

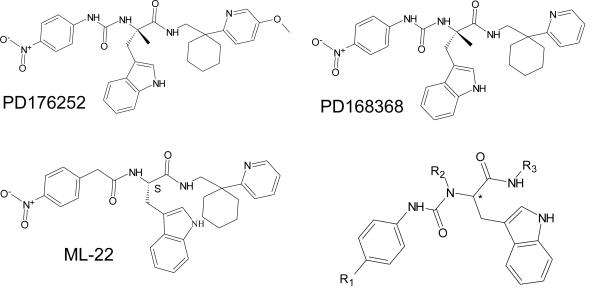
Abstract
N-formyl peptide receptors (FPRs) are G protein-coupled receptors (GPCRs) that play critical roles in inflammatory reactions, and FPR-specific interactions can possibly be used to facilitate the resolution of pathological inflammatory reactions. Recent studies indicated that FPRs have stereo-selective preference for chiral ligands. Here, we investigated the structure-activity relationship of 24 chiral ureidopropanamides, including previously reported compounds PD168368/PD176252 and their close analogs, and used molecular modeling to define chiral recognition by FPR2. Unlike previously reported 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones, whose R-forms preferentially activated FPR1/FPR2, we found that four S-enantiomers in the seven ureidopropanamide pairs tested preferentially activated intracellular Ca2+ flux in FPR2-transfected cells, while the R-counterpart was more active in two enantiomer pairs. Thus, active enantiomers of FPR2 agonists can be in either R- or S- configurations, depending on the molecular scaffold and specific substituents at the chiral center. Using molecular modeling approaches, including field point methodology, homology modeling, and docking studies, we propose a model that can explain stereoselective activity of chiral FPR2 agonists. Importantly, our docking studies of FPR2 chiral agonists correlated well with the FPR2 pharmacophore model derived previously. We conclude that the ability of FPR2 to discriminate between the enantiomers is the consequence of the arrangement of the three asymmetric hydrophobic subpockets at the main orthosteric FPR2 binding site with specific orientation of charged regions in the subpockets.
Keywords: Enantiomer, N-formyl peptide receptor, G protein-coupled receptor, agonist, chiral recognition
1. Introduction
Formyl peptide receptors (FPRs) are G protein-coupled receptors (GPCR) that play an important role in leukocyte activation and chemotaxis [reviewed in [1]]. These receptors were originally identified by their ability to bind and be stimulated by N-formyl peptides, which are produced by bacteria but can also be released from damaged mitochondria during tissue injury [2]. It has been proposed that a primary function of FPRs is to promote trafficking of phagocytic myeloid cells to sites of infection and tissue damage, where they exert antibacterial effector functions and clear cell debris [3]. In human and other primates, three FPR subtypes have been identified (FPR1, FPR2 and FPR3), which are expressed on a variety of cell types, including neutrophils, macrophages, T lymphocytes, epithelial cells, hepatocytes, fibroblasts, astrocytes, and other cells. These receptors have been reported to participate in a variety of regulatory functions during host defense responses [1;4;5].
Agonist binding and stimulation of FPRs induces a variety of responses, such as intracellular Ca2+ mobilization, chemotaxis, and generation of reactive oxygen species, which are dependent on agonist structure, cell type, receptor subtype, and species involved [1]. In last decade, a large number of peptide and synthetic non-peptide small molecules with a wide range of chemical diversity have been shown to be potent FPR agonists [6-15]. Some FPR2 agonists have shown to promote resolution of inflammatory processes, including Quin-C1, a highly specific non-peptide FPR2 agonist that demonstrated anti-inflammatory properties in a mouse model of lung injury [16]. Similarly, the FPR2 peptide agonist Trp-Lys-Tyr-Met-Val-D-Met (WKYMVm) protected against death by enhancing bactericidal activity and inhibiting vital organ inflammation and apoptosis in a sepsis mouse model [17].
Stereochemical information plays an important role in GPCR recognition processes [18;19]. Stereoselectivity suggests direct and specific receptor targeting, and identification of such stereospecific interactions may have important consequences in drug discovery and development, such as improvement of pharmacokinetic properties and removing undesirable side effects of agents by virtue of the unique activity of enantiomers [20-22]. To date, little is known regarding the stereoselectivity of FPR-ligand interactions. For example, R-enantiomers of N-substituted benzimidazole and pyridazin-3(2H)-one FPR1/FPR2 agonists were found to be more potent than their S-counterparts [9;15]. Previously, we found that antagonists of gastrin-releasing peptide /neuromedin B receptors (bombesin receptors), PD168368/PD176252 and related chiral derivatives, were potent FPR1/FPR2 agonists [12]. However, a systematic study of enantiomer pairs to determine the impact of enantiomer orientation on FPR agonist activity was not performed. In the present studies, we evaluated 22 structural derivatives of PD168368/PD176252 including seven enantiomer pairs for their ability to activate human neutrophils and HL-60 cells transfected with human FPR1 or FPR2. While none of the compounds had affinity for bombesin receptors, 15 of the compounds stimulated Ca2+ flux in FPR1/FPR2 transfected cells. Based on the results, we propose a molecular model of enantiomeric recognition at FPR2 that can explain stereoselective activity of the compounds identified in the present study, as well as other enantiomers reported previously as FPR2 agonists.
2. Materials and Methods
2.1. Materials
Dimethyl sulfoxide (DMSO), fMLF, HEPES, and Histopaque 1077 were purchased from Sigma Chemical Co. (St. Louis, MO). RPMI 1640 medium and penicillin-streptomycin solution were purchased from Mediatech (Herdon, VA). Fetal bovine serum was purchased from Atlas Biologicals (Fort Collins, CO). Peptides Trp-Lys-Tyr-Met-Val-D-Met (WKYMVm) and Trp-Lys-Tyr-Met-Val-L-Met (WKYMVM) were from Calbiochem (San Diego, CA) and Tocris Bioscience (Ellisville, MO), respectively. The antagonist WRW4 (WRWWWW) was from Genscript Corporation (Scotch Plains, NJ). Hanks’ balanced salt solution (HBSS; 0.137 M NaCl, 5.4 mM KCl, 0.25 mM Na2HPO4, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 5.56 mM glucose, and 10 mM HEPES, pH 7.4), G418, and probenecid were from Life Technologies (Grand Island, NY). HBSS containing 1.3 mM CaCl2 and 1.0 mM MgSO4 is designated as HBSS+. Selected N’-phenethylureas (compounds AG-10/16 through AG-10/22) were purchased from ChemDiv (San Diego, CA).
2.2. Compound preparation and analysis of compound purity
The 22 structural derivatives of PD168368 and PD176252 were synthesized as pure enantiomers using stereospecific methods starting from the enantiomerically pure N-BOC-R-tryptophan or N-BOC-S-tryptophan and following previously reported methods [23;24]. Enantiomeric purity of the compounds was assessed by chiral high-performance liquid chromatography (HPLC) analysis on a Perkin-Elmer series 200 LC instrument using a Daicel ChiralCell OD column (250 × 4.6 mm, 5 μm particle size) and equipped with a Perkin-Elmer 785A UV/VIS detector setting λ= 230 nm. The compounds were eluted with n-hexane/EtOH, 4:1, v/v at a flow rate of 0.8 mL/min. All compounds showed enantiomeric excesses (e.e.) ≥ 95%.
2.3. Cell culture
Human promyelocytic leukemia HL-60 cells stably transfected with FPR1 (FPR1-HL-60 cells) or FPR2 (FPR2-HL-60 cells) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 10 mM HEPES, 100 μg/ml streptomycin, 100 U/ml penicillin, and G418 (1 mg/mL), as described previously [11]. Wild-type HL-60 cells were cultured under the same conditions, but without G418.
2.4. Isolation of human neutrophils
Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at Montana State University. Neutrophils were purified from the blood using dextran sedimentation, followed by Histopaque 1077 gradient separation and hypotonic lysis of red blood cells, as previously described [8]. Isolated neutrophils were washed twice and resuspended in HBSS without Ca2+ and Mg2+ (HBSS-). Neutrophil preparations were routinely > 95 % pure, as determined by light microscopy, and > 98 % viable, as determined by trypan blue exclusion.
2.5. Bombesin receptor binding assays
The compounds were evaluated in binding assays to test their ability to displace [125I][Tyr4]bombesin using bombesin receptor (BB2)-transfected HEK-293 cells (compounds ML-8, EMY-87, ML-11, EMY-89, ML-18, EMY-98, ML-16, and EMY-96) or PC-3 cells overexpressing BB2 (compounds PD168368, PD-360, PD-362, PD-361, ML-22, EMY-124, and PD-59). Ki value for PD176252 was 66 nM in the assay with PC3 cells and Ki for human gastrin-releasing peptide was 28 pM in the assay with HEK-293 cells. Binding assays with BB2-transfected HEK-293 cells were performed by the CEREP Corp. (France) and binding assays with PC-3 cells were performed as described by Gourni et al. [25] with minor modifications.
2.6. Ca2+ mobilization and Fluo-4 efflux assays
Changes in intracellular Ca2+ were measured with a FlexStation II scanning fluorometer using a FLIPR 3 calcium assay kit (Molecular Devices, Sunnyvale, CA) for human neutrophils and HL-60 cells, as described previously [11]. All active compounds were evaluated in parent (wild-type) HL-60 cells for supporting that the agonists are inactive in non-transfected cells. Human neutrophils or HL-60 cells, suspended in HBSS- containing 10 mM HEPES, were loaded with Fluo-4 AM dye (Invitrogen) (1.25 μg/mL final concentration) and incubated for 30 min in the dark at 37 °C. After dye loading, the cells were washed with HBSS- containing 10 mM HEPES, resuspended in HBSS containing 10 mM HEPES and Ca2+ and Mg2+ (HBSS+), and aliquotted into the wells of a flat-bottomed, half-area-well black microtiter plates (2 × 105 cells/well). If indicated, 2 mM probenecid was added 5 min before the assay. The compound of interest was added from a source plate containing dilutions of test compounds in HBSS+, and changes in fluorescence were monitored (λex = 485 nm, λem = 538 nm) every 5 s for 240 s at room temperature after automated addition of compounds. Maximum change in fluorescence, expressed in arbitrary units over baseline, was used to determine agonist response. Responses were normalized to the response induced by 5 nM fMLF for FPR1-HL-60 cells and neutrophils, or 5 nM WKYMVM for FPR2-HL-60 cells, which were assigned a value of 100%. Curve fitting (5-6 points) and calculation of median effective concentration values (EC50) were performed by nonlinear regression analysis of the dose-response curves generated using Prism 5 (GraphPad Software, Inc., San Diego, CA).
For evaluation of Fluo-4 efflux, human neutrophils were loaded with Fluo-4 AM dye, washed, and resuspended in HBSS+, as described above. Compounds EMY-96 (25 μM), ML-16 (25 μM), and ST-6 (25 μM) or vehicle (DMSO) were added. After a 5 min incubation at room temperature, the samples were centrifuged to pellet cells (1 min, 1400 g), and fluorescence in the cell supernatants was measured (λex = 485 nm, λem = 538 nm).
2.7. β-Arrestin recruitment assay
The PathHunter® eXpress β-arrestin assay was performed according to the manufacturer's protocol using CHO-K1 cells transfected with FPR1 (FPR1-CHO-K1) or FPR2 (FPR2-CHO-K1) (DiscoveRx Corporation, Fremont, CA). These cell lines monitor GPCR activity by detecting the interaction of β-arrestin with the activated GPCR using β-galactosidase (β-gal) enzyme fragment complementation [26]. Briefly, frozen cells were thawed and resuspended in DiscoveRx Optimized Cell Culture Medium (OCCM), provided by the manufacturer. Assay plates [96-well half area plates with clear bottom (Greiner Bio-One, Monroe, NC)] were prepared with 5000 cells/well in 50 μl of OCCM. Serial dilutions of test compounds were prepared in OCCM, contained DMSO as a solvent. For each dilution, the final concentration of DMSO remained constant. After incubation at 37°C (5% CO2, 95% relative humidity) for 48 h, 5.5 μl of test compound was added, and the incubation was continued at 37°C for 90 min. Detection agent (25 μl) was added, and the incubation was continued at room temperature for 60 min. Chemiluminescene was monitored using a Fluoroskan Ascent FL microtiter plate reader (Thermo Fisher Scientific, Waltham, MA). Maximum change in luminescence, expressed in arbitrary units over baseline, was used to determine agonist response. Responses were normalized to the response induced by 5 nM WKYMVm for both FPR1-CHO-K1 and FPR2-CHO-K1 cells, which was assigned a value of 100%. Curve fitting (5-6 points) and calculation of median effective concentration values (EC50) were performed by nonlinear regression analysis of the dose-response curves generated using GraphPad Prism 5.
2.8. Chemotaxis assay
Human or murine neutrophils were suspended in HBSS+ containing 2% (v/v) heat-inactivated fetal bovine serum (2 × 106 cells/ml), and chemotaxis was analyzed in 96-well ChemoTx chemotaxis chambers (Neuroprobe, Gaithersburg, MD), as described previously [8]. In brief, lower wells were loaded with 30 μl of HBSS+ containing 2% (v/v) fetal bovine serum and the indicated concentrations of test compounds, DMSO (negative control), or 1 nM fMLF as a positive control. Neutrophils were added to the upper wells and allowed to migrate through the 5.0-μm pore polycarbonate membrane filter for 60 min at 37°C and 5% CO2. The number of migrated cells was determined by measuring ATP in lysates of transmigrated cells using a luminescence-based assay (CellTiter-Glo; Promega, Madison, WI), and luminescence measurements were converted to absolute cell numbers by comparison of the values with standard curves obtained with known numbers of neutrophils. Curve fitting (at least eight to nine points) and calculation of median effective concentration values (EC50) were performed by nonlinear regression analysis of the dose-response curves generated using Prism 5.
2.9. Molecular modeling
FPR2 active enantiomers and their counterparts were imported into FieldAlign program (FieldAlign Version 2.0.1; Cresset Biomolecular Discovery Ltd., Hertfordshire, UK) in Tripos MOL2 format and superimposed onto the template obtained previously by FieldTemplater software on the base of five FPR2 agonists [12]. The template contains field points of different types in the positions of energy extrema for probe atoms. Positive probes give “negative” field points, whereas energy extrema for negative and neutral probe atoms correspond to “positive” and steric field points, respectively. Hydrophobic field points were also generated with neutral probes capable of penetrating into the molecular core and reaching extrema in the centers of hydrophobic regions (e.g., benzene rings). The size of a field point depends on magnitude of an extremum [27]. For the enantiomers imported into FieldAlign, conformation hunter algorithm was used to generate representative sets of conformations corresponding to local minima of energy calculated within the extended electron distribution force field [28;29]. This algorithm incorporated in the FieldTemplater and FieldAlign software allowed us to obtain up to 200 independent conformations that were passed to further calculation of field points surrounding each conformation of each molecule. To decrease the number of rotatable bonds during the conformation search, the “force amides trans” option was enabled in the program. Field point calculations were performed for each conformation, as described above. Conformations with the best fit to the geometry and field points of the template were identified, and their superimpositions were refined by the simplex optimization algorithm incorporated in FieldAlign. The measure of similarity was derived from the field point spatial distribution and geometric overlap between a molecule and the template according to the FieldAlign program.
For homology modeling FPR2, the primary amino acid sequence of FPR2 was submitted to the Phyre2 (Protein Homology/analogy Recognition Engine V2.0) protein fold recognition server (http://www.sbg.bio.ic.ac.uk/phyre2) [30]. The server used one template with known crystal structure for homology modeling per protein sequence. We obtained 18 predicted protein structure models with the highest level of confidence, based on dissolved crystal structures of GPCRs, including bovine and squid rhodopsins, human adenosine receptor A2A, turkey β1 adrenoceptor, human β2 adrenoceptor, human histamine receptor H1, human dopamine D3 receptor, and human chemokine receptor CXCR4. Two homology FPR2 models were pre-selected from the set of predicted models. One model, based on the CXCR4 structure, has a maximal sequence identity of 28%, but with a low crystal structure resolution (3.2 Å) for the template. The second model has a template with sequence identity of 16%, but the highest resolution crystal structure (2.2 Å) known to date for a GPCR. Side chain conformations of eight residues in FPR2 (His102, Val105, Asp106, Leu109, Trp254, Phe257, Ser288, Phe292), which were previously identified as belonging to the binding site [31], were optimized in both models using a corresponding module of Molegro software. Since our pre-docking studies indicated that the rhodopsin-based model gave the best docking positions for FPR2 agonists used previously for pharmacophore modeling [12], we propose that these data justify use of the bovine rhodopsin structure as a template for the FPR2 homology model vs. the CXCR4 template. Thus, further modeling was based on the rhodopsin-based model of the FPR2.
Taking into account a lack of structural information about any ligand-receptor complex with FPR2, we tried to locate cavities in the macromolecule obtained by homology modeling in order to identify the search space for docking. Use of the MVD “Detect cavity” module with probe size 1.2 Å gave two cavities with volumes of 241 and 25 Å3 in the region of the ligand binding site. Positions of these two cavities obviously reflect a bottle-neck shape of the binding site. Hence, for FPR2, we also chose a spherical search space with a default radius of 15 Å centered at the terminus of the larger cavity directed to the smaller one.
Before docking, structures of the compounds were pre-optimized using HyperChem 8.0 software with MM+ force field and saved in Tripos MOL2 format. The ligand structures were then imported into the MVD with the options “Create explicit hydrogens”, “Assign charges (calculated by MVD)”, and “Detect flexible torsions in ligands” enabled. Selected molecules were docked into FPR2 using the search spaces indicated above with a rigid receptor structure. Ligand flexibility was accounted for with respect to torsion angles auto-detected in MVD. MolDock score functions were used with 0.3 Å grid resolution. The “Internal HBond” option was activated in the “Ligand evaluation” menu of Docking Wizard. Thirty docking runs were performed for each molecule, while 60 docking runs were performed for the peptide. The option “Return multiple poses for each run” was enabled, and the post-processing options “Energy minimization” and “Optimize H-bonds” were applied after docking. Similar poses were clustered at a RMSD threshold of 1 Å.
Atom charges were calculated by semi-empirical AM1 method with full geometry optimization of the molecules using HyperChem 8.0 software.
3. Results
3.1. Activity of PD168368/PD176252 derivatives at bombesin receptors and FPRs
Previously, we found that bombesin receptor antagonists PD168368 and PD176252 were potent dual FPR1/FPR2 agonists [12]. In the present study, 22 structural derivatives of PD168368 and PD176252 were synthesized as pure enantiomers using stereospecific methods, and structures of the compounds are shown in Table 1. All synthesized compounds showed enantiomeric excesses (e.e.) ≥ 95%, as determined by chiral HPLC analysis.
Table 1.
| |||
|---|---|---|---|
| Compound (Enantiomer Type) | R1 | R2 | R3 |
| ST-12 (S) | OCH3 | H |
|
| ST-13 (S) | CF3 | H | |
| ST-14 (S) | CN | H | |
| ST-15 (S) | Br | H | |
| ST-16 (S) | CH3 | H | |
| PD-362 (S) | NO2 | H | |
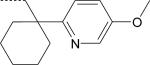
| ST-6 (R) | NO2 | H | |
| PD-361 (S) | NO2 | CH3 | |
| EMY-124 (S) | NO2 | CH3 |
|
| PD-359 (R) | NO2 | CH3 | |
| ML-8 (S) | NO2 | H |
|
| EMY-87 (R) | NO2 | H | |
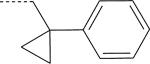
| ML-11 (S) | NO2 | H |
|
| EMY-89 (R) | NO2 | H | |
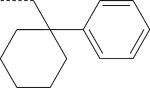
| ST-11 (S) | NO2 | H |
|
| ST-9 (R) | NO2 | H | |
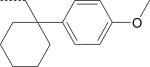
| ML-18 (S) | NO2 | H |
|
| EMY-98 (R) | NO2 | H | |
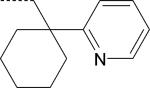
| ML-16 (S) | NO2 | H |
|
| EMY-96 (R) | NO2 | H | |
| PD-360 (S) | NO2 | CH3 | |
Unlike PD168368/PD176252, none of the enantiomers had affinity for bombesin receptor BB2 (data not shown). However, several of enantiomers demonstrated agonist activity in FPR1-HL-60 and FPR2-HL-60 cells, but not in wild-type non-transfected HL-60 cells (Table 2). Among the 15 active compounds, 9 were found to be FPR2-specific agonists, with the most active compounds being ST-6 and EMY-96. The active compounds caused a rapid increase in Ca2+ mobilization that peaked by 40-60 sec after treatment (data not shown). Moreover, the specificity of EMY-96 was confirmed using the FPR2-specific antagonist WRW4 (Figure 1). The other 6 compounds were dual FPR1/FPR2 agonists, with the most active compounds being ST-12 and ST-16 (Table 2). ML-11, ML-22, ST-11, ST-13, ST-14, and PD-362 were partial FPR2 agonists and ML-8 and ML-16 were partial FPR1/FPR2 agonists, with reduced efficacies (≤65%). All other active compounds were full agonists with efficacies close to those of 5 nM fMLF (in FPR1-HL-60 cells) or 5 nM WKYMVM (in FPR2-HL-60 cells) (Table 2).
Table 2.
Effect of the compounds on Ca2+ mobilization in FPR1/FPR2 transfected cells and chemotaxis and Ca2+ mobilization in human neutrophils
| Compound (Enantiomer Type) | Ca2+ Mobilization in HL-60 cells | Neutrophils | ||||
|---|---|---|---|---|---|---|
| FPR1 | FPR2 | Chemotaxis | Ca2+ Mobilizationa | |||
| w/o Prob. | with Prob. | |||||
| EC50 (μM) and Efficacy (%)b | ||||||
| PD176252 (S) | 0.33 ±0.12 (100) | 1.3 ± 0.35 (90) | 0.81 ± 0.22 | 0.72 ± 0.21 | 0.21 ± 0.067 | |
| PD168368 (S) | 0.57 ± 0.19 (95) | 0.62 ± 0.26 (75) | 0.7 ± 0.15 | 0.91 ± 0.26 | 0.19 ± 0.07 | |
| PD-360 (S) | N.A.c | N.A. | N.A. | N.A. | N.A. | |
| PD-361 (S) | N.A. | 17.6 ± 5.7 (75) | N.A. | N.A. | N.A. | |
| ML-22 (S) | N.A. | 17.4 ± 5.2 (30) | 1.4 ± 0.37 | N.A. | 5.5 ± 1.5 | |
| ST-12 (S) | 0.26 ± 0.08 (130) | 0.19 ± 0.06 (110) | 0.030 ± 0.002 | 0.086 ± 0.03 | 0.025 ± 0.008 | |
| ST-13 (S) | N.A. | 0.21 ± 0.05 (65) | 0.041 ± 0.02 | 0.082 ± 0.02 | 0.035 ± 0.01 | |
| ST-14 (S) | N.A. | 0.36 ± 0.11 (30) | 1.8 ± 0.52 | N.A. | 0.022 ± 0.006 | |
| ST-15 (S) | 0.23 ± 0.07 (95) | 0.35 ± 0.09 (95) | 0.010 ± 0.008 | 0.089 ± 0.03 | 0.015 ± 0.004 | |
| ST-16 (S) | 0.041 ± 0.01 (120) | 0.21 ± 0.06 (120) | 0.029 ± 0.004 | 0.026 ± 0.008 | 0.0035 ± 0.001 | |
| Enantiomer Pairs | PD-362 (S) ST-6 (R) |
N.A. N.A. |
4.1 ± 1.2 (50) 0.023 ± 0.01 (85) |
N.A. 0.051 ± 0.006 |
N.A. N.A. |
0.29 ± 0.09 0.0032± 0.001 |
| ST-11 (S) ST-9 (R) |
N.A. N.A. |
0.7 ± 0.22 (85) N.A. |
0.22 ± 0.07 N.A. |
N.A. N.A. |
0.05 ± 0.02 N.A. |
|
| EMY-124 (S) PD-359 (R) |
N.A. N.A. |
N.A. N.A. |
N.A. 4.2 ± 1.2 |
N.A. N.A. |
N.A. 1.1 ± 0.29 |
|
| ML-8 (S) EMY-87 (R) |
22.3 ± 6.9 (45) N.A. |
23.2 ± 7.4 (45) N.A. |
4.9 ± 1.4 N.A. |
N.A. N.A. |
0.42 ± 0.17 N.A. |
|
| ML-11 (S) EMY-89 (R) |
N.A. N.A. |
7.6 ± 2.4 (50) N.A. |
22.3 ± 5.4 N.A. |
N.A. N.A. |
2.3 ± 0.51 N.A. |
|
| ML-18 (S) EMY-98 (R) |
14.8 ± 4.6 (40) N.A. |
10.4 ± 3.5 (80) N.A. |
12.4 ± 3.8 N.A. |
N.A. N.A. |
5.6 ± 1.6 N.A. |
|
| ML-16 (S) EMY-96 (R) |
12.9 ± 3.4 (25) N.A. |
2.0 ± 0.65 (60) | 2.3 ± 0.62 4.3 ± 1.3 |
N.A. N.A. |
0.68 ± 0.21 0.46 ± 0.13 |
|
| 0.19 ± 0.06 (110) | ||||||
| WKYMVM | 0.50 | 0.001 | 0.002 | 0.0016 | 0.0014 | |
| fMLF | 0.01 | 20.4 | 0.0005 | 0.005 | 0.0012 | |
Ca2+ mobilization in human neutrophils was monitored without (w/o) or in the presence of 2.5 mM probenecid (Prob.).
EC50 values are presented as the mean ± S.D. of three independent experiments and efficacy (in parentless) is expressed as the percentage of the response induced by 5 nM fMLF (FPR1) or 5 nM WKYMVM (FPR2); values of efficacy are presented as average of three independent experiments.
N.A., non-active compound (<10% of positive control level over a concentration range of 0-30 μM for Ca2+ mobilization in HL-60 cells and neutrophils.
Figure 1.

Effect of the FPR2 antagonist WRW4 on Ca2+ mobilization induced by EMY-96. FPR2 HL60 cells were pretreated for 30 min with DMSO (control) or WRW4 (2 μM), followed by the addition of 10 μM compound EMY-96, and Ca2+ flux was monitored, as described under Materials and Methods. Control samples were treated with 1 nM of WKYMVM. The data are presented as % of response induced by WKYMVM and are the mean ± S.D. of triplicate samples from one experiment that is representative of three independent experiments.
Elimination of the methyl group at the chiral center in the parent compounds PD168368 and PD176252 was associated with loss of the ability to stimulate Ca2+ mobilization in FPR1-HL-60 cells (compound PD-362) or decreased activity in both FPR1-HL-60 and FPR2-HL-60 cells (compound ML-16). In confirmation of these results, PD-362 also failed to induce β-arrestin recruitment in FPR1 and FPR2 CHO-K1 cells (Figure 2), whereas compound ML-16 had EC50 values of 18.5 ± 5.1 and 12.1 ± 3.4 μM in FPR1-CHO-K1 and FPR2-CHO-K1 cells, respectively, in the β-arrestin recruitment assay. In comparison, EC50 values for PD168368 were 2.8 ± 0.73 and 11.1 ± 2.8 μM, and for PD176252, these values were 3.8 ± 1.1 and 8.8 ± 2.0 μM in FPR1-CHO-K1 and FPR2-CHO-K1 cells, respectively. Generally, EC50 values for β-arrestin recruitment were ~5-fold higher than EC50 values for Ca2+ mobilization, which confirms our previous observations using different FPR agonists [15].
Figure 2.

Analysis of β-arrestin recruitment in cells treated with PD176252 and PD-362. FPR1-CHO-K1 (○,□) and FPR2-CHO-K1 (■,▲) cells were incubated with the indicated concentrations of PD176252 (○,■) or PD-362 (□,▲) and analyzed, as described under Materials and Methods. Representative of three independent experiments.
Introduction of an additional CH3 group at R2 in ML-16, forming the S-enantiomer PD-360, resulted in a loss of FPR1/FPR2 agonist activity in HL-60 transfected cells. The same modification of the FPR2-specific agonist PD-362, forming S-enantiomer PD-361, resulted in a partial loss of agonist activity. Likewise, replacing the carbamide NH group closest to the nitro-benzene ring in molecule ML-16 with a methylene (compound ML-22) also resulted in a loss of activity in FPR2 HL-60 cells (Table 2). Substitution of the nitro group at the para-position of the phenyl ring in S-enantiomer PD-362 with OCH3, CF3, CN, Br, and CH3 resulted to agonists ST-12 through ST-16, which had mixed FPR1/FPR2 (ST-12, ST-15, and ST-16) or FPR2-specific (ST-13 and ST-14) agonist activity (Table 2).
There were seven enantiomer pairs among the 22 PD168368/PD176252 derivatives evaluated. Both enantiomers of the pair EMY-124/PD-359, which contain a cyclohexylmethyl substituent at R3 and a CH3 group at R2, were inactive at FPR1/FPR2 (Table 2). Although different variations of R3 in three enantiomer pairs with unsubstituted R2 were evaluated, including tetrahydronaphthyl (compounds ML-8 and EMY-87), (1-phenylcyclopropyl)methyl (ML-11 and EMY-89), (1-phenylcyclohexyl)methyl (ST-11 and ST-9), and [1-(4-methoxyphenyl)cyclohexyl]methyl (ML-18 and EMY-98), only the S-enantiomers of these pairs (ML-8, ML-11, ST-11, and ML-18) were active FPR agonists (Table 2). Conversely, both the S- and R- enantiomers from two pairs (ML-16/EMY-96 and PD-362/ST-6) were active at FPR2 (Table 2). Representative dose–response curves for Ca2+ flux induced in HL-60 FPR2 cells by S- (ML-8) and R- (EMY-87) enantiomers are shown in Figure 3.
Figure 3.

Analysis of Ca2+ mobilization in FPR2 transfected HL-60 cells treated with S- (ML-8) and R- (EMY-87) enantiomers. FPR2 HL-60 cells were loaded with Fluo-4 AM dye, and Ca2+ flux in response to the indicated concentrations of compounds or control WKYMVM peptide (5 nM) was analyzed, as described under Materials and Methods. The data are presented as % of response induced by WKYMVM and are the mean ± S.D. of triplicate samples from one experiment that is representative of three independent experiments.
Six enantiomers had no agonist activity for either FPR1 or FPR2. Thus, we considered whether such compounds might be FPR antagonists. FPR1-HL-60 and FPR2-HL-60 cells were pretreated with the selected compounds and then evaluated for subsequent responses to control peptide agonists (5 nM fMLF for FPR1 and 1nM WKYMVM for FPR2). Pretreatment of cells for 30 min with a dose range (1–50 μM) of selected compounds that were inactive in the Ca2+ mobilization assay (PD-360, ST-9, and EMY-124) had no inhibitory effect on Ca2+ flux induced by either fMLF or WKYMVM, suggesting that these compounds were not receptor antagonists. In contrast, pretreatment of FPR2-HL-60 cells with compounds EMY-89 and EMY-98 resulted in a dose-dependent loss of the response induced by subsequent treatment with WKYMVM, although with relatively low potency (IC50 ~17-25 μM). Compound EMY-87 was able to antagonize both FPR1 and FPR2 responses (IC50 ~14-15 μM).
3.2. Activity of the enantiomers in human neutrophils
PD168368/PD176252 and their 22 analogs were evaluated for their ability to stimulate chemotaxis and Ca2+ mobilization in human neutrophils. The majority of compounds found to be FPR1/FPR2 agonists in FPR-transfected HL-60 cells stimulated human neutrophil chemotaxis, with only two exceptions (compounds PD-361 and PD-362). Likewise, all compounds found to be inactive in FPR-transfected HL-60 cells were also inactive in the neutrophil chemotaxis assay (Table 2).
While ST-12, ST-13, ST-15, and ST-16 dose-dependently stimulated Ca2+ mobilization in human neutrophils (Table 2), which peaked by 40-60 sec after treatment, ten of the compounds found to induce Ca2+ flux in FPR-transfected HL-60 cells unexpectedly failed to simulate this response in human neutrophils. Of note, these compounds all contained NO2 or CN groups in the para position of the phenyl ring (Table 1). On the other hand, these compounds were able to desensitize neutrophil Ca2+ mobilization induced by chemotactic peptides. For example, pretreatment of neutrophils with EMY-96, the most potent FPR2 agonist in transfected cell lines, dose–dependently inhibited Ca2+ mobilization induced by WKYMVm and the FPR2-specific agonist WKYMVM but not fMLF (Figure 4).
Figure 4.

Desensitization of formyl peptide-induced Ca2+ mobilization in human neutrophils by EMY-96. Neutrophils were pretreated with the indicated concentrations of EMY-96 for 30 min, and Ca2+ mobilization was monitored after addition of fMLF (10 nM), WKYMVm (1 nM), or WKYMVM (10 nM). Representative of three independent experiments.
In previous studies investigating FPR agonists, we observed differential activity between FPR-transfected cells and primary neutrophils [10;11;15], although neutrophils still responded to all agonists that activated FPR-expressing HL-60 cells. Thus, the NO2- and CN-substituted compounds reported here seem to have properties that affect their ability to stimulate Ca2+ flux or that interfere with the assay system. Indeed, we found that pretreatment of human neutrophils with probenecid restored the Ca2+ flux response in neutrophils treated with all of these PD168368/PD176252 derivatives except PD-361 (Table 2). Because pretreatment of neutrophils with probenecid, an anion exchange protein inhibitor used for slowing dye leakage from cells [32;33], restored the Ca2+ flux response, we considered whether the chiral ureidopropanamides might be activating cellular efflux of Fluo-4, the fluorescent dye used for monitoring Ca2+ current, and thereby interfering with the assay. However, direct experiments assessing dye efflux showed that the most potent compounds EMY-96, ML-16, and ST-6 did not induce significant Fluo-4 efflux in human neutrophils (data not shown).
Because substitution of OCH3, CF3, Br, and CH3 in the para position of the phenyl ring with CN or NO2 and elimination of CH3 at the chiral center of PD168368 and PD176252 resulted in loss of ability to activate neutrophil Ca2+ flux without probenecid pretreatment, it appears that the requirement for probenecid is somehow related to compound structure. To investigate this idea, we further evaluated if probenicid could alter agonist potency for Ca2+ mobilization in neutrophils using a series of previously described achiral (AG-10/14 through AG-10/18) and chiral (AG-10/19 through AG-10/22) FPR agonists with an N’-phenethylurea scaffold [12] and found that for some of compounds and unrelated to their chirality/achirality, agonist potency was greatly increased (>80-fold) in the presence of probenecid (e.g., AG-10/15), but for other compounds (e.g., AG-10/18, AG-10/19, and AG-10/20) agonist activity only changed slightly (~1.5-fold). Thus, the reason for the selective sensitivity of the Ca2+ flux response to probenecid for these previously reported compounds and the chiral ureidopropanamides reported here is still unclear, and future studies will be necessary to elucidate this phenomenon.
3.3. Molecular modeling
Interaction of FPRs with most known enantiomer agonists has been found to be stereoselective. For example, Frohn et al. [9] identified an enantiomer pair of N-substituted benzimidazoles as potent FPR2 ligands and found that the R-enantiomer was significantly more potent than the S-enantiomer. Similarly, we recently found that among chiral pyridazin-3(2H)-one FPR1/FPR2 agonists, R-enantiomers were active, while their S-enantiomer counterparts were less active or inactive [15]. In the present study, we found that among chiral FPR2 agonists with an ureidopropanamide scaffold, the S- enantiomers were generally preferred over the R-enantiomers. Although several FPR1-specific chiral agonists were described previously [12;15] and in the present work (Tables 1-2), we focused here on FPR2 and its chiral agonists for molecular modeling based on the potential therapeutic properties of FPR2 agonists.
To investigate the enantiomer preference observed for FPR2 agonists with different scaffolds and chiral center substituents, we used a previously refined pharmacophore model for FPR2 [12]. The model template was created on the basis of five potent small-molecule non-peptide FPR2 agonists, four of which were chiral molecules (both S- and R- forms), including (S)-PD168368, (S)-AG-10/5, (S)-AG-10/8, and (R)-Frohn-11, as described previously [12]. The hydrophobic field surface of the 5-molecule FPR2 template includes space where positioning of a neutral “hydrophobic” probe atom is energetically favorable (see Materials and Methods). The surface consists of three regions (H1, H2, and H3), which correlate with subpockets I, II, and III in the FPR2 binding site, respectively ([12] and Figure 5). Enantiomeric FPR2 agonists and their active/inactive counterparts reported here and published previously [9;13;15] were then compared to the pharmacophore model. All molecules were overlaid in the three-subpocket model, and the highest-score superimpositions together with values of calculated similarity are shown in Table 3. The similarity values depend equally on geometric and field similarity of a molecule and template. These field descriptors (or field points) are extrema of electrostatic, steric and hydrophobic fields [27]. For most 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones [15], the similarity between aligned molecule and the template for R-enantiomers [compounds R-(-)-5b, R-(-)-5d, R-(-)5e, and R-(-)5f] was higher than that of their S-enantiomer counterparts. In the case of R-(-)5c, the active configuration was characterized by a smaller similarity value compared to that of the S-enantiomer, which is probably due to the high flexibility of the propyl chain, resulting in inadequate conformational sampling achieved by the “conformation hunt” algorithm of FieldAlign. Conversely, the active S-enantiomers of the ureidopropanamides reported here (ML-8, ML-11, ML-18, and ST-11) had better alignments than the corresponding inactive R-enantiomers. For two enantiomer pairs (ML-16/EMY-96 and PD-362/ST-6), the R-enantiomer forms were more active and had better alignments than their S-enantiomer counterparts. This finding suggests that not only the general scaffold, but also specific chiral center substituents influence the FPR2 agonist activity of R- or S-enantiomers.
Figure 5.

Overlay of molecular conformations of enantiomer pair (S)-ML-18/(R)-EMY-98 with the best fit to the geometry of the FPR2 template. Superimpositions of the conformations to the template were refined by the simplex optimization algorithm incorporated in FieldAlign. Field points are colored as follows: blue, electron-rich (negative); red, electron-deficient (positive); yellow, van der Waals attractive (steric). All field points belonging to FPR templates are tetrahedral shaped, and field points, belonging to ML-18/EMY-98 are spherical. The hydrophobic field surface is colored in orange. The main groups of negative field points are marked as “A” and “B”, the main group of positive field points is marked as “C”, and the three hydrophobic surfaces are marked as H1, H2, and H3 in accordance with our previously reported FPR2 pharmacophore model [12]. The green arrows point to positions of the large negative field points (blue spheres) corresponding to both carbonyl groups of compound ML-18 and an incomplete geometric overlap of EMY-98 with the template.
Table 3.
Location of substituents from representative conformations obtained for the 5-molecule FPR2 Field Point pharmacophore template in hypothetical hydrophobic subpockets and alignments on this template of enantiomeric FPR2 agonists.
| Compd. | Enantiomer | FPR2 HL-60 (EC50, μM)a | Sim.b | Submolecule | ||
|---|---|---|---|---|---|---|
| Subpocket Ic | Subpocket II | Subpocket III | ||||
| ML-8 EMY-87 |
S
R |
22.3 N.A. |
0.622 0.590 |
4-Nitrophenyl 4-Nitrophenyl |
Tetrahydronaphthyl Indolyl |
Indolyl Tetrahydronaphthyl |
| ML-11 EMY-89 |
S
R |
7.6 N.A. |
0.591 0.571 |
4-Nitrophenyl 4-Nitrophenyl |
1-Phenylcyclopropyl Indolyl |
Indolyl 1-Phenylcyclopropyl |
| ML-18 EMY-98 |
S
R |
10.4 N.A. |
0.560 0.535 |
4-Nitrophenyl 4-Nitrophenyl |
Indolyl Indolyl |
[1-(4-Methoxyphenyl)cyclohexyl]methyl 1-(4-Methoxyphenyl)cyclohexyl |
| ML-16 EMY-96 |
S
R |
2.0 0.19 |
0.589 0.592 |
4-Nitrophenyl 4-Nitrophenyl |
Indolyl Indolyl |
1-(2-Pyridyl)cyclohexyl 1-(2-Pyridyl)cyclohexyl |
| PD-362 ST-6 |
S
R |
4.1 0.023 |
0.606 0.608 |
4-Nitrophenyl 4-Nitrophenyl |
Indolyl Indolyl |
1-(4-Methoxy-2-pyridyl)cyclohexyl 1-(4-Methoxy-2-pyridyl)cyclohexyl |
| ST-11 ST-9 |
S
R |
0.7 N.A. |
0.602 0.595 |
4-Nitrophenyl 4-Nitrophenyl |
1-Phenylcyclohexyl Indolyl |
Indolyl 1-Phenylcyclohexyl |
| (±)-5b |
S
R |
22.2 2.3 |
0.554 0.560 |
4-Bromophenyl 4-Bromophenyl |
6-Methyl-3(2H)-pyridazinone Ethyl |
3-Methoxyphenyl 3-Methoxyphenyl |
| (±)-5c |
S
R |
N.A. 0.84 |
0.602 0.585 |
4-Bromophenyl 4-Bromophenyl |
6-Methyl-3(2H)-pyridazinone n-C3H7 |
3-Methoxyphenyl 3-Methoxyphenyl |
| (±)-5d |
S
R |
N.A. 5.4 |
0.548 0.561 |
4-Bromophenyl 4-Bromophenyl |
6-Methyl-3(2H)-pyridazinone i-Propyl |
3-Methoxyphenyl 3-Methoxyphenyl |
| (±)-5e |
S
R |
7.0 0.089 |
0.553 0.591 |
4-Bromophenyl 4-Bromophenyl |
3-Methoxyphenyl n-C4H9 |
n-C4H9 3-Methoxyphenyl |
| (±)-5f |
S
R |
N.A. 0.54 |
0.537 0.576 |
4-Bromophenyl 4-Bromophenyl |
3-Methoxyphenyl Phenyl |
Phenyl 3-Methoxyphenyl |
| (±)-5g |
S
R |
N.A. 13.7 |
0.550 0.550 |
4-Bromophenyl 4-Bromophenyl |
6-Methyl-3(2H)-pyridazinone Ethyl |
3-Methoxyphenyl 3-Methoxyphenyl |
| Frohn-12 Frohn-11 |
S
R |
3.5 0.034 |
0.576 0.630 |
5-Methoxyindolyl 5-Methoxyindolyl |
Ethyl (subpockets II and III) Benzene ring of benzimidazole |
Benzene ring of benzimidazole Ethyl (between subpockets II and III) |
| Comp. 1 Comp. 2 |
S
R |
0.035 0.030 |
0.770 0.712 |
4-Bromophenyl 4-Bromophenyl |
Benzyl Benzyl |
Piperidyl Piperidyl |
Potency of compounds were used as reported previously [9;13;15] or determined in the present work (see Table 2).
Similarity (Sim.) between aligned molecule and the template was calculated using the MVD program. Highest values of the similarity, which correlate with agonistic activity at FPR2 are in bold.
Subpockets I, II, and III in the notation of our previously published pharmacophore model [12].
A visual inspection of the molecule overlays on the 5-molecule FPR2 template showed that in most cases the active enantiomers had alignment modes with non-polar molecular fragments located in subpocket II, while bromo- or nitro-substituted phenyl rings were always positioned in subpocket I (Table 3). In particular, moieties without polar atoms (i.e., ethyl, propyl, i-propyl, butyl, or phenyl for R-enantiomers 5b, 5c, 5d, 5e, 5f, and 5g; tetrahydronaphthyl or phenylcyclopropyl for S-enantiomers ML-8 and ML-11, respectively) occupied subpocket II, while polar substructures (methoxy in benzene rings or indole NH groups) were located in subpocket III for these alignments. Similarly, for enantiomers Frohn-11 and Frohn-12 [9], the non-polar part of the benzimidazole moiety (i.e., benzene ring) was localized in subpocket II for the active Frohn-11 (R-form), while this ring was localized in subpocket III for the low-activity enantiomer Frohn-12 (S-form). It should be noted that for the enantiomeric pairs of Compound 1/Compound 2 [13], ML-16/EMY-96, and PD-362/ST-6, a similar location of their submolecules in subpockets II and III was obtained in the superimpositions (Table 3). Accordingly, all compounds in these pairs were active FPR2 agonists (Table 2).
For the enantiomer pair EMY-98/ML-18, all three chiral center substituents were located in the same subpockets for both molecules in our modeling study, but only the S-enantiomer ML-18 was active. This is likely due to the high flexibility of molecular chains connecting the asymmetric carbon with key moieties and, in spite of the reverse configuration of the chiral center, the molecular fragments could theoretically bind within the same three hydrophobic subpockets. On the other hand, the inverted S-enantiomer configuration appears to lead to undesirable steric and electronic characteristics, resulting in a loss of FPR2 agonist activity. Indeed, these enantiomers do have different positions of electropositive and electronegative field points around the periphery of their skeletons despite having similar hydrophobic center positions. For example, a group of negative field points of the template (blue tetrahedra) coincide well with a large negative field point (blue sphere) corresponding to both carbonyl groups of compound ML-18 (see green arrow in Figure 5, upper panel). In contrast, the conformation of inactive enantiomer EMY-98 aligned with the template has two carbonyl groups located on opposite sides of the molecular skeleton. This leads to a lower coincidence of negative field points originated from molecule EMY-98 and the template. Additionally, EMY-98 is characterized by an incomplete geometric overlap with the template (see green arrow in Figure 5, lower panel). Obviously, such a difference between ML-18 and EMY-98 is determined by the chiral character of the template. Hence, the group of negative field points (blue tetrahedra) around the chiral center can be regarded as an important pharmacophore feature, along with hydrophobic centers H1, H2, and H3, which allows differentiation between two highly flexible enantiomers in three-dimensional space. Indeed, the following molecular modeling supported a role of electro-negative and -positive groups in binding of FPR2 agonists.
Since the geometry and location of the FPR2-binding site were not considered explicitly in the field point methodology described above, a homology model of FPR2 was created (see Materials and Methods). Using the MVD “Detect cavity” module, we found that the ligand binding site(s) of FPR2 had a non-symmetric dumb-bell shape with two cavities of different sizes (Figure 6A). The smaller cavity (volume 25 Å3) is located deep within the binding site and is surrounded by residues Val105, Asp106, Leu109, Phe110, Arg201, Trp254, and Gln258. The larger cavity of the docking site (volume 241 Å3) opens outside the receptor and has a complex shape with two regions (Figures 6B and 7, center panel). A narrow channel connecting the two cavities is bounded by residues Phe257, Val260, Ala261, Thr177, Phe178, and Phe180. Thus, virtual detection of the receptor binding sites and subsequent visual inspection showed that the FPR2 binding site has 3 well-defined subpockets. To determine if these subpockets could accommodate the FPR2 agonist pharmacophore, receptor-docking poses of two molecules used previously to construct the FPR2 pharmacophore template (compounds AG-10/5 and AG-10/8) were determined.
Figure 6.

Homology model of FPR2 agonist docking. Panel A. A PDB file of the homology model for FPR2, based on bovine rhodopsin template, was loaded into MVD software and the “Detect cavity” feature was applied with probe size 1.2 Å to identify potential areas of the protein where ligands could be docked. Two cavities were found with volumes 241 Å3 and 25 Å3 (indicated by arrows). The docking pose of PD176252 in the FPR2 lignd-binding site is shown. Panel B. Overlapping docking poses of FPR2 agonists S-(-)-5e (red), AG-10/5 (magenta), AG-10/8 (green), and EMY-96 (yellow) in the FPR2 ligand-binding site with schematic representation of the three receptor subpockets (yellow dashed line) described previously [12].
Figure 7.

Model of chiral compound docking to FPR2. Geometry of the hydrophobic field surface of the pharmacophore model, but not its mirrored (chiral) template matches to the binding site geometry of FPR2. An FPR2 agonist can approach the FPR2 binding site from the top (“mouth”) of the cavity, shown by dashed yellow line around the agonist template (hydrophobic regions H2 and H3) and around the cavity mouth, which includes subpockets II and III. Field points are colored as follows: blue, electron-rich (negative); red, electron-deficient (positive); yellow, van der Waals attractive (steric). Hydrophobic region H1 (usually associated with 4-nitrophenyl or 4-bromophenyl groups in FPR2 agonists) should properly fit into subpocket I of the FPR2 ligand-binding site. The cavity of the FPR2 binding site shows the position of side chain tails of EMY-96 in subpockets II and III. Surface coloring was made according to electrostatic properties, whereby negatively and positively charged areas are shown in red and blue, respectively. It should be noted, that blue (positively charged) surface areas of the receptor correspond to blue field points obtained with positive probe atom and red (negatively charged) surface areas of the receptor correspond to red field points obtained with negative probe atom. Areas of subpockets are indicated with light-blue arrows. Numeration of subpockets and the hydrophobic surface of the FPR2 pharmacophore model are as described previously [12].
According to our docking study, the pharmacophore subpocket I is bound by His102, Val105, Asp106, Leu109, Trp254, Phe257, Ser288, and Phe292 and lies in the smaller 25 Å3 cavity. As reported previously by Fujita et al. [31] this FPR2 domain is occupied by highly hydrophobic bromo-substituted phenyl rings of the FPR agonists (Figure 6). Pharmacophore subpockets II and III form the “mouth” of the FPR2 binding site and lie within the larger 241 Å3 cavity (see Materials and Methods). Pharmacophore subpocket II is bound by Thr168, Ile169, Pro170, Asn179, and Ala181, and this subpocket is occupied mainly by non-polar hydrocarbon groups of the FPR agonists. Molecular subgroups with electronegative heteroatoms can be identified as occupying subpocket III, which is bound by Gly264, Tyr277, Ile279, and Ala181. It should be noted that the FPR2-specific peptide agonist WKYMVM, in its best docking pose, occupies all three subpockets, with the N-terminal indole moiety located in subpocket I (data not shown).
Overall, the geometric configuration of the FPR2 binding site is in a good agreement with the shape of the hydrophobic field obtained for the FPR2 agonist pharmacophore model. It is also clear that the hydrophobic field surface reflecting the shape of the “enantiomeric” (mirrored) template for FPR2 agonist enantiomers does not correspond well to the binding site shape of FPR2 receptor (Figure 7). Additionally, extrema of negative and positive fields (“blue” and “red” field points of the template are shown by icosahedra in Figure 7, upper and lower panels) correspond well to colored areas of the binding site surface calculated by the MVD program with the use of charged probe atoms. Thus, this correspondence can be important for proper orientation of an agonist molecule for penetration into the binding site.
Subsequent modeling with pyridazin-3(2H)-one FPR agonists showed that our FPR2 pharmacophore model is in good agreement with the docking results. For example, a docking pose of S-(-)-5e, completely overlapping with poses of compounds AG-10/5 and AG-10/8, is shown in Figure 6B. The docking study of ten FPR2-active ureidopropanamides showed that the 4-nitrophenyl group of PD168368, ML-16, and ML-8 lies outside of subpocket I, whereas the remaining seven compounds had the 4-nitrophenyl moiety located in subpocket I in their docking poses where it could form H-bonds with Arg201 and Gln258. Other chiral center substituents of these molecules were located within subpockets II and III of the FPR2 binding site similarly to the p-bromo-substituted pyridazine derivatives, and carbonyl and NH-groups of the amide bridge of these molecules form H-bonds with Thr177, Phe1789 and Phe257 residues.
4. Discussion
FPRs are GPCRs that are able to recognize many ligands, often of very different chemical nature [1]. Phylogenetic analysis has revealed that FPRs belong to family of chemosensory GPCR, which also includes vomeronasal receptors, trace-amine associated receptors, and odorant receptors [34]. Recently it was postulated that FPRs expressed in the vomeronasal organs of mammals have an olfactory function associated with the identification of pathogenic states [35], and Bufe et al. [36] found that these receptors exhibited stereo-selective preference for peptides containing D-amino acids. Although numerous GPCR have been characterized as enantioselective receptors [18;19;37], including odorant receptors [38], only one example of enantioselective recognition of non-peptide ligands by FPRs has been observed previously [9]. The growing evidence implicating anti-inflammatory and tissue-protective effects of FPR agonists [16;17] and the recent development of novel chiral ligands as potential therapeutics and agonists of various GPCR [18;19;39] prompted us to search for novel non-peptide small-molecule enantiomeric FPR agonists [12;15].
Previously we found that bombesin receptor antagonists PD168368 and PD176252 and their chiral derivatives were potent FPR1/FPR2 agonists [12]; however, a systematic study of enantiomer pairs was not performed. In the present studies, we evaluated a small library of 22 structural derivatives of PD168368/PD176252, including seven enantiomer pairs, for their ability to activate FPR1/FPR2. We showed that among the ureidopropanamides tested, most of the S-enantiomers preferentially activated FPR2. Because both R- and S- enantiomers in the pairs EMY-96/ML-16 and ST-6/PD-362 induced Ca2+ flux, we propose that it is not only the general chemical scaffold that plays a role in determination of FPR agonist activity for S- or R-counterparts, but also that the specific structure of the chiral center substituents can contribute to ligand recognition. Indeed, analysis of the enantiomers of known chiral FPR2 agonists showed that they could be inactive, less active, or have similar activity at this receptor (see Table 3). Furthermore, three R-enantiomers exhibited antagonist activity at FPR2 (compounds EMY-89 and EMY-98) or at both FPR1 and FPR2 (compound EMY-87), while their S-counterparts (compounds ML-11, ML-18, and ML-8) exhibited FPR agonist activity. Although there are a couple examples of enantiomer pairs with counteractive properties at different GPCRs reported in the literature (e.g., [18]), to date there are no reports of antagonist activity for enantiomers of any known FPR agonist.
Surprisingly, and in contrast to the activity found with the parent compounds PD168368 and PD176252, we found that NO2 and CN derivatives which active in FPR1/FPR2-transfected cells were inactive in our conventional Ca2+ mobilization assay using human neutrophils. This finding is supported by our previous findings that some chiral FPR agonists and their closely related achiral derivatives are much less active for activation of Ca2+ mobilization in human neutrophils than in the FPR-transfected cells (up to 600 fold by EC50 value) [12]. This activity profile could be related to the more flexible structures of the analogs compared to PD168368 and PD176252, where an intramolecular H-bond between an amino group and the carbamide carbonyl atom could significantly restrict conformational freedom of these molecules. Thus, increased flexibility of the chiral agonists could be analogous to conformation changes in FPR2 peptide. Alternatively, because OCH3, CF3, Br, and CH3 groups in para-position of phenyl ring of the FPR2 agonists ST-12, ST-13, ST-15, and ST-16, respectively, are less electronegative than NO2 and CN groups [40] in compounds PD-362 and ST-14, potency of electrostatic and H-bond interactions in subpocket I could be important characteristics in activation of G-protein coupling and Ca2+ flux in human neutrophils.
The possibility that nitro compounds could enter cells with subsequent reduction of their nitro group during the first minute of the Ca2+ mobilization assay in primary cell suspension (neutrophils), but not in HL-60 cells, seems unlikely. Indeed, although enzymatic reduction of nitro compounds could be a relatively fast at hypoxic conditions, their bioreduction at normoxic conditions, which is relevant to the present Ca2+-mobilization assay, is slow [41;42]. The nitro derivatives under investigation (Table 1) are closely related to parent compounds PD168368 and PD176252, which are active under experimental conditions in vivo and ex vivo, at least for several hours [43-45]. For example, PD176252 was previously reported to be active during prolonged (65 h) incubation with myometrial explants [45]. Structures of the nitro compounds (Table 1) differ in substituents remote from the nitro-phenyl moiety, and results obtained by the semi-empirical AM1 method show that changes in charge distribution in the nitro group were negligible when varying substituents in other moieties of the molecules (data not shown). Hence, it is reasonable to consider that these nitro derivatives are active in their initial (not metabolized) forms, at least during the first minute of the Ca2+-mobilization assay. Indeed, these compounds were stable in aqueous solution stored for up to week during testing. Finally, the ureidopropanamides that stimulated Ca2+ flux in FPR-transfected cells still activated other human neutrophil responses, such as chemotaxis, and desensitized FPR2 responsiveness to WKYMVM. Furthermore, pretreatment with probenecid, an anion exchange protein inhibitor [32;33], restored the Ca2+ flux response in human neutrophils treated with these agonists.
Analysis of the literature indicates that probenecid is actually a non-specific inhibitor of multidrug resistance-associated proteins and can have different effects on several other cellular targets. For example, probenecid is able to activate transient receptor potential V2 (TRPV2), Ca2+-permeable nonselective cation channel [46], and transient receptor potential channel subtype A member 1 (TRPA1), a nonselective cation channel [47], and it has been recently reported that probenecid can directly modulate interaction of GPCR and G-proteins [48]. In addition, Prossnitz et al. [49] proposed that primary myeloid cells maintain a subpopulation of FPR in a low-affinity, possibly G protein-free state, which is not a feature of FPR-transfected HL-60 cells. Because allosteric communication between the ligand-binding orthosteric site and the cytoplasmic G-protein-binding surface is a fundamental feature of GPCRs [40], it is possible that certain FPR2 agonists, such as the ureidopropanamides reported here, could stabilize this receptor in a G-protein-free state, and additional agents (e.g., probenecid) could reactivate G-protein coupling. Alternatively, because non-differentiated HL-60 cells and mature neutrophils have different sets of G-proteins able to couple with FPRs [50;51], the unique property of these chiral FPR2 agonists could be their ability to modulate conformational changes in FPRs and functional interactions with different G-proteins. In addition, other known FPR cofactors, such as ADP-ribosyl cyclase CD38 or macrophage receptor with collagenous structure (MARCO) [52;53] in HL-60 cells, could behave somewhat differently from those of primary neutrophils that can be a potential factor to explain the observed results.
We suggest that, unlike the parent compounds PD168368/PD176252, NO2- or CN - substituted phenyl ring derivatives lacking a methyl group at the chiral center may uniquely bind FPR2 and modulate receptor activity as biased agonists. Based on this concept, a biased agonist exhibits differential ability to activate various signaling pathways [54]. The molecular basis of this theory is that chemically-distinct agonists of a particular GPCR can induce a variety of conformation changes in this GPCR, which then promotes distinct affinity and coupling efficiencies to the various G proteins and also possibly to other cofactors that interact with the receptor and leads to the activation of different pathways [54;55]. Although there are many examples of biased activity for other GPCR agonists (reviewed in [54]), the phenomenon of biased activity at FPRs has not been clearly elucidated (e.g., [56]). Thus, future experiments will be necessary to characterize these potential features of chiral ureidopropanamides by evaluation of various FPR-dependent intercellular pathways.
Our previous modeling experiments suggested that FPR2 agonists might not precisely occupy all three proposed receptor subpockets [12]. For example, achiral FPR2 agonists occupied subpockets I and II (compounds AG-09/3 and AG-09/4) or subpockets I and III (compound AG-09/10) only. Nevertheless, the present pharmacophore modeling assumes that chiral FPR2 agonists should occupy all three subpockets. On the other hand, docking poses for several chiral FPR2 agonists (i.e., PD168368, ML-16, and ML-8) occupied subpockets II and III, while the 4-nitrophenyl group of these molecules did not access subpocket I. Hence, the docking poses of these molecules are quite different as compared to the overlay on the field point pharmacophore model. Perhaps, docking was restricted by the bulkiness of compound substituents. Hence, the narrow channel between subpocket I and the rest of the binding site may not allow molecules to penetrate into subpocket I, which is located deep in the FPR2 macromolecule. Although there are significant differences between EC50 for these agonists at FPR2, the relevance of virtual docking study to functional effects is unclear. It should be noted, that our docking studies were completed for a rigid FPR2 structure. Thus, we suggest that geometric differences between docked poses of the molecules and conformations of their best-fit overlays on the FPR2 pharmacophore model can also be due to a flexibility of the receptor itself. Improved results could be obtained with X-ray structures of ligand-receptor complexes, which are not available for FPRs so far. However, efforts are now in progress to isolate crystals of such complexes with quality sufficient for X-ray study [57]. Success in these efforts will eventually allow computational modeling and docking with higher precision.
One of the most important outcomes of this work is the finding that the FPR2 homology modeling and ligand-based pharmacophore modeling are in good agreement with each other. These aspects of the computational investigation were performed independently of each other and show that the first interaction of an agonist with FPR2 could fit well with the lock and key hypothesis [58]. Using field point methodology, homology modeling, and virtual docking, we proposed a molecular model that can discriminate between active and non-active enantiomers and explain stereoselective activity of chiral FPR2 agonists. The fact that FPR2 is able to discriminate between the enantiomers is the consequence of the presence of three asymmetric hydrophobic subpockets at the main well-buried orthosteric FPR2-binding site with specific orientation of charged regions. Hence, active enantiomers can be in either R- or S-configurations, depending on the molecular scaffold and specific chiral center substituents. This model could provide guidance for the rational design of novel potent and selective FPR agonists with unique properties.
Acknowledgements
This work was supported in part by the National Institutes of Health grant GM103500, an equipment grant from the M. J. Murdock Charitable Trust, and the Montana State University Agricultural Experimental Station.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) Family. Pharmacol Rev. 2009;61:119–61. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schiffmann E, Corcoran BA, Wahl SM. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc Natl Acad Sci USA. 1975;72:1059–62. doi: 10.1073/pnas.72.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao JL, Murphy PM. Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J Biol Chem. 1993;268:25395–401. [PubMed] [Google Scholar]
- 4.Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17:501–19. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Gavins FN. Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia-reperfusion injury? Trends Pharmacol Sci. 2010;31:266–76. doi: 10.1016/j.tips.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christophe T, Karlsson A, Dugave C, Rabiet MJ, Boulay F, Dahlgren C. The synthetic peptide Trp-Lys-Tyr-Met-Val-Met-NH2 specifically activates neutrophils through FPRL1/lipoxin A4 receptors and is an agonist for the orphan monocyte-expressed chemoattractant receptor FPRL2. J Biol Chem. 2001;276:21585–93. doi: 10.1074/jbc.M007769200. [DOI] [PubMed] [Google Scholar]
- 7.Nanamori M, Cheng X, Mei J, Sang H, Xuan Y, Zhou C, Wang MW, Ye RD. A novel nonpeptide ligand for formyl peptide receptor-like 1. Mol Pharmacol. 2004;66:1213–22. doi: 10.1124/mol.104.004309. [DOI] [PubMed] [Google Scholar]
- 8.Schepetkin IA, Kirpotina LN, Khlebnikov AI, Quinn MT. High-throughput screening for small-molecule activators of neutrophils: Identification of novel N-formyl peptide receptor agonists. Mol Pharmacol. 2007;71:1061–74. doi: 10.1124/mol.106.033100. [DOI] [PubMed] [Google Scholar]
- 9.Frohn M, Xu H, Zou X, Chang C, McElvaine M, Plant MH, Wong M, Tagari P, Hungate R, Bürli RW. New ‘chemical probes’ to examine the role of the hFPRL1 (or ALXR) receptor in inflammation. Bioorg Med Chem. 2007;17:6633–7. doi: 10.1016/j.bmcl.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 10.Cilibrizzi A, Quinn MT, Kirpotina LN, Schepetkin IA, Holderness J, Ye RD, Rabiet MJ, Biancalani C, Cesari N, Graziano A, Vergelli C, Pieretti S, Dal P, V, Giovannoni MP. 6-Methyl-2,4-disubstituted pyridazin-3(2H)-ones: A novel class of small-molecule agonists for formyl peptide receptors. J Med Chem. 2009;52:5054–7. doi: 10.1021/jm900592h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirpotina LN, Khlebnikov AI, Schepetkin IA, Ye RD, Rabiet MJ, Jutila MA, Quinn MT. Identification of novel small-molecule agonists for human formyl peptide receptors and pharmacophore models of their recognition. Mol Pharmacol. 2010;77:159–70. doi: 10.1124/mol.109.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schepetkin IA, Kirpotina LN, Khlebnikov AI, Jutila MA, Quinn MT. Gastrin-releasing peptide/neuromedin B receptor antagonists PD176252, PD168368, and related analogs are potent agonists of human formyl-peptide receptors. Mol Pharmacol. 2011;79:77–90. doi: 10.1124/mol.110.068288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forsman H, Kalderen C, Nordin A, Nordling E, Jensen AJ, Dahlgren C. Stable formyl peptide receptor agonists that activate the neutrophil NADPH-oxidase identified through screening of a compound library. Biochem Pharmacol. 2011;81:402–11. doi: 10.1016/j.bcp.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Khlebnikov AI, Schepetkin IA, Kirpotina LN, Brive L, Dahlgren C, Jutila MA, Quinn MT. Molecular docking of 2-(benzimidazol-2-ylthio)-N-phenylacetamide-derived small-molecule agonists of human formyl peptide receptor 1. J Mol Model. 2012;18:2831–43. doi: 10.1007/s00894-011-1307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cilibrizzi A, Schepetkin IA, Bartolucci G, Crocetti L, Dal P, V, Giovannoni MP, Graziano A, Kirpotina LN, Quinn MT, Vergelli C. Synthesis, enantioresolution, and activity profile of chiral 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones as potent N-formyl peptide receptor agonists. Bioorg Med Chem. 2012;20:3781–92. doi: 10.1016/j.bmc.2012.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He M, Cheng N, Gao WW, Zhang M, Zhang YY, Ye RD, Wang MW. Characterization of Quin-C1 for its anti-inflammatory property in a mouse model of bleomycin-induced lung injury. Acta Pharmacol Sin. 2011;32:601–10. doi: 10.1038/aps.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SD, Kim YK, Lee HY, Kim YS, Jeon SG, Baek SH, Song DK, Ryu SH, Bae YS. The agonists of formyl peptide receptors prevent development of severe sepsis after microbial infection. J Immunol. 2010;185:4302–10. doi: 10.4049/jimmunol.1001310. [DOI] [PubMed] [Google Scholar]
- 18.Valentine WJ, Kiss GN, Liu J, E S, Gotoh M, Murakami-Murofushi K, Pham TC, Baker DL, Parrill AL, Lu X, Sun C, Bittman R, Pyne NJ, Tigyi G. (S)-FTY720-vinylphosphonate, an analogue of the immunosuppressive agent FTY720, is a pan-antagonist of sphingosine 1-phosphate GPCR signaling and inhibits autotaxin activity. Cell Signal. 2010;22:1543–53. doi: 10.1016/j.cellsig.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patil PN, Li C, Kumari V, Hieble JP. Analysis of efficacy of chiral adrenergic agonists. Chirality. 2008;20:529–43. doi: 10.1002/chir.20506. [DOI] [PubMed] [Google Scholar]
- 20.Agrawal YK, Bhatt HG, Raval HG, Oza PM, Gogoi PJ. Chirality--a new era of therapeutics. Mini Rev Med Chem. 2007;7:451–60. doi: 10.2174/138955707780619617. [DOI] [PubMed] [Google Scholar]
- 21.Bielory L, Leonov A. Stereoconfiguration of antiallergic and immunologic drugs. Ann Allergy Asthma Immunol. 2008;100:1–8. doi: 10.1016/S1081-1206(10)60396-1. [DOI] [PubMed] [Google Scholar]
- 22.Campo VL, Bernardes LS, Carvalho I. Stereoselectivity in drug metabolism: molecular mechanisms and analytical methods. Curr Drug Metab. 2009;10:188–205. doi: 10.2174/138920009787522188. [DOI] [PubMed] [Google Scholar]
- 23.Augelli-Szafran CE, Horwell DC, Kneen C, Ortwine DF, Pritchard MC, Purchase TS, Roth BD, Trivedi BK, Hill D, Suman-Chauhan N, Webdale L. Cholecystokinin B antagonists. Synthesis and quantitative structure-activity relationships of a series of C-terminal analogues of CI-988. Bioorg Med Chem. 1996;4:1733–45. doi: 10.1016/0968-0896(96)00185-x. [DOI] [PubMed] [Google Scholar]
- 24.Tannert R, Milroy LG, Ellinger B, Hu TS, Arndt HD, Waldmann H. Synthesis and structure-activity correlation of natural-product inspired cyclodepsipeptides stabilizing F-actin. J Am Chem Soc. 2010;132:3063–77. doi: 10.1021/ja9095126. [DOI] [PubMed] [Google Scholar]
- 25.Gourni E, Bouziotis P, Benaki D, Loudos G, Xanthopoulos S, Paravatou-Petsotas M, Mavri-Vavagianni M, Pelecanou M, Archimandritis SC, Varvarigou AD. Structural assessment and biological evaluation of two N3S bombesin derivatives. J Med Chem. 2009;52:4234–46. doi: 10.1021/jm900360d. [DOI] [PubMed] [Google Scholar]
- 26.McGuinness D, Malikzay A, Visconti R, Lin K, Bayne M, Monsma F, Lunn CA. Characterizing cannabinoid CB2 receptor ligands using DiscoveRx PathHunter b arrestin assay. J Biomol Screen. 2009;14:49–58. doi: 10.1177/1087057108327329. [DOI] [PubMed] [Google Scholar]
- 27.Cheeseright T, Mackey M, Rose S, Vinter A. Molecular field extrema as descriptors of biological activity: definition and validation. J Chem Inf Model. 2006;46:665–76. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- 28.Vinter JG. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J Comput Aided Mol Des. 1994;8:653–68. doi: 10.1007/BF00124013. [DOI] [PubMed] [Google Scholar]
- 29.Cheeseright TJ, Mackey MD, Scoffin RA. High content pharmacophores from molecular fields: a biologically relevant method for comparing and understanding ligands. Curr Comput Aided Drug Des. 2011;7:190–205. doi: 10.2174/157340911796504314. [DOI] [PubMed] [Google Scholar]
- 30.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 31.Fujita H, Kato T, Watanabe N, Takahashi T, Kitagawa S. Stimulation of human formyl peptide receptors by calpain inhibitors: homology modeling of receptors and ligand docking simulation. Arch Biochem Biophys. 2011;516:121–7. doi: 10.1016/j.abb.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 32.Merritt JE, McCarthy SA, Davies MP, Moores KE. Use of fluo-3 to measure cytosolic Ca2+ in platelets and neutrophils. Loading cells with the dye, calibration of traces, measurements in the presence of plasma, and buffering of cytosolic Ca2+. Biochem J. 1990;269:513–9. doi: 10.1042/bj2690513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abrahamse SL, Rechkemmer G. Identification of an organic anion transport system in the human colon carcinoma cell line HT29 clone 19A. Pflugers Arch. 2001;441:529–37. doi: 10.1007/s004240000437. [DOI] [PubMed] [Google Scholar]
- 34.Riviere S, Challet L, Fluegge D, Spehr M, Rodriguez I. Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature. 2009;459:574–7. doi: 10.1038/nature08029. [DOI] [PubMed] [Google Scholar]
- 35.Liberles SD, Horowitz LF, Kuang D, Contos JJ, Wilson KL, Siltberg-Liberles J, Liberles DA, Buck LB. Formyl peptide receptors are candidate chemosensory receptors in the vomeronasal organ. Proc Natl Acad Sci USA. 2009;106:9842–7. doi: 10.1073/pnas.0904464106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bufe B, Schumann T, Zufall F. Formyl peptide receptors from immune and vomeronasal system exhibit distinct agonist properties. J Biol Chem. 2012;287:33644–55. doi: 10.1074/jbc.M112.375774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waelbroeck M, Lazareno S, Pfaff O, Friebe T, Tastenoy M, Mutschler E, Lambrecht G. Stereoselective recognition of the enantiomers of phenglutarimide and of six related compounds by four muscarinic receptor subtypes. Br J Pharmacol. 1996;119:1319–30. doi: 10.1111/j.1476-5381.1996.tb16041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bohbot JD, Dickens JC. Characterization of an enantioselective odorant receptor in the yellow fever mosquito Aedes aegypti. PLoS One. 2009;4:e7032. doi: 10.1371/journal.pone.0007032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soudjin W, van W, I, IJzerman AP. Stereoselectivity of drug-receptor interactions. IDrugs. 2003;6:43–56. [PubMed] [Google Scholar]
- 40.Marriott S, Reynolds WF, Taft RW, Topsom RD. Substituent electronegativity parameters. J Org Chem. 1984;49:959–65. [Google Scholar]
- 41.Schepetkin IA, Cherdyntseva NV, Kagiya VT. Sanazole as substrate of xanthine oxidase and microsomal NADPH/cytochrome P450 reductase. Pathophysiology. 2001;8:119–27. doi: 10.1016/s0928-4680(01)00073-6. [DOI] [PubMed] [Google Scholar]
- 42.Wisniewska A, Niemira M, Jagiello K, Potega A, Swist M, Henderson C, Skwarska A, Augustin E, Konopa J, Mazerska Z. Diminished toxicity of C-1748, 4-methyl-9-hydroxyethylamino-1-nitroacridine, compared with its demethyl analog, C-857, corresponds to its resistance to metabolism in HepG2 cells. Biochem Pharmacol. 2012;84:30–42. doi: 10.1016/j.bcp.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 43.Merali Z, Bedard T, Andrews N, Davis B, McKnight AT, Gonzalez MI, Pritchard M, Kent P, Anisman H. Bombesin receptors as a novel anti-anxiety therapeutic target: BB1 receptor actions on anxiety through alterations of serotonin activity. J Neurosci. 2006;26:10387–96. doi: 10.1523/JNEUROSCI.1219-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park HJ, Kim SR, Kim MK, Choi KS, Jang HO, Yun I, Bae SK, Bae MK. Neuromedin B receptor antagonist suppresses tumor angiogenesis and tumor growth in vitro and in vivo. Cancer Lett. 2011;312:117–27. doi: 10.1016/j.canlet.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 45.Tattersall M, Cordeaux Y, Charnock-Jones DS, Smith GC. Expression of gastrin-releasing peptide is increased by prolonged stretch of human myometrium, and antagonists of its receptor inhibit contractility. J Physiol. 2012;590:2081–93. doi: 10.1113/jphysiol.2012.228239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bang S, Kim KY, Yoo S, Lee SH, Hwang SW. Transient receptor potential V2 expressed in sensory neurons is activated by probenecid. Neurosci Lett. 2007;425:120–5. doi: 10.1016/j.neulet.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 47.McClenaghan C, Zeng F, Verkuyl JM. TRPA1 agonist activity of probenecid desensitizes channel responses: Consequences for screening. Assay Drug Dev Technol. 2012 doi: 10.1089/adt.2012.447. [DOI] [PubMed] [Google Scholar]
- 48.Greene TA, Alarcon S, Thomas A, Berdougo E, Doranz BJ, Breslin PA, Rucker JB. Probenecid inhibits the human bitter taste receptor TAS2R16 and suppresses bitter perception of salicin. PLoS One. 2011;6:e20123. doi: 10.1371/journal.pone.0020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prossnitz ER, Quehenberger O, Cochrane CG, Ye RD. Signal transducing properties of the N-formyl peptide receptor expressed in undifferentiated HL60 cells. J Immunol. 1993;151:5704–15. [PubMed] [Google Scholar]
- 50.Amatruda TT, III, Steele DA, Slepak VZ, Simon MI. Ga16, a G protein a subunit specifically expressed in hematopoietic cells. Proc Natl Acad Sci USA. 1991;88:5587–91. doi: 10.1073/pnas.88.13.5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tenailleau S, Corre I, Hermouet S. Specific expression of heterotrimeric G proteins G12 and G16 during human myeloid differentiation. Exp Hematol. 1997;25:927–34. [PubMed] [Google Scholar]
- 52.Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nature Med. 2001;7:1209–16. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 53.Brandenburg LO, Konrad M, Wruck CJ, Koch T, Lucius R, Pufe T. Functional and physical interactions between formyl-peptide-receptors and scavenger receptor MARCO and their involvement in amyloid beta 1-42-induced signal transduction in glial cells. J Neurochem. 2010;113:749–60. doi: 10.1111/j.1471-4159.2010.06637.x. [DOI] [PubMed] [Google Scholar]
- 54.Zheng H, Loh HH, Law PY. Agonist-selective signaling of G protein-coupled receptor: mechanisms and implications. IUBMB Life. 2010;62:112–9. doi: 10.1002/iub.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Urban JD, Clarke WP, von ZM, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 56.Bena S, Brancaleone V, Wang JM, Perretti M, Flower RJ. Annexin A1 Interaction with the FPR2/ALX Receptor: Identification of distinct domains and downstream associated signaling. J Biol Chem. 2012;287:24690–7. doi: 10.1074/jbc.M112.377101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Zhang S. Production of a bioengineered G-protein coupled receptor of human formyl peptide receptor 3. PLoS One. 2011;6:e23076. doi: 10.1371/journal.pone.0023076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caceres RA, Pauli I, Timmers LF, de Azevedo WFJ. Molecular recognition models: a challenge to overcome. Curr Drug Targets. 2008;9:1077–83. doi: 10.2174/138945008786949414. [DOI] [PubMed] [Google Scholar]