encompassing K222 (light blue), an acetylated p53 peptide (PDB ID: 1MA3) (green), and an acetylated histone H4 peptide (PDB ID: 1Q1A) (yellow) onto Sir2, with the Sir2–Sir4SID structure, in which a shortened Sir4SID segment I is shown in magenta, and Sir2 is shown in white ribbon models. The residue bound in the substrate-binding channel and one preceding and two following residues of the peptide are shown in stick models, and their carbon bonds are colored the same as the corresponding ribbon representation.
The above structural features of Sir2 in our Sir2–Sir4 complex are independent of the N-terminal domain of Sir2 or Sir4 binding, as these features are well maintained in the Sir2 structure lacking most of the N-terminal domain residues (Fig. 3B). The two structures can be superimposed with a root-mean-squared deviation (RMSD) of 0.75 Å using the Cα atoms of the C-terminal domain residues (amino acids 237–555) for alignment. The extra residues belonging to the N-terminal domain of Sir2 in the 2HJH structure form an extended loop structure, presumably due to a lack of a stabilizing core in the crystallized fragment. Curiously, Lys222 in the extended N-terminal loop is bound in the substrate-binding channel, much like authentic substrate lysine residues and Met747 of Sir4SID (Fig. 3C). This cannot occur with a folded N-terminal domain of Sir2, as Lys222 is located in the middle of a helix (Fig. 3B).
Sir2–Sir4 interaction
An analysis of the interaction between Sir2 and Sir4SID shows that a total surface area of 5433 Å2 is buried. To put the number in perspective, a survey of protein complex structures in PDB reveals an average total buried surface area of 1600 Å2 in typical protein–protein recognitions (Lo Conte et al. 1999). Hence, the large protein contact areas indicate extensive and stable interactions between Sir4SID and Sir2.
Sir4SID can be roughly divided into three segments based on their distinct contact regions on Sir2. The first Sir4SID segment (segment I) is a short stretch of amino acids (amino acids 740–752) connected to αA via a long disordered loop (amino acids 753–805). Segment I interacts with Sir2C only, and the binding mode closely resembles that of substrate peptides bound to the catalytic core of sirtuins (Avalos et al. 2002; Zhao et al. 2003b), including the binding of Met747 in the acetyl-lysine-binding channel (Figs. 2B,C, 3C). The significance of this binding is unclear, as serendipitous occupation of the substrate-binding site was observed previously, such as the binding of a methionine in Hst2 (PDB ID: 1Q14) and Lys222 from the unfolded N-terminal domain of Sir2 (Fig. 3B,C).
Segment II of Sir4SID encompasses helices αA and αB, and they make exclusive contacts with Sir2N. αA packs against the N-terminal half of Sir2's helix α5, and αB is situated in a wedged area formed by α2 and α5 of Sir2. The interactions between this region of Sir4SID and Sir2N are mostly hydrophobic, with notable exceptions of hydrogen bonds between Glu837 and Asp810 of Sir4SID and His148 and Lys222 of Sir2N, respectively (Fig. 4A).
Figure 4.

Sir2–Sir4 interactions. (A) Three distinct Sir2-binding regions of Sir4. The N-terminal tail of Sir4SID (segment I; blue) binds Sir2C, a middle portion encompassing αA and αB (segment II; yellow) binds Sir2N, and the C-terminal portion (segment III; magenta) binds the domain interface between Sir2N and Sir2C. (B) Segment III of Sir4SID (magenta) interacts with Sir2 residues important for silencing of mating type and telomeric loci. The involved residues are shown as stick models, where the Sir2 locus specificity residues are shown with carbon bonds colored yellow and labeled with black letters; the carbon bonds of Sir4 residues interacting with the Sir2 locus specificity residues are colored in magenta and indicated with magenta labels. The carbon bonds of a Sir2 eso residue, R139, are colored gray.
Segment III (amino acids 840–893) is a long loop with a short helix (αC) in the middle. A stretch of 10 residues at the N-terminal end of segment III continues from αB and makes contacts to Sir2N only. The rest of segment III snakes around the narrow “neck” of the interface between Sir2N and Sir2C, making extensive and mostly hydrophobic interactions with both Sir2 domains. Importantly, locus specificity mutants of Sir2 for the mating type and telomeric loci (Cuperus et al. 2000) are all located in the region that interacts with segment III. The mutated Sir2 residues Asp223, Cys233, Arg235, Met485, Gln492, and His502, together with the Sir4 residues involved in interaction with them are shown in Figure 4B. Interestingly, an enhancer of sir one (eso) mutant of Sir2, R139K, is also located in the segment III-interacting region (Fig. 4B; Garcia and Pillus 2002).
Sir4 binding stabilizes Sir2 domains and enhances its enzymatic activity
The limited contact area of the N-terminal and C-terminal lobes of Sir2 prompted us to reason that the two domains may not be stably positioned with each other in the absence of Sir4SID. To gain an insight into the dynamic behavior of the two Sir2 domains, we carried out a 75-nsec molecular dynamics simulation (MDS) of Sir2 alone and in complex with Sir4SID, both in the presence of an ADPr molecule. The distance between the centers of mass (COMs) of Sir2 N and C domains was chosen as an indicator of domain movement. Figure 5A shows that, in the absence of Sir4SID, the COM distance swings violently; in <5 nsec, the distance can change >0.6 nm. Figure 5B shows the superposition of a representative simulated Sir2 structure with that from the Sir2–Sir4 crystal structure. A similar MDS in the presence of Sir4SID shows that the COM distance changes more gradually and with a smaller range of variations (∼0.2 nm) in the entire 75-nsec simulation. Thus, the MDS results support the notion that the two Sir2 domains are dynamically positioned, and the binding of Sir4SID stabilizes the relative positioning of the two Sir2 domains.
Figure 5.

Sir4 binding stabilizes Sir2 domains and stimulates the deacetylase activity. (A) Plot showing changes of the distance between COMs of the N-terminal and C-terminal domains of Sir2 during a 75-nsec MDS. Black and red lines indicate MDS results obtained with the structures of the Sir2–Sir4 complex and with Sir4 removed, respectively. The distances were sampled at a 0.1-nsec time interval. (B) Different conformation of the crystal and representative MDS structures of the Sir2 N-terminal domain. The MDS structures were taken at 62.2 nsec, and the figure was generated with the C-terminal catalytic domain Sir2 aligned. The crystal structure is shown in green, the Sir2 MDS structure from the Sir2–Sir4 complex is colored yellow, and the MDS structure with Sir4 removed is shown in orange. (C) Histone deacetylase activity of Sir2 complexed with various Sir4 fragments.
We hypothesized that the stabilization of the two Sir2 domains might have a beneficial effect for its histone deacetylase activity. Indeed, a fragment of Sir4 encompassing Sir4SID (fragment I+II+III, amino acids 737–897) stimulates the histone deacetylase activity by approximately twofold (Fig. 5C). A similar degree of stimulation by Sir4SID was observed when tested on a C543S mutant of Sir2 that was used for refinement of the structure or using a Sir4SID fragment lacking segment I (II+III) (Fig. 5C). The observed degree of stimulation is somewhat smaller than the approximately fivefold stimulation reported using a much longer fragment of Sir4 (Tanny et al. 2004). To see whether the domain-stabilizing Sir4 region (segment III) is important for the observed stimulation effect, we made two Sir4SID derivatives: one missing segment III (fragment I+II), and another missing both segments I and III (fragment II). The Sir2 complexes with these fragments were prepared the same way as for the wild-type Sir2–Sir4SID complex. The result shows that both Sir4 fragments without segment III lost the ability to stimulate the Sir2 activity (Fig. 5C). In fact, the binding of these fragments reduced the Sir2 activity somewhat, and the addition of segment III can rescue the activity in a concentration-dependent manner (Supplemental Fig. S2), qualitatively in agreement with previous findings (Tanny et al. 2004). Furthermore, a MDS calculation with Sir2 and the Sir4SID fragment lacking segment III (i.e., fragment I+II) indicates large movements between the two Sir2 domains (Supplemental Fig. S3), reinforcing the notion that interdomain positioning affects the deacetylase activity of Sir2.
Discussion
It is not obvious how the binding of segment III of Sir4—namely, stabilization of the two Sir2 domains—stimulated the activity of Sir2. An examination of the Sir2 structures from MDS shows a dynamic substrate-binding channel in the absence of Sir4. Figure 6A shows that in the presence of Sir4SID, the geometry of the substrate-binding channel remains essentially the same in the crystal and MDS structures. In the MDS structure without Sir4SID bound, three major changes occurred: (1) Helix α15 is tilted toward segment III of Sir4SID, (2) the loop forming the ceiling of the substrate-binding channel (loop-sc) moves away from the lysine-binding position, and (3) helix α8 moves down toward the substrate-binding site (Fig. 6B). As a consequence, two key residues of the substrate-binding channel, Phe445 and His364, undergo major conformational changes, and Gln293 on α8, which normally is not part of the substrate-binding channel, moves in and completely blocks the substrate-binding channel (Figs. 2B, 6A,B). It should be emphasized that the MDS structure represents a transient state of the enzyme, meaning that in an ensemble of Sir2 molecules, many conformations of Sir2 can coexist, and some may be in an inactive conformation, such as that in Figure 6. The MDS results suggest that Sir2 can toggle between active and inactive conformations in the absence of Sir4, and the binding of Sir4 increases the population of Sir2 in an active conformation. The binding of segment I of Sir4SID may help stabilize the substrate-binding channel but at the same time competes with the substrate for binding to Sir2. Thus, the impact on the enzymatic activity of Sir2 upon the binding of segment I of Sir4SID is difficult to predict. On the other hand, the binding of segment III of Sir4SID does not interfere with substrate binding and stabilizes the position of α15, which should favor an active conformation of the adjacent loop-sc. The results of the enzymatic activity assays clearly indicate that the enhancement function of segment III overcomes potential inhibitory effects by segment I of Sir4SID (Fig. 5C). Therefore, the stabilization of the two Sir2 domains by Sir4SID couples helix α15 of Sir2 in a conformation favorable for productive deacetylation.
Figure 6.

Conformational difference of the substrate-binding channel in the MDS structures of Sir2 with and without Sir4SID bound. (A) Superposition of the crystal and MDS structures of the Sir2–Sir4SID complex. The MDS structure was taken at 62.2 nsec of simulation. For viewing clarity, the MDS Sir4SID structure is not shown. Sir2 from the crystal structure is colored green and that from MDS is shown in yellow. Segment I of Sir4SID is colored light blue, whereas segment III, which is shown both in ribbon and mesh representations, is colored magenta. Met747 of Sir4 and Phe445, His364, and Gln293 of Sir2 are shown in stick models. The Sir2 residues are also decorated with dot models. (B) The MDS structure of Sir2 (at 62.2 nsec) without Sir4SID bound is superimposed with the crystal structure of the Sir2–Sir4SID complex. The figure was generated from the same viewing direction as in A. The MDS Sir2 structure is colored brown. Sir2 and Sir4SID fragments from the crystal structure are colored the same as in A. The blue arrows indicate the direction of movement of the relevant structural elements between the crystal and MDS structures.
The significance of the Sir2–Sir4 complex structure is twofold. First, it provides the structural basis for Sir2–Sir4 interaction. Previous studies have loosely defined the regions in both proteins required for their interaction, and a set of Sir2 mutants affecting silencing of the mating type and telomeric loci has been identified (Cuperus et al. 2000). The structure precisely defines the Sir2 and Sir4 regions required for their interaction and directly shows that the Sir2 locus specificity mutants involve residues normally interacting with Sir4. Interestingly, an eso mutant of Sir2, which has no obvious connection to interaction with Sir4, is also located in the Sir4-interacting region of Sir2 (Garcia and Pillus 2002). It is known that establishment of a silenced chromatin domain in the mating type loci involves a cascade of protein–protein interactions, including the binding of Sir1 to the silencer-bound ORC1 and the recruitment of the Sir2–Sir4 complex by the interaction between Sir1 and Sir4 (Triolo and Sternglanz 1996; Bose et al. 2004; Hou et al. 2005; Hsu et al. 2005). Thus, a weakened or disrupted Sir2–Sir4 interaction can undermine the Sir1 function of recruiting a histone deacetylase to the targeted chromatin region, and our discovery that a Sir2 mutant affecting Sir1 function is located in the Sir4 interaction region is testimony to the network of protein–protein interaction required for transcriptional silencing in yeast.
Second, our result shows that the multidomain structure of Sir2 provides a means for allosteric regulation of the enzymatic activity by the binding of a modulator protein. Most sirtuins have no obvious extra domains beyond the conserved catalytic domain; notable exceptions are yeast Sir2 and Hst1 and human SIRT1 (Frye 2000). Hence, the structure of the Sir2–Sir4 complex serves as a first example for understanding how sirtuin activities may be regulated by protein–protein interactions. The structure shows that Sir2 has an independently folded N-terminal domain in addition to the conserved catalytic domain. The structure implicates that the two domains are not stably positioned in the absence of Sir4, and the MDS results confirm the structural prediction. Furthermore, our enzymatic activity assay shows that Sir4 binding per se is not sufficient for allosteric activation of Sir2—it requires the domain-stabilizing fragment of Sir4. MDS shows that the substrate-binding channel of Sir2 can toggle between an open and closed conformation, and Sir4 binding stabilizes the relative positioning of the two Sir2 domains and maintains a crucial helix in a productive conformation. This finding has important implications, as mammalian SIRT1 is also a multidomain NAD-dependent deacetylase, and molecular modeling predicted the presence of a four-helix allosteric domain immediately N-terminal to the Rossmann fold catalytic domain (Autiero et al. 2009) and suggested an allosteric mechanism by a small protein, AROS, which binds the N-terminal domain of SIRT1 and stimulates the deacetylase activity of SIRT1 (Kim et al. 2007). However, it should be mentioned that Sir4 only exists in S. cerevisiae and its closely related yeasts, such as Kluyveromyces lactis, and no obvious sequence homology is found between the N-terminal domain of Sir2 and the regulatory regions of SIRT1. Nevertheless, the concept that altering the spatial positioning of the regulatory domain in sirtuins can modulate its enzymatic activity may be generally applicable. It would be interesting to find out whether a similar allosteric mechanism is operating in SIRT1.
Materials and methods
Protein expression and purification
The S. cerevisiae Sir2 protein lacking N-terminal 86 amino acids was expressed as a polyhistidine-tagged protein using a pACYCDuet-1 vector. The coding sequence for the Sir2 fragment, together with an in-frame PreScission protease cleavage site at the 5′ end, was amplified by PCR and cloned between the EcoRI and SalI sites. A C543S mutant of the same Sir2 fragment, useful for reducing crystal anisotropy, was engineered by PCR. GST-tagged Sir4 fragments (Sir4SID, amino acids 737–893; I+II+III, amino acids 737–897; I+II, amino acids 737–839; II, amino acids 786–839) were expressed using a pGEX-KG vector (cloned between BamHI and SalI sites). All expression plasmids were verified by DNA sequencing.
Binary complexes of Sir2 and Sir4 fragments were obtained by coexpression in the BL21(DE3) strain of Escherichia coli. Cells with both plasmids were grown in LB medium with 100 mg/mL ampicillin and 34 mg/mL chloramphenicol. Once the cell density reached OD600 ∼0.7, protein production was induced with 0.5 mM IPTG for 16 h at 18°C. Then, cells were harvested by centrifugation and lysed by passing through an Emulsiflex C3 cell disruptor in 10 mM sodium phosphate (pH 7.5), 10 mM imidazole, and 0.25 M NaCl. The homogenate was clarified by spinning at 27,000g, and the supernatant was applied to a Ni-NTA (Qiagen) column pre-equilibrated with the lysis buffer. The His-tagged complexes were eluted with a 0–500 mM imidazole gradient, in 10 mM sodium phosphate (pH 7.5) and 0.25 M NaCl. Eluted fractions enriched with both proteins were pooled and applied to a glutathione fast flow column pre-equilibrated with 10 mM sodium phosphate (pH 7.5) and 0.25 M NaCl. After extensive washing with the equilibrating buffer, the GST tag was cleaved with the treatment of thrombin for 2 h at 22°C, followed by removal of the His tag by incubation with the PreScission protease for 16 h at 4°C. The tag-free proteins were applied to a Hi-Trap Q column pre-equilibrated with 10 mM sodium phosphate (pH 7.5) and 50 mM NaCl and eluted with a 100–400 mM NaCl gradient. The Sir2–Sir4 complexes were further purified on a Superdex 200 HR column (GE Healthcare) in a buffer containing 10 mM sodium phosphate (pH 7.5), 1 mM dithiothreitol, and 0.25 M NaCl.
Crystallization and structure determination
The purified Sir2–Sir4SID complex was concentrated to ∼20 mg/mL and was added at a 5:1 NAD:protein molar ratio, and the mixture was incubated for 30 min on ice to allow efficient formation of the Sir2–Sir4SID–NAD complex. Orthorhombic crystals were obtained at 16°C in 100 mM sodium phosphate (pH 7.5), 0.6 M sodium citrate, and 2 mM dithiothreitol by the hanging-drop vapor diffusion method. These crystals with the wild-type proteins diffracted with a high degree of anisotropy. Introducing the C543S mutation in Sir2 largely overcame the anisotropic problem, and later crystallographic analyses were all done using the C543S mutant of Sir2. To use the MAD method for phasing, SeMet-substituted proteins were expressed in a minimal medium supplemented with SeMet using the metabolic inhibition method. Two sets of three-wavelength SeMet MAD data (one to 3.6 Å and another to 3.1–3.3 Å) and one 2.9 Å native data set were all collected at the X29A beamline of the National Synchrotron Light Source (NSLS), Brookhaven National Laboratory (BNL). All data were processed using HKL2000 (Otwinowski and Minor 1997). SHELX was used to locate the Se sites, and phasing was carried out using autoSHARP (Vonrhein et al. 2007) and PHENIX (Adams et al. 2010). The MAD phased 3.6-Å map allowed unambiguous building of the model of the Sir2 catalytic domain, but assignment of side chains to Sir2 NTD and Sir4 was difficult. The problem was overcome by (1) phase extension to 3.0 Å and (2) locating the position of Leu167 of Sir2 NTD and Leu768 and Leu813 of Sir4 using the SeMet anomalous difference peaks of the Sir2 L167M and Sir4 L768M and L813M mutant complexes. Model building was done with Coot (Emsley and Cowtan 2004), and refinement was performed using PHENIX. A section of the electron density map is shown in Supplemental Figures S4 and S5.
NAD hydrolysis assay
Reactions were carried out as described previously (Wang et al. 2008) using 31 μM H4 peptide or H4K16 acetylated peptide, 40 mM of NAD+ containing 0.02 μCi [4-3H]NAD+ (0.37 Ci/mmol), and 100–150 ng of purified Sir2 or different copurified Sir2–Sir4 complexes. Reactions were incubated for 1 h at 37°C and terminated by the addition of 13.5 μL of 0.5 M sodium borate (pH 8.0) and quenching on ice. Released nicotinamide was extracted by the addition of 0.5 mL of water-saturated ethyl acetate. After vortexing, samples were centrifuged at 16,000g for 2 min. A fraction of the ethyl acetate (0.35 mL) was transferred into 3 mL of Ecoscint scintillation fluid, and the released nicotinamide was quantified in a liquid scintillation counter.
MDS
MDSs were performed using the GROMACS package (version 4.5.3) (Berendsen et al. 1995; Lindahl et al. 2001; Van Der Spoel et al. 2005; Hess et al. 2008) with Amber-ff03 force field (Duan et al. 2003) under constant number, pressure, temperature, and periodic boundary conditions. MDSs were performed on two models: one with the Sir2–Sir4SID complex and another with the Sir4SID molecule removed. The two models were separately solvated with the simple point charge (SPC) water model (Berendsen et al. 1981). A constant temperature of 310 K was maintained using the velocity rescale (v-rescale) (Bussi et al. 2007) algorithm with a coupling time of 0.1 psec, coupling the water molecules and the proteins independently to an external bath. The pressure was maintained by coupling to a reference pressure of 1 bar. All bond lengths, including hydrogen atoms, were constrained by the LINCS algorithm (Hess et al. 1997). Cutoff distances of 14 Å and 9 Å were chosen for van der Waals and Coulomb interactions, respectively. The simulation cell was a rectangular periodic box, and the minimum distance between the protein and the box walls was set to >10 Å to avoid direct interaction between the protein and its own periodic image. Na+ and Cl− ions were added to neutralize the modeled systems. During the simulation, the cysteines that coordinate zinc ions were deprotonated. All water molecules and ions in the simulation models were first minimized with the protein structures fixed using the steepest descent algorithm. Then, energy minimizations were performed on the whole systems but with protein main chain and Cα atoms fixed, followed by the minimization of the whole systems. The first 100-psec NVT equilibration was carried out to stabilize the whole system to 310 K with the protein atoms fixed. Following that, the second 100-psec NPT equilibration was performed to stabilize the pressure of the whole system with the protein atoms fixed. The rest of the 75-nsec simulation was carried out with the whole system relaxed.
Atomic coordinates of the structure and diffraction data have been deposited with PDB under accession code 4IAO. Structural figures were prepared using Pymol (http://www.pymol.org).
Supplementary Material
Acknowledgments
We thank the NSLS beamline scientists for technical support during data collection. The work was supported by grants from the Ministry of Science and Technology (2009CB825501 to R.M.X., and 2012CB910702 to N.Y.), the Natural Science Foundation of China (90919029 and 3098801 to R.M.X.), the Chinese Academy of Sciences (CAS), and the NIH (GM28220 to R.S.). R.M.X. holds a CAS-Novo Nordisk Great Wall Professorship.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.208140.112.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autiero I, Costantini S, Colonna G 2009. Human sirt-1: Molecular modeling and structure-function relationships of an unordered protein. PLoS ONE 4: e7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C 2002. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell 10: 523–535 [DOI] [PubMed] [Google Scholar]
- Avalos JL, Boeke JD, Wolberger C 2004. Structural basis for the mechanism and regulation of Sir2 enzymes. Mol Cell 13: 639–648 [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J 1981. Interaction models for water in relation to protein hydration. In Intermolecular forces (ed. B Pullman), pp. 331–342. Reidel, Dordrecht, The Netherlands [Google Scholar]
- Berendsen HJC, van der Spoel D, van Drunen R 1995. GROMACS: A message-passing parallel molecular dynamics implementation. Comput Phys Commun 91: 43–56 [Google Scholar]
- Bose ME, McConnell KH, Gardner-Aukema KA, Muller U, Weinreich M, Keck JL, Fox CA 2004. The origin recognition complex and Sir4 protein recruit Sir1p to yeast silent chromatin through independent interactions requiring a common Sir1p domain. Mol Cell Biol 24: 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD 1995. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev 9: 2888–2902 [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015 [DOI] [PubMed] [Google Scholar]
- Bussi G, Donadio D, Parrinello M 2007. Canonical sampling through velocity rescaling. J Chem Phys 126: 014101. [DOI] [PubMed] [Google Scholar]
- Cockell MM, Perrod S, Gasser SM 2000. Analysis of Sir2p domains required for rDNA and telomeric silencing in Saccharomyces cerevisiae. Genetics 154: 1069–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperus G, Shafaatian R, Shore D 2000. Locus specificity determinants in the multifunctional yeast silencing protein Sir2. EMBO J 19: 2641–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denu JM 2005. The Sir 2 family of protein deacetylases. Curr Opin Chem Biol 9: 431–440 [DOI] [PubMed] [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. 2003. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 24: 1999–2012 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Pavletich NP 2001. Structure of the histone deacetylase SIRT2. Nat Struct Biol 8: 621–625 [DOI] [PubMed] [Google Scholar]
- Frye RA 2000. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273: 793–798 [DOI] [PubMed] [Google Scholar]
- Garcia SN, Pillus L 2002. A unique class of conditional sir2 mutants displays distinct silencing defects in Saccharomyces cerevisiae. Genetics 162: 721–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L 2011. Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med 364: 2235–2244 [DOI] [PubMed] [Google Scholar]
- Hecht A, Laroche T, Strahl-Bolsinger S, Gasser SM, Grunstein M 1995. Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: A molecular model for the formation of heterochromatin in yeast. Cell 80: 583–592 [DOI] [PubMed] [Google Scholar]
- Hecht A, Strahl-Bolsinger S, Grunstein M 1996. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature 383: 92–96 [DOI] [PubMed] [Google Scholar]
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM 1997. LINCS: A linear constraint solver for molecular simulations. J Comput Chem 18: 1463–1472 [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E 2008. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4: 435–447 [DOI] [PubMed] [Google Scholar]
- Hou Z, Bernstein DA, Fox CA, Keck JL 2005. Structural basis of the Sir1-origin recognition complex interaction in transcriptional silencing. Proc Natl Acad Sci 102: 8489–8494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Stillman B, Xu RM 2005. Structural basis for origin recognition complex 1 protein-silence information regulator 1 protein interaction in epigenetic silencing. Proc Natl Acad Sci 102: 8519–8524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800 [DOI] [PubMed] [Google Scholar]
- Kim EJ, Kho JH, Kang MR, Um SJ 2007. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell 28: 277–290 [DOI] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R 2000. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci 97: 5807–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E, Hess B, van der Spoel D 2001. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J Mol Model 7: 306–317 [Google Scholar]
- Lo Conte L, Chothia C, Janin J 1999. The atomic structure of protein-protein recognition sites. J Mol Biol 285: 2177–2198 [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W 2001. Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107: 137–148 [DOI] [PubMed] [Google Scholar]
- Min J, Landry J, Sternglanz R, Xu RM 2001. Crystal structure of a SIR2 homolog–NAD complex. Cell 105: 269–279 [DOI] [PubMed] [Google Scholar]
- Moazed D, Kistler A, Axelrod A, Rine J, Johnson AD 1997. Silent information regulator protein complexes in Saccharomyces cerevisiae: A SIR2/SIR4 complex and evidence for a regulatory domain in SIR4 that inhibits its interaction with SIR3. Proc Natl Acad Sci 94: 2186–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Freeman K, Coodly L, Shore D 1994. Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev 8: 2257–2269 [DOI] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L 2004. Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P 2005. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434: 113–118 [DOI] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, Rine J 2003. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72: 481–516 [DOI] [PubMed] [Google Scholar]
- Sanders BD, Jackson B, Marmorstein R 2010. Structural basis for sirtuin function: What we know and what we don't. Biochim Biophys Acta 1804: 1604–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Wolberger C, Schramm VL, Boeke JD 2006. The biochemistry of sirtuins. Annu Rev Biochem 75: 435–465 [DOI] [PubMed] [Google Scholar]
- Shankaranarayana GD, Motamedi MR, Moazed D, Grewal SI 2003. Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr Biol 13: 1240–1246 [DOI] [PubMed] [Google Scholar]
- Shou W, Seol JH, Shevchenko A, Baskerville C, Moazed D, Chen ZW, Jang J, Charbonneau H, Deshaies RJ 1999. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 97: 233–244 [DOI] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, et al. 2000. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci 97: 6658–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M 1997. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev 11: 83–93 [DOI] [PubMed] [Google Scholar]
- Straight AF, Shou W, Dowd GJ, Turck CW, Deshaies RJ, Johnson AD, Moazed D 1999. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell 97: 245–256 [DOI] [PubMed] [Google Scholar]
- Tanny JC, Kirkpatrick DS, Gerber SA, Gygi SP, Moazed D 2004. Budding yeast silencing complexes and regulation of Sir2 activity by protein–protein interactions. Mol Cell Biol 24: 6931–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triolo T, Sternglanz R 1996. Role of interactions between the origin recognition complex and SIR1 in transcriptional silencing. Nature 381: 251–253 [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ 2005. GROMACS: Fast, flexible, and free. J Comput Chem 26: 1701–1718 [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D 2004. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 16: 93–105 [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA 2001. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107: 149–159 [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G 2007. Automated structure solution with autoSHARP. Methods Mol Biol 364: 215–230 [DOI] [PubMed] [Google Scholar]
- Wang CL, Landry J, Sternglanz R 2008. A yeast sir2 mutant temperature sensitive for silencing. Genetics 180: 1955–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Chai X, Clements A, Marmorstein R 2003a. Structure and autoregulation of the yeast Hst2 homolog of Sir2. Nat Struct Biol 10: 864–871 [DOI] [PubMed] [Google Scholar]
- Zhao K, Chai X, Marmorstein R 2003b. Structure of the yeast Hst2 protein deacetylase in ternary complex with 2′-O-acetyl ADP ribose and histone peptide. Structure 11: 1403–1411 [DOI] [PubMed] [Google Scholar]
- Zhao K, Harshaw R, Chai X, Marmorstein R 2004. Structural basis for nicotinamide cleavage and ADP-ribose transfer by NAD+-dependent Sir2 histone/protein deacetylases. Proc Natl Acad Sci 101: 8563–8568 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.