

Abstract
Over 77 million dogs and 93 million cats share our households in the United States. Multiple studies have demonstrated the importance of pets in their owners' physical and mental health. Given the large number of companion animals in the United States and the proximity and bond of these animals with their owners, understanding and preventing the diseases that these companions bring with them are of paramount importance. Zoonotic protozoal parasites, including toxoplasmosis, Chagas' disease, babesiosis, giardiasis, and leishmaniasis, can cause insidious infections, with asymptomatic animals being capable of transmitting disease. Giardia and Toxoplasma gondii, endemic to the United States, have high prevalences in companion animals. Leishmania and Trypanosoma cruzi are found regionally within the United States. These diseases have lower prevalences but are significant sources of human disease globally and are expanding their companion animal distribution. Thankfully, healthy individuals in the United States are protected by intact immune systems and bolstered by good nutrition, sanitation, and hygiene. Immunocompromised individuals, including the growing number of obese and/or diabetic people, are at a much higher risk of developing zoonoses. Awareness of these often neglected diseases in all health communities is important for protecting pets and owners. To provide this awareness, this review is focused on zoonotic protozoal mechanisms of virulence, epidemiology, and the transmission of pathogens of consequence to pet owners in the United States.
INTRODUCTION
There are over 77 million dogs and 93 million cats in our households in the United States alone. Approximately 62% of households have at least one pet, and over half of these households have multiple pets (1). Various studies have demonstrated the importance of pets in overall health and well-being and for providing social support (2–5). Consistent with this devotion to pets, owners in the United States spend approximately $10.94 billion annually on pet supplies and over-the-counter pet medications and $14.11 billion annually on veterinary care (1). Given the number of companion animals in the United States and the bond with their owners, awareness and prevention of the zoonotic diseases of our companions are of paramount importance. Protozoal diseases, such Chagas' disease and leishmaniasis, are insidious, with large numbers of asymptomatic animals being able to transmit disease. Giardia duodenalis and Toxoplasma gondii, endemic to the United States, have high prevalences in companion animals (6, 7) (Fig. 1). Leishmania species and Trypanosoma cruzi are regional and have low prevalences in the United States but are significant sources of human disease worldwide and are reemerging and expanding their geographic distribution in companion animals in the United States (8, 9). Thankfully, we are generally protected by intact immune systems, and our health is bolstered by good nutrition, sanitation, and hygiene, but immunocompromised individuals, including the growing number of obese and/or diabetic individuals, in the United States are at a much higher risk of developing any zoonosis (10, 11). As such, an awareness of these often neglected diseases in veterinary and human health communities is important for protecting pet health and preventing human disease. In this article, we review mechanisms of virulence, epidemiology, transmission, and clinical signs of zoonotic protozoal pathogens of consequence to pet owners in the United States.
Fig 1.

Global burden of zoonotic protozoal disease in humans. (Panels D and E are adapted from references 349 and 350, respectively, with permission of Elsevier.)
TOXOPLASMOSIS
Life Cycle and Mechanisms of Virulence
Toxoplasma gondii has a high prevalence globally and is capable of infecting all species of animals and birds (12). Definitive hosts for T. gondii are members of the family Felidae (Fig. 2) (12, 13). Felids are the only animals capable of shedding oocysts in their feces and transmitting the parasite by this means. In other host species, the ingestion of infective oocysts from cat feces or contaminated soil, water, or other materials can lead to the formation of tissue cysts that are infective via the secondary consumption of infected tissues (12, 14). Oocysts are shed in large numbers by acutely infected cats once for approximately 2 weeks, except in cases of feline immunosuppression, such as coinfection with feline immunodeficiency virus (FIV) or feline leukemia virus (FeLV), which can result in secondary shedding (15). After shedding, parasite sporulation takes place in 1 to 5 days, providing infective oocysts (13, 16). T. gondii rapidly excysts within the environment of the intestine, dependent upon temperature, pH, bile salts, and trypsin, developing into the highly infective tachyzoite form (17, 18). Cellular infection is rapidly established, resulting in bradyzoite-containing tissue cysts (Fig. 3) (17, 18). The consumption of infected tissue or fecal material by naïve, primarily young, felines results in their infection and subsequent shedding of infectious oocysts (19). People become infected through the accidental consumption of feline fecal material, through food or water with fecal contamination, through the consumption of undercooked meat containing infective cysts, through transplantation, or transplacentally from mother to fetus (7, 20). Cases of congenital infection and human immunodeficiency virus (HIV) coinfection cause the most serious morbidity, resulting in severe neurologic and ocular diseases or miscarriage (21). T. gondii tachyzoite virulence is dependent upon multiple parasite factors, including those necessary for motility, cellular invasion, and immune evasion.
Fig 2.

Toxoplasma gondii life cycle. Domesticated and wild cats are the definitive hosts of Toxoplasma gondii and become infected after the consumption of animals containing infective tissue cysts. Fecal oocysts are shed in large numbers by acutely infected cats for approximately 2 weeks. After shedding, parasite sporulation into infective oocysts takes place in 1 to 5 days. The ingestion of oocysts by other species leads to the formation of tissue. T. gondii rapidly excysts within the intestine, developing into the highly invasive tachyzoite form. Cellular infection results in bradyzoite-containing tissue cysts.
Fig 3.
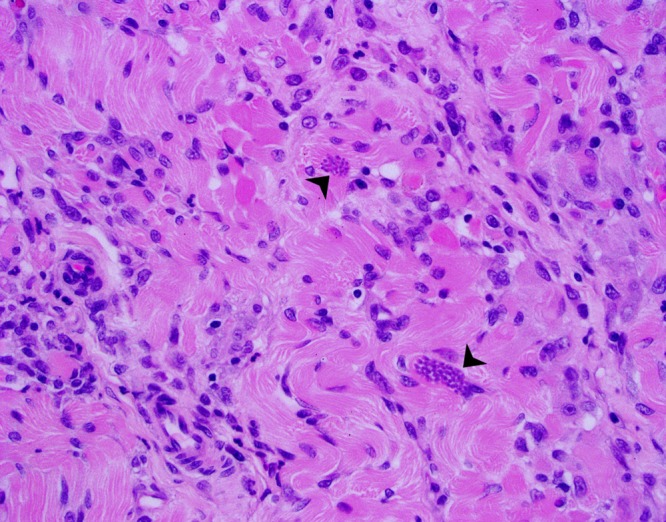
Canine toxoplasmosis. This section of canine skeletal muscle contains numerous lymphocytes and macrophages with myofibrillar necrosis and fibrosis and a myriad of Toxoplasma gondii tachyzoites (arrowheads) confirmed by immunohistochemistry (magnification, ×40). (Reproduced from a slide by Alexandria University, Department of Veterinary Pathology, Egyptian Society for Comparative and Clinical Pathology, Alexandria, Egypt, from the Armed Forces Institute of Pathology Wednesday Slide Conference 2007-2008, Conference 8, Case 2.)
T. gondii motility and cellular invasion were thoroughly reviewed by Carruthers and Boothroyd (18). A brief synopsis is provided here. T. gondii locomotion requires linear myosin, F-actin filaments, and gliding-associated proteins anchored between the plasma membrane and the inner membrane complex (17, 18). Invasion and the formation of a parasitophorous vacuole (PV) occur through apical parasite polarization and the adhesion of micronemal proteins and apical membrane antigen 1 (AMA-1) (18). Rhoptry proteins, which complex with AMA-1, are expelled by the parasite to form a moving junction, which migrates along the parasite surface during invasion to envelope the parasite in a PV (18).
After invasion, T. gondii utilizes multiple mechanisms of immune evasion to facilitate parasite survival and persistence within the host. The interaction of T. gondii and the immune system was thoroughly reviewed by Lang et al. (22). One mechanism of evasion is via rapid cellular invasion by T. gondii, which minimizes parasite exposure to host complement and antibodies. The moving junction and the incorporation of the host cell membrane into the PV membrane create an immune-privileged site for the parasite, devoid of transmembrane proteins and inhibited from fusion with phagolysozomes (23). Once within the PV, T. gondii continues its stealth-like state by inducing the production of the anti-inflammatory cytokines interleukin 10 (IL-10) and transforming growth factor beta (TGF-β), inhibiting the production of the proinflammatory cytokines interleukin 12 (IL-12) and tumor necrosis factor alpha (TNF-α), and slowing its recognition by blocking major histocompatibility complex class II (MHC-II) expression (24–28). Apoptotic body mimicry through the parasite surface expression of phosphatidylserine may facilitate the production of TGF-β and the degradation of induced nitric oxide synthase (iNOS) (29). Additionally, T. gondii maintains a favorable host cell state by inhibiting the programmed cell death of infected cells and inducing leukocyte apoptosis (30–32). In a fascinating virulence approach to maintain primary host infection, T. gondii induces behavioral changes in infected rats, causing an altered avoidance of cats and increased signaling in reproductive regions of the medial amygdala, which result in an attraction to cat urine, increasing the likelihood of feline consumption and subsequent feline infection (33).
Epidemiology and Transmission Dynamics
Felines are very susceptible to T. gondii infection; a single bradyzoite from a tissue cyst can cause the establishment of infection and the subsequent shedding of millions of oocysts (13). Cats are, however, significantly less susceptible to infection through the ingestion of oocysts (13). Oocysts are highly infective to most nonfeline mammalian hosts, indicating adaptations for fecal-oral transmission in these species, including people (13). Cats are the only species demonstrated to actively shed oocysts. However, coprophagic animals, including our “best friends,” dogs, can transport and subsequently appear to shed T. gondii oocysts (34). Cats are generally thought to actively shed oocysts only after an initial exposure, but immune-suppressive conditions caused by comorbidity or immunosuppressive therapy can result in shedding a second time in young cats after reexposure (15). Seroprevalence rates in U.S. cats vary by location, ranging from 18 to 80%, primarily dependent on climate, with higher seroprevalences in more humid regions (14). The estimated worldwide prevalence of feline infection with T. gondii is 30 to 40% (14).
The feeding of undercooked or raw-food diets to cats has been associated with an increased risk for infection with T. gondii (35). Cats with outdoor access and those from rural areas, with predation as a main food source, are more likely than indoor cats to be seropositive, with rates of 39% in outdoor cats versus 26% in indoor-dwelling cats in one study and up to 69% in rural outdoor cats (35).
Diagnosis in cats is difficult, especially as a means to predict human exposure. The shedding of fecal oocysts is transient and generally occurs only once in the life of a cat, temporarily after the primary exposure. Therefore, testing via fecal flotation or centrifugation concentration has a poor sensitivity for the diagnosis of feline infection (14, 35). Serologic testing is equally poor as a predictor of possible human exposure, as seroconversion occurs after active infection and shedding (14, 35). Due to limitations in the diagnosis and prevention of T. gondii in cats and its extensive distribution, the prevention of human infection is targeted toward risk avoidance.
Human exposure to T. gondii occurs frequently, with an estimated serologic prevalence of 9.0% in the United States in persons 6 to 49 years of age, based on 1999-2004 National Health and Nutrition Examination Survey (NHANES III) data (36), although the rate of T. gondii serologic positivity decreased from 14% in 1998 to 9% in 2004 (36). In other areas of the world, the serologic prevalence of T. gondii is much higher, ranging between 8 and 50% (37, 38), where again, serologic prevalence in humans is closely associated with variability in climate. T. gondii exposure occurs commonly throughout the world and across economic and social strata.
Human exposure occurs secondary to exposure to fecal oocysts present in feces of T. gondii-infected cats through many routes, including contaminated water or food, contaminated soil (gardening and sandboxes, etc.), the cleaning of litter boxes, or the consumption of improperly cooked or processed meats, dairy products, or shellfish (7). Shellfish have become infected as filter feeders exposed to contaminated water containing cat feces (7). Off the coast of California, T. gondii has been found in ocean water likely contaminated from rivers containing cat feces, which either survived or bypassed sewage treatment facilities (39, 40). Jones et al. (7) conducted a comprehensive case-control study of 148 U.S. toxoplasmosis cases and 413 control patients, and they found the following risk factors for human infection: (i) eating raw ground beef, (ii) eating rare lamb, (iii) eating locally grown and processed cured meats, (iv) working with meat, (v) drinking unpasteurized goat's milk, (vi) owning three or more kittens, and (vii) eating raw oysters, clams, or mussels (7). The U.S. food supply was evaluated by Dubey et al., who found no indication of oocysts in U.S. beef and chicken and low prevalences in pork (41). All Toxoplasma gondii risk factors are related to either contact with cat fecal material or contact with meats potentially containing tissue cysts. While all of the above-mentioned exposures are important, a recent confounding study by Boyer et al. demonstrated that 31% of patients transmitting toxoplasmosis congenitally to their child indicated none of these common risk factors for T. gondii exposure. While exposure to cat feces was a significant source of exposure to oocysts, ownership of a pet cat was not a significant risk factor (42). While risk avoidance is paramount for the prevention of toxoplasmosis, up to one-third of patients have unrecognized routes of infection. It is likely that only comprehensive screening or effective vaccination programs, which do not currently exist, may help further prevent congenital toxoplasmosis.
A small percentage of people and animals exposed to T. gondii develop clinical disease. However, largely due to its global distribution, the morbidity rate due to T. gondii is high. For instance, the incidence of toxoplasmic retinochoroiditis ranges from approximately 0.4 to 2.46 cases per 100,000 people, making T. gondii the most common identifiable cause of posterior uveitis in many regions of the world (43). Congenital toxoplasmosis impacts approximately 500 to 5,000 of 4.2 million live births per year in the United States (36). National surveillance in France in 2007 estimated the prevalence of congenital toxoplasmosis to be approximately 3.3 per 10,000 live births (44). Considerations for immunosuppressed individuals must be made, as immunosuppression may cause disease recrudescence or a susceptibility to new infection. HIV-associated toxoplasmosis resulted in 1.25% (2,985 cases) of total HIV-related hospitalizations in 2008, making it a significant comorbidity of HIV-infected individuals (45). HIV-associated toxoplasmosis accounted for the vast majority of adult primary disease due to T. gondii, with 83.3% of toxoplasmosis-associated hospitalizations occurring in HIV-positive individuals (45).
Prevention
Toxoplasmosis is, for the most part, preventable by the avoidance of exposure to cat feces and the careful handling and preparation of food. Cat owners can reduce cat exposure to T. gondii by following a few simple measures: (i) do not feed pet cats raw meat, (ii) limit and monitor pet cats' outdoor activity to prevent the ingestion of potentially infected birds or rodents, (iii) control intermediate hosts such as rodents, and (iv) maintain routine vaccination for common viral diseases (FeLV, feline rhinotracheitis virus, and feline parvovirus), deworming, and other routine veterinary care for pet cats to reduce the risk of comorbidities. Dogs can serve as mechanical carriers of T. gondii and should be kept away from litter boxes; should not be fed raw meat, rodents, or game; and should have routine veterinary care. Human infection can be prevented by consuming only pasteurized dairy products and meat which has been properly cooked to 145°F (63°C) for whole cuts of meat, excluding poultry; to 160°F (71°C) for ground meats; and to 165°F (74°C) for poultry (14). Second, all cutting boards, utensils, and hands should be washed with soap after use with uncooked meat and unwashed produce and before cutting or preparing “ready-to-eat” foods. Finally, the freezing of meats to 10.4°F (−12°C) for 24 h reduces the chance of infection by T. gondii (14). Environmentally acquired infections may also be prevented by the following measures: (i) wash hands and teach children the importance of washing hands often; (ii) cover outdoor sandboxes; (iii) follow means to prevent cats and dogs from becoming infected; (iv) if pregnant or immunocompromised, wear gloves when gardening or handling sand or soil; (v) avoid drinking untreated water; (vi) change litter boxes daily to prevent the sporulation of T. gondii within the litter; and (vii) if pregnant or immunocompromised, do not handle unknown or stray cats or kittens. Multiple serologic and epidemiologic surveys have noted that currently known risk factors cannot account for 14 to 48% of infections, suggesting additional unknown areas of risk or difficulty in recall by respondents (42). Nonetheless, basic hygiene precautions will greatly reduce the risk of toxoplasmosis.
GIARDIASIS
Life Cycle and Mechanisms of Virulence
Giardiasis, caused by Giardia duodenalis (synonyms [syn.], G. lamblia and G. intestinalis) infection, is the most common pathogenic parasitic infection of humans. There are an estimated 280 million cases of symptomatic giardiasis worldwide annually, with approximately 20,000 cases reported annually in the United States (46, 47). There are currently seven genotypic assemblages (assemblages A to F), which are distinct evolutionary lineages, as defined by phylogenetic analysis and enzyme electrophoresis; humans can be infected with assemblage A or B (48). Dogs and cats become infected with canine-oriented assemblages C and D, feline-oriented assemblage F, or potentially zoonotic assemblages A and B (6, 48). The distribution of zoonotic forms depends, to a degree, on the animal housing environment as well as host adaptation. All companion animals, including those housed in kennels, catteries, and households, are infected predominantly with assemblages C, D, and F. Household pets and feral dog and cat populations can be infected with zoonotic assemblages AII and, less commonly, B (6, 49).
The life cycle of G. duodenalis is conserved whether in a canine, feline, or human host (Fig. 4). Both Giardia cysts and trophozoites are shed in the feces of infected humans or animals, and cysts are infectious (Fig. 4). Infection occurs either after the ingestion of cysts through the fecal-oral route or after the ingestion of contaminated food or water (Fig. 4). Cysts are environmentally resistant and can persist for months in soil or water (50). Excystation occurs within the small intestine after parasite ingestion, with each cyst releasing two trophozoites (Fig. 4) (50). Trophozoites remain either free in the intestinal lumen or attached to villous enterocytes, which causes clinical signs (50). Trophozoites encyst upon movement toward the colon and become infectious oocysts by the time of fecal excretion (Fig. 5) (50).
Fig 4.

Giardia sp. life cycle. Giardia cysts shed in the feces are infectious. Infection occurs after the ingestion of cysts either through the fecal-oral route or through the ingestion of contaminated water or food. Cysts are environmentally resistant and can persist for months in soil or water (50). Excystation occurs within the small intestine. Trophozoites remain either free in the intestinal lumen or attached to villous enterocytes, causing clinical signs. Trophozoites encyst upon movement toward the colon, becoming infectious oocysts, and are shed in the feces.
Fig 5.
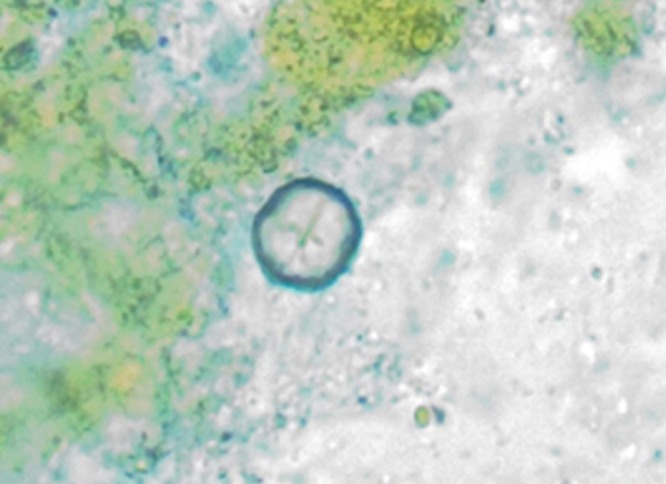
Giardia cyst observed in a fecal flotation from a patient dog at the Iowa State University College of Veterinary Medicine.
Giardia virulence is dependent on both parasite and host factors. The infectious dose of G. duodenalis is estimated to be 10 cysts (51). Once ingested, the cyst becomes metabolically active, and excystation occurs within as little as 15 min (52). Excystation is dependent upon the gastric acid of the host stomach, cysteine proteases produced by G. duodenalis peripheral vesicles, the phosphorylation/dephosphorylation of cyst wall proteins, and Ca2+ signaling (52–55). The released excyzoite undergoes two rounds of binary fission while upregulating processes related to mobility and the organization of the adhesive disc (56). The adhesive disc provides nonspecific adhesion to enterocytes (57). Trophozoite virulence is highly dependent upon mobility, mediated by eight flagella present in four pairs, and host cell attachment functions of the adhesive disc (50, 57).
In addition to the adhesion disc (58), host cell contact is facilitated by a complex cytoskeletal contractile network comprised of giardin proteins, microtubular proteins, and up to 30 disc-associated proteins (DAPs) comprising the ventral disc and lateral crest, many of which have unknown functions (50, 57). Attachment to enterocytes triggers a poorly understood intracellular cascade, resulting in osmotic changes, diarrhea, and other clinical signs of giardiasis. The induction of enterocyte apoptosis is one well-studied virulence mechanism of G. duodenalis (59, 60). Enterocytes exposed to trophozoites rapidly activate pathways of apoptosis, including increased levels of activation of caspase 8 (60), caspase 9, and caspase 3 (59, 60); increased expression levels of Bax; and decreased expression levels of Bcl-2 (60). The activation of caspase 3 as well as other unknown factors modulated intestinal epithelial barrier permeability by the disruption of F-actin, zonula occludens 1, claudin-1, and α-actinin, altering paracellular flow and enterocyte tight junctions, with resultant diarrhea due to the malabsorption of Na+ and glucose and the hypersecretion of Cl− (61–63). G. duodenalis performs a unique mechanism of immune evasion via antigenic variation, different from those recognized for Trypanosoma brucei and Plasmodium falciparum. The mechanism of Giardia sp. variation is not due to recombination or sequence alterations but to variable control via epigenetic mechanisms and, possibly, RNA interference (RNAi) (64). Many questions remain regarding the multifactorial pathogenesis and virulence of G. duodenalis and have been extensively reviewed elsewhere (50, 64, 65).
Immune protection from G. duodenalis is dependent upon multiple host immune response mechanisms, and immunocompromised, malnourished, or agammaglobulinemic people or animals may be severely affected (66). Extensive reviews of current research into the immune response to Giardia are available elsewhere (66, 67). The understanding of immunity against Giardia is cursory at this point, with important ongoing research into the human immune response, mechanisms of immune protection, the effects of variable microflora, and how comorbidities impact Giardia colonization and survival.
Epidemiology and Transmission Dynamics
The epidemiologic distribution of human assemblages of Giardia duodenalis is due largely to exposure to and ingestion of infectious cysts through contaminated food or water (Fig. 5). Other means of transmission include person-to-person and direct zoonotic transmissions, which account for a significantly smaller number of cases (46). Giardiasis surveillance by the CDC documented 19,000 cases of giardiasis in the United States from 2006 to 2008, excluding five states in which giardiasis is not a reportable disease (46, 47). Common age distributions indicate that children less than 10 years old and adults aged 35 to 44 years have elevated incidences of giardiasis (46, 47). In the United States, cases are clustered geographically, with northern states typically having higher incidences of giardiasis (46, 47). Infections follow a seasonal trend, with a 2-fold increase in numbers of cases from June through October, likely due to increased exposure to recreational water contaminated by human sources (46, 47). Outbreaks due to Giardia duodenalis are common in the United States and elsewhere, and outbreaks are generally associated with the consumption of contaminated surface water or improperly filtered and sanitized water from spray fountains in summer play areas or contaminated swimming pools (46). In Florida in 2006, an interactive fountain contaminated with Giardia spp. and Cryptosporidium was responsible for 57 cases (68). In 2007, a failure of water treatment in a community in New Hampshire resulted in 31 illnesses from mixed infections with Giardia spp. and pathogenic Escherichia coli (68, 69). Clusters of patients with giardiasis commonly occur after the consumption of untreated water from freshwater streams (70, 71). Giardiasis caused 86% of drinking water-associated outbreaks of illness from 1971 to 2006, and outbreaks of more than 1,000 cases have occurred due to contaminated drinking water (72). In South America, rural areas of India, Southeast Asia, and numerous other areas of the world, the incidence of giardiasis may be much higher, with rates of infection ranging from 6% to more than 50% in children under 12 years of age, with a high risk in low-income populations (73–78).
The zoonotic transmission of Giardia duodenalis has been a topic of debate, given the distribution of assemblages between different species of animals and humans. The overall prevalence rate of giardiasis in both dogs and cats in the United States is between 2 and 15%, with the highest rates being found in the northern and northwest United States (79). In the United States, kennels typically have high rates of infection with canine-specific assemblage D, and client-owned pets, while still infected predominantly with canine-specific assemblages, do have increased rates of infection with zoonotic assemblages A and B (6). In contrast to those studies, Covacin et al. demonstrated that client-owned dogs presenting with giardiasis to veterinary clinics in the western United States had a greater variety of Giardia duodenalis assemblages, with 28% and 41% having potentially zoonotic assemblages A and B, respectively, and 15% and 16% having host-specific assemblages C and D, respectively (49). Although controversial, such data suggest the possibility of zoonotic transmission from dogs to humans as well as a potential for the transmission of non-canine-specific assemblages from owners to their pets. Other recent studies have found the presence of zoonotic assemblages in livestock species such as cattle and sheep (48, 80–83). In these cases, species-specific strains predominate, but studies have found zoonotic assemblages in less than 10% up to more than 20% of Giardia-positive animals (48, 80–83). Mark-Carew et al. found that 100% of Giardia isolates from New York dairy calves less than 84 days old were of zoonotic assemblage A, indicating the possibility of a greater zoonotic potential of young calves, although subtyping to definitively establish that these isolates were human-adapted Giardia spp. was not performed (80). While no definitive transmission between animals and humans has been documented, data from cross-sectional surveillance studies and evaluations during giardiasis epidemics imply bidirectional interspecies transmission from animals to humans (48). The potential zoonotic risk and high rates of infection in animals and humans make Giardia duodenalis a major target of disease prevention efforts.
Risk factors for Giardia duodenalis infection include any type of activity that would increase the likelihood of the consumption of infective cysts. In the United States, these activities commonly include camping, backpacking, and participation in recreational water activities in streams, lakes, and rivers, which would increase risk due to the consumption of untreated surface water (84). Interestingly, one meta-analysis demonstrated only a weak association of recreational surface water consumption with giardiasis in North America, suggesting that other sanitary measures for campers, such as hand washing and waste removal, may be inadequate (85). Contact with animals or livestock also increased the risk of giardiasis (86, 87). Children in child care centers and individuals working in child care centers have an increased risk of infection by both the amount of time in day care centers and the duration of attendance (88). Finally, failures in water treatment, either in swimming pools, in recreational fountains, or in community water treatment, have resulted in multiple epidemics in the past (46, 47, 72, 89). In developing countries, giardiasis is due largely to the consumption of inadequately treated surface water, more often due to failures of infrastructure rather than recreational exposure (73). Risk factors in cross-sectional studies included the education level of the parents, homes with self-drainage of sewage, or dysbiosis caused by the presence of Helicobacter pylori (90, 91).
Prevention
The prevention of giardiasis hinges upon the proper sanitation of water sources and the avoidance of fecal-oral exposures. Effective preventive measures include the adequate treatment of water for consumption and appropriate sanitary practices such as hand washing, the proper disposal and handling of human and animal waste, and not allowing children with diarrhea to participate in recreational water activities. Hand washing for the prevention of giardiasis or any fecal-oral pathogen is a universal precaution and should be performed regularly after handling soil, diapers, animal feces, or garbage; treating a wound; or going to the bathroom. Special precautions in day care facilities include removing sick children from day care settings, properly handling diapers, and taking children to the bathroom and/or changing diapers often. Surface water for drinking should be boiled at a rolling boil for 1 min or filtered with an approved water filtration device with a National Safety Foundation Standard 53 or Standard 58 rating for cyst reduction (http://www.nsf.org/business/drinking_water_treatment/standards.asp). Fresh fruits and vegetables should be adequately washed prior to consumption. Travelers in areas where water treatment capabilities are unknown should avoid consuming water or ice in drinks and drink only bottled beverages.
In dogs, prevention depends upon the prompt removal of fecal material, preventing dogs from consuming contaminated surface water or feces, and the disinfection and cleaning of kennels. The disinfection of kennels can be accomplished with 1% sodium hypochlorite (20% commercial bleach), 2% glutaraldehyde, or quaternary ammonium compounds (92). Cysts are relatively resistant to chlorination, and levels of chlorine in drinking water are inadequate to inactivate cysts (92). Giardia cysts are susceptible to desiccation, and cleaning and thorough drying will kill them (92).
BABESIOSIS
Life Cycle and Mechanisms of Virulence
Babesiosis is caused by intracellular erythrocyte infection with Babesia complex species. Infection with Babesia spp. was originally recognized by Babes in 1888 as microorganisms present in erythrocytes of cattle and sheep (93). The completion of the Babesia life cycle requires a mammalian host and Ixodes ticks, the same genus known for the transmission of Borrelia burgdorferi, the causative agent of Lyme disease (Fig. 6). When taking a blood meal, infected ticks infect a mammalian host with sporozoites (94). These sporozoites enter erythrocytes and reproduce through asynchronous binary fission, resulting in two, or sometimes four, merozoites (95, 96). Once present in a reservoir host, parasites will develop into male and female gametes (95, 96). Zoonotic Babesia species have diverse reservoirs, including the white-footed mouse, cattle, wild ruminants, canids, shrews, and, possibly, cottontail rabbits (Table 1). Reservoirs are unknown for some human Babesia pathogens (Table 1) (96).
Fig 6.

Babesia sp. life cycle. Sporozoite-carrying ticks infect a mammalian host while taking a blood meal. Sporozoites enter erythrocytes (RBCs) and reproduce through asynchronous binary fission, resulting in two, or sometimes four, merozoites. Once present in a reservoir host (for B. microti, the reservoir is the white-footed mouse), parasites will develop into male and female gametes. When an ixodid tick feeds upon a competent reservoir, blood-stage gametes are introduced into the gut, where these gametes are fertilized to become zygotes. Zygotes enter the tick salivary gland and undergo a sporogonic cycle, forming infectious sporozoites. Humans are generally an intermediate host of Babesia species, although blood transfusion transmission does occur. Dogs are intermediate hosts, much like humans, although they may have a domestic reservoir role in the human transmission of the newly emerging species Babesia conradae.
Table 1.
Geographic locations and host seroprevalences of Babesia spp.
| Disease entity | Species | Area(s) of endemicity | Predominant reservoir(s) | Human seroprevalence and case severity | Animal seroprevalence | Reference(s) |
|---|---|---|---|---|---|---|
| American babesiosis | Babesia microti | NY, NJ, MA, NH, MN, CT, WI, DE, RI, VT, MD | White-footed mouse, rodents, shrews | 1,092 cases in 2011; 0.9–1.1% in CT and 1.4% in MA | 25% in white-footed mouse in CT | 97–100 |
| B. duncani | WA, CA | Unknown | 7 confirmed cases, subclinical to severe; 2% in WA and 2.04% in multiple regions of the U.S. | 101–105 | ||
| CA1–CA4 | WA, CA | Unknown | 4 confirmed cases, severe to fatal | 106 | ||
| B. divergens-like | KY, MO, WA | Cottontail rabbits | 3 confirmed cases, severe to fatal | 50% in Texas cottontail rabbits, 27.8% in jackrabbits | 107–110 | |
| B. conradae | CA | Dog | 9 suspected cases | 1.1% in dogs in CA shelters | 111 | |
| European babesiosis | B. divergens | Europe | Cattle | 40 confirmed cases, 42% mortality, severe to fatal | 10.7–20% in cows in Belgium, 27% in cows in Norway, 7% in cows in France, 17.4% in cows in Italy, 28.3% in deera | 112–116 |
| B. microti | Germany, Africa | Meadow vole, rodents, shrews | 1 confirmed case, moderate severity, 5.4% in Germany | 22% in nonhuman primates in Kenya | 117, 118 | |
| B. venatorum | Austria, Italy, Germany, France, Slovenia | Deer | 3 confirmed cases, moderate to severe | 23% in deer in Francea, 21.6% in deer in Slovenia, 0.9% in deer in Italy | 115, 119–122 | |
| B. divergens-like | Portugal | Unknown | 1 reported case, asplenic, fatal | 123 | ||
| B. bovis | Africa, America, Asia, Australia, Europe | Cattle, buffalo | 2 reported cases, 100% mortality, Spain and the former Yugoslaviab | 45.4% in cows in Italy, 35.3% in cows in South Africa, 73.8% in cows in Thailand, 79% in cows in Portugal, 26% in cows in Puerto Rico, 63.7–95.5% in cows in Brazil | 116, 124–130 | |
| B. canis canis | Europe | Dog, cat? | 1 reported case, nonfatalb | 34% in dogs in Italy, 2.4% in dogs in Croatia, 4–33% in dogs in France | 125, 131–133 |
The prevalences in these studies were quantified by using PCR rather than serology.
Unverified cases reviewed previously by Gorenflot et al. (125).
When an ixodid tick feeds upon a competent reservoir, blood-stage gametes are introduced into the gut, where these gametes are fertilized to become zygotes (96). Zygotes enter the tick salivary gland and undergo a sporogonic cycle, forming infectious sporozoites (94). Prior to entry into the salivary gland, Babesia spp. migrate to the tick ovary, resulting in transovarial transmission and the maintenance of infection in subsequent generations of ixodid ticks (94). However, a recently exiled genus of Babesia-like protozoa, Theileria, does not achieve transovarial transmission (94). The common human pathogen Babesia microti also does not achieve transovarial transmission, even though this species is still classified as a Babesia sp. These nontransovarial species have larger gametes called ookinetes and migrate directly to the salivary gland (Fig. 6) (94). Humans are generally a dead-end host for Babesia species, and conventional transmission via infected humans is unlikely. However, the transmission of babesiosis via blood transfusion is not uncommon and is a source of concern, especially for immunocompromised/splenectomized individuals, who are susceptible to severe clinical babesiosis (134).
Specific molecular mechanisms of pathogenesis, virulence, and host adaptation are poorly understood for Babesia species. Human babesiosis cases are relatively rare (Table 1), and little is known about pathogenic mechanisms in humans specifically. However, a number of domestic animal species have higher incidences of babesiosis and/or theileriosis, and the mechanisms of pathogenesis and immunity discussed here are largely derived from data for these domestic animal species. In cattle and other mammalian species, mechanisms which allow the organism to evade the immune system and invade and persist within the erythrocyte have been identified. First, immediately after introduction into the blood from infectious ticks, merozoites gain entry to erythrocytes to avoid complement and other mechanisms of innate immunity.
Erythrocyte adhesion and infection are facilitated through a number of variable merozoite surface antigens (VMSAs), which contain carboxy-terminal glycosylphosphatidylinositol (GPI) anchor signal sequences (135–138). Antibodies targeting VMSA prevent the attachment of sporozoites to erythrocytes and cellular invasion by merozoites (137, 138). While the mammalian receptors for these proteins and the mechanisms of VMSA-facilitated invasion remain unknown, their importance in pathogenicity has been established. After attachment, tight junction formation and erythrocytic invasion occur via cytoskeletal reorganization as well as rhoptry proteins, rhoptry-associated proteins, and microneme proteins, similarly to T. gondii. Cellular invasion and the establishment of infection are dependent largely on host susceptibility, and certain breeds of animals differ in their susceptibilities to the establishment of infection by Babesia species. While the mechanisms are poorly established, differences in susceptibilities of inbred strains of mice strongly suggest a genetic basis for susceptibility and resistance (139).
Immunity to Babesia species is dependent upon immune responses toward infected erythrocytes or free merozoites. Immunity during primary infection is dependent upon innate mechanisms. Macrophages, including splenic macrophages, and a pronounced proinflammatory response early in infection are necessary for parasite clearance and the prevention of clinical disease (140). This necessity explains the susceptibility of splenectomized and immunosuppressed patients, breed and individual differences due to differences in inflammatory cytokine production, and the relative resistance of children and younger animals to infection, as younger animals have peak levels of production of interleukin 12 (IL-12) and gamma interferon (IFN-γ) 3 days earlier than adults (141).
Babesia species have adapted for long-term survival in the vertebrate host by immune avoidance and modulation strategies. In addition to VMSA, Babesia spp. produce variable erythrocyte surface antigen (VESA), which is transported to the erythrocyte surface much like Plasmodium knob proteins, causing adhesion to the endothelial cells of small vessels (95). This serves the function of sequestering infected erythrocytes in the peripheral microvasculature and away from the spleen. Babesia species are evolutionarily well adapted for long-term survival and replication both in the healthy host and in the ixodid tick, causing minimal disease in most cases. However, with comorbidity, immunosuppressive therapy, or splenectomy, infections with this protozoon can be devastating.
Epidemiology and Transmission Dynamics
The worldwide distribution of Babesia spp. is dependent largely upon the geographic distribution of competent Ixodes vectors. Cases of babesiosis occur throughout Europe and across the Eastern Seaboard of the United States and the U.S. West Coast, with foci of infection in Wisconsin and Minnesota. Babesia spp. are incredibly divergent, with over 100 known species, and there is limited knowledge regarding reservoirs and predominant vectors for many of them. Major species resulting in human infection are B. divergens, B. divergens-like species, B. duncani, and B. microti. B. divergens-like species are those genetically similar to Babesia divergens but are not classified as being of this species or of their own species. Most European infections result from B. divergens, and at least three species are present in the United States: B. microti, present on the East Coast and in Wisconsin and Minnesota; B. duncani and B. conradae, present on the West Coast; and B. divergens-like organisms, having a widespread distribution (142).
European babesiosis.
B. divergens is primarily a cattle parasite transmitted by ticks of the species Ixodes ricinus, discovered in 1911 by M'Fadyean and Schein (143). Research in the 1950s and 1960s demonstrated the susceptibility of rhesus macaques to B. divergens infection. Splenectomized primates were susceptible to severe clinical disease resulting in intravascular hemolysis and hemoglobinuria (144, 145). Subsequent studies demonstrated that B. divergens has low host susceptibility, with resistance in most laboratory animal species, with the exception of Meriones unguiculatus, the Mongolian gerbil (146), which serves as a laboratory model for B. divergens (147). Numerous human cases have been reported throughout Europe, primarily in splenectomized individuals. Clinical infection continues to be rare in Europe, with approximately 40 acquired cases to date, with a 42% mortality rate (Table 1) (148–150). The severity of infection is due to the vast majority of cases of B. divergens babesiosis occurring in splenectomized or immunocompromised patients. A recent report suggested that immunocompetent individuals may become infected, exhibit only mild clinical disease, and recover (150). The seroprevalence of B. divergens in Europe suggests that the rate of exposure is much higher, highlighting the necessity of detection to prevent transmission to immunocompromised individuals via blood transfusion (117).
Two studies in midwestern Germany demonstrated B. divergens seroprevalence rates of 3.6% and 5.4% for B. microti in people with significant tick exposure and a seroprevalence of less than 0.5% in populations without tick exposure (117, 151). Animal populations also bear high seroprevalences: roe deer in France had a seroprevalence of B. divergens of 58%, and cattle in Norway had a seroprevalence of B. divergens of 27% (112, 152). In addition to reports of both B. divergens and B. microti in multiple locales in Europe, additional B. divergens-like species, EU-1 and B. capreoli, have been identified, which may have been previously mistaken as B. divergens. The surveillance completed thus far indicates a significant presence of pathogenic Babesia species throughout many areas of Europe, and immunocompromised and splenectomized patients are susceptible to severe, possibly fatal, clinical babesiosis. Surveillance efforts to prevent iatrogenic transmission via blood transfusion are of importance, due to the likelihood of transfusion to immunocompromised patients (Fig. 6).
North American babesiosis.
In North America, rare instances of B. divergens-like babesiosis occur, including isolated cases in Missouri, Kentucky, and Washington (107–109). A B. divergens-like parasite was designated MO-1 after a Missouri case and was found to be maintained within cottontail rabbit populations, with close homology to bovine-derived B. divergens (153). Cases in Washington and California (101, 106) were described as being caused by B. duncani, a species closely related to the canine species B. gibsoni. Babesia spp. from cases in Southern California were related to Babesia spp. of deer and other wildlife and have been named B. conradae (Fig. 7) (154–156). These foci have resulted in 9 cases: 4 in splenectomized patients, 4 through blood transfusion, and 1 in an apparently healthy patient (101, 102, 106). B. conradae has been shown to cause more virulent disease in dogs than observed for B. gibsoni-infected dogs. Canine B. conradae was shown to be most closely related to human piroplasms recently detected in the western United States (156). The disease in these patients was consistent with B. divergens-like disease in Europe, indicating that numerous opportunistic strains of Babesia exist, which can infect humans under immunocompromised conditions and may be the same pathogen circulating in dogs.
Fig 7.
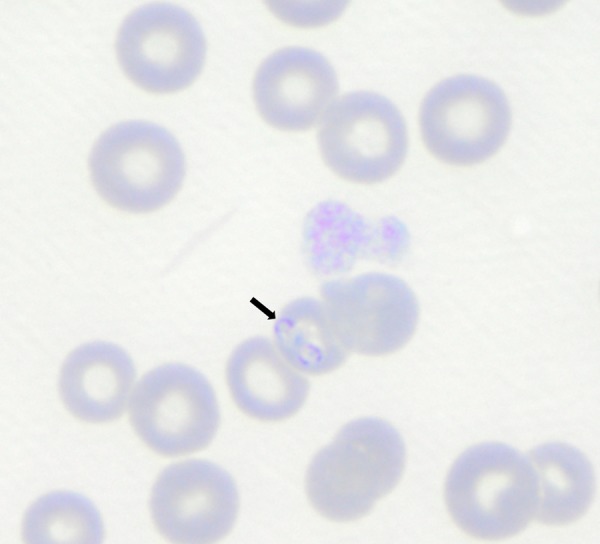
B. conradae piroplasms. Parasites are indicated by an arrow. Babesia conradae was present in erythrocytes from a canine patient of the veterinary medical teaching hospital at the University of California, Davis. (Courtesy of Jane Sykes.)
While B. divergens-like infections occur sporadically in the United States, B. microti has a much higher rate of incidence, causing babesiosis in both immunocompromised and immunocompetent individuals. The definitive host of B. microti is the white-footed mouse and is transmitted by Ixodes scapularis (deer tick), for which deer assist in the maintenance of vector populations (Fig. 6). While B. microti does not preferentially infect splenectomized individuals over healthy individuals, splenectomized patients are more susceptible to severe clinical disease. Disease is highly endemic in foci along the East Coast and within Wisconsin and Minnesota. Blood transfusion-transmitted babesiosis (TTB) has resulted in a greater geographic distribution of cases, causing 162 reported cases from 141 donors, with 18% mortality (157). The number of TTB cases increased during each decade from 1979 to 2009, with 91 of the 162 cases occurring from 2005 to 2009 (157). The increase in the occurrence of TTB cases resulted in babesiosis in the United States becoming a nationally reported disease in 2011 (157). Vertical transmission of Babesia microti has been reported (158–162). Given the overlap of competent vectors, coinfection with Borrelia burgdorferi is common, and Lyme disease patients have a significantly higher risk of babesiosis in both Europe and the United States (117). Serologic surveys of blood donors for B. microti from 2000 to 2007 indicated that seroprevalence rates were 1.4% and 1.1% in Massachusetts and Connecticut, respectively (97). There were small geographic areas of higher seroprevalence in counties where B. microti is considered hyperendemic (97). Babesia spp. are able to survive up to 35 days at 4°C in refrigerated blood and indefinitely under conditions of cryopreservation (163). Serologic studies of reservoir animals suggested seroprevalences of approximately 25% for B. microti in the white-footed mouse and approximately 35% in mule deer (98, 164) and between 16 and 28% PCR positivity in peripheral blood of rabbits in areas of endemicity (110, 153).
Prevention
The prevention of autochthonous cases of babesiosis includes avoiding heavily wooded and grassy areas during the seasons of highest tick activity, from May to September. If hiking or performing other activities in these areas, long pants and long-sleeved shirts should be worn, with the shirt and pants being tucked in. Additionally, permethrin has a repellent effect on ticks and can be applied to clothing but should not be applied to the skin. N,N-Diethyl-meta-toluamide (DEET)-based insect repellents do have some degree of repellent effect on ticks. Infection through tick bites requires at least 24 h of attachment, and the body surface of people entering areas where transmission is likely should be inspected daily for ticks after participating in activities that have a high risk for tick exposure.
While human babesiosis has been attributed directly to B. conradae or feline Babesia spp., companion animals do serve as a source of tick exposure for their owners. The use of monthly topical insecticides on pets will reduce the likelihood of ticks entering the home environment as well as prevent tick- and flea-borne diseases in companion animals.
The control of TTB is another important target for community public health intervention. TTB has been recognized as an important risk to the U.S. blood supply. Currently, any patient with a previous diagnosis of babesiosis is not allowed to donate blood. TTB is a challenge for prevention, and current methods of reporting and donor exclusion appear to be minimally effective. Leiby provided a thorough review of TTB and strategies for prevention and mitigation (142). Babesiosis is a widespread disease with numerous infective species, reservoir hosts, and vectors. While the number of cases of this disease has been small, within certain immunocompromised populations, the mortality rate is high. Babesiosis remains an important and challenging emerging zoonotic disease.
AMERICAN TRYPANOSOMIASIS (CHAGAS' DISEASE)
Life Cycle and Mechanisms of Virulence
American trypanosomiasis (AT), or Chagas' disease (CD), a vector-borne protozoal disease caused by Trypanosoma cruzi, occurs in North, Central, and South America. T. cruzi is transmitted via infected feces from numerous different triatome insect species. The burden of the parasite and life cycle were discovered and largely described in 1909 by Carlos Chagas (165). The World Health Organization has estimated that approximately 10 million people had CD in 2004, primarily in Latin America, making CD the most important parasitic disease in the Americas by disability-adjusted life years (DALYs) (166). T. cruzi is an indiscriminate parasite with demonstrated infections of over 100 mammalian species and is capable of infecting all mammalian species. Avian species are resistant to infection (167).
An infected triatome vector takes a blood meal from a mammalian host, triggering the release of infective trypomastigotes in feces (Fig. 8). Infective trypomastigotes (Fig. 9) enter the mammalian host through a bite wound or by penetrating intact mucous membranes, including conjunctiva and the intestinal tract. The genera Triatoma, Rhodnius, and Panstrongylus are of importance for AT vector transmission. Trypomastigotes invade cells and replicate near the site of infection, differentiating into intracellular amastigotes. Amastigotes replicate via binary fission within parasitophorous vacuoles, escape into the cytoplasm, and are released as trypomastigotes into the extracellular matrix, reaching the bloodstream. During initial replication, CD8+ T cell infiltration can be delayed as long as 10 to 12 days, facilitating parasite survival (168). Trypomastigotes are indiscriminately infective to host cells, infecting a variety of cell types, with tropism for smooth and cardiac myocytes. Trypomastigotes within the bloodstream are nonreplicating, a difference from Trypanosoma brucei, the trypanosome species which causes African trypanosomiasis. Triatome insects become infected through the ingestion of circulating trypomastigotes in mammalian blood. To complete the life cycle, trypomastigotes transform into epimastigotes within the triatome midgut, multiplying and differentiating, with final differentiation back into infective trypomastigotes within the insect hindgut.
Fig 8.

Life cycle of Trypanosoma cruzi. An infected triatome vector or “kissing bug” takes a blood meal from a mammalian host, releasing infective trypomastigotes in feces near the bite wound or mucosae. Infective trypomastigotes enter the mammalian host, penetrating intact mucous membranes, including conjunctiva, or orally through the intestinal tract after food-borne exposure. Trypomastigotes invade cells and replicate near the site of infection, differentiating into intracellular amastigotes. Amastigotes replicate via binary fission within parasitophorous vacuoles, escape into the cytoplasm, and differentiate into trypomastigotes. Trypomastigotes are released from the cell, reaching the bloodstream. Triatome insects become infected through the ingestion of circulating trypomastigotes in mammalian blood meals, transform into epimastigotes within the triatome midgut, and undergo final differentiation into infective trypomastigotes within the insect hindgut.
Fig 9.
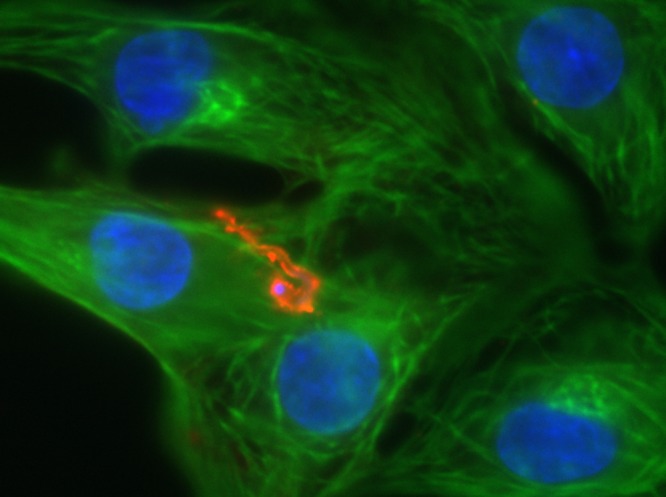
Neonatal rat cardiomyocytes and a Trypanosoma cruzi trypomastigote with fluorescent immunolabeling. Actin myofilaments are labeled in green, the T. cruzi trypomastigote is labeled in red, and nucleic acids are labeled in blue (DAPI).
The pathogenic mechanisms and molecular means of cellular invasion by T. cruzi have been an active area of research for decades and are relatively well described. Upon entry into the mammalian host, the infective trypomastigotes quickly infect local macrophages, fibroblasts, muscle cells, and adipocytes. Intracellular amastigotes undergo binary fission every 15 to 18 h. Amastigotes replicate for approximately 5 to 6 days until they consume a high percentage of the cytoplasmic compartment of a cell before differentiation into trypomastigotes, resulting in the rupture of the plasma membrane and dissemination. Numerous cellular adhesion molecules, including trans-sialidases (TSs), GPI-anchored proteins, mucins, mucin-associated proteins (MAPs), dispersed gene family 1 (DGF-1), and GP-63, facilitate parasite entry (169). More than 50% of the T. cruzi genome is repetitive sequences encoding an enormously heterogeneous population of surface molecules (170). Glycoproteins of the TS family are important for numerous functions of the parasite, including extracellular matrix and cellular adhesion, cellular invasion, and pathogenicity (168, 171). Recent research demonstrated the ability of T. cruzi to utilize the low-density lipoprotein (LDL) receptor (LDLr) for entry into fibroblasts and cardiomyocytes (172). The accumulation of LDLs within the heart may be a factor contributing to the pathogenesis of Chagas' disease-associated cardiac lesions (172). Lysosome-dependent and -independent invasion pathways required host cell phosphotidylinisitol-3-kinase (PI3-kinase), as the blockage of PI3-kinase with wortmannin inhibited parasite entry (173). Trypanosomal escape into the cytoplasm is dependent upon lysosomal acidification causing the release of the hemolysin T. cruzi toxin (TC-Tox) (174, 175). The exposure of the lysosomal membrane to TC-Tox is thought to be facilitated by the expression of numerous TS enzymes, which desialylate lysosome-associated membrane proteins (LAMPs), resulting in the disruption of the parasitophorous vacuole (176, 177). The multiple mechanisms of invasion, pathogenesis, and persistence of T. cruzi highlight the reasons for its chronicity in many hosts and the establishment of AT in many areas of endemicity.
An understanding of effective immunity against Trypanosoma cruzi has also been a research focus for some time, with goals of vaccine development and disease prevention. Vaccine development has remained difficult, due to the complex nature of CD. Correlates of protective immunity and parasite control within the host have been extensively studied and are dependent upon the infectious dose and strain of T. cruzi and the innate and acquired immune responses of the host. Macrophages commonly become infected with T. cruzi and may be one of the first cell types encountered by the parasite during natural infection (167). The recognition of T. cruzi by macrophages is through numerous surface Toll-like receptors (TLRs) and lectin receptors (178, 179). Repetitive CPG DNA motifs and mRNA from intracytoplasmic T. cruzi have also been shown to engage TLR9 and TLR7, respectively, and are necessary for the control of T. cruzi infection (180–182). The acquired immune response is also necessary for parasite control and clearance. Humoral immunity is thought to be important in early acute infection, with IgG2b and B cells conferring protection, although the exact mechanism is not understood (183, 184). Cellular immunity is thought to be the largest and most important component of the immune response to infection by T. cruzi and the main target for effective vaccination strategies against AT. The immune response to T. cruzi is a complex interplay of nearly every aspect of innate and adaptive immunity, and the suppression of any one of these aspects can result in parasite survival and chronic infection.
Epidemiology and Transmission Dynamics
Chagas' disease is widespread throughout the Americas, with endemic triatome transmission occurring in all countries of South America, Central America, and Mexico and with limited autochthonous transmission in the United States (185). Trypanosoma cruzi is found throughout these areas in numerous triatome vectors and in over 100 mammalian species. It is believed that T. cruzi has been a human pathogen in the Americas for as long as humans have inhabited the continent, based on studies of archeological samples from the United States, Chile, and Peru from approximately 9,000 years ago (186). There are multiple means of infection by T. cruzi, including classical vector-borne transmission, congenital infection, transfusion-associated CD, and oral/food-borne exposures. The wide distribution, numbers of possible vectors and reservoir species, and multiple means of transmission all contribute to the immense burden of CD.
Classical sylvatic and domestic transmission in areas of endemicity.
(i) Vector species and risk factors.
There are over 41,000 estimated new CD cases due to vector-borne transmission each year (185). Over 130 known triatome species exist within the Americas, with most of them being considered capable of vectoring T. cruzi (187). Sylvatic enzootic cycles result in human infection when adult sylvatic triatomes are attracted to light or other characteristics of human domiciles (188). With deforestation and the introduction of companion animals, sylvatic vector species can initiate domestic infection. Members of the family Triatominae are hematophagous throughout their life cycle, developing a close evolutionary relationship with their host species, primarily small mammals and rodents. There are a few triatome species with both domiciliary and peridomiciliary cycles in the Americas, resulting in greater human transmission. In the United States, 11 species of triatomes are present, and infection with T. cruzi has been identified in all but one of these triatomes. These species are found in the entire southern half of the United States, including Texas, Southern California, and Arizona, and one can be found in Florida. Fifty-eight percent of Triatoma gerstaeckeri insects, commonly trapped in New Mexico and Texas, were found to be positive for T. cruzi (188). All seropositive U.S. blood donors identified through screening lived in areas of the United States with documented T. cruzi infection, and 80% had no travel history in areas of endemicity outside the United States (189). In almost all cases, in households with dogs and positive humans, dogs were positive as well, highlighting the common vector source. This strongly suggests vector-associated transmission in the United States, requiring vigilance by U.S. public health services and disease cognizance by local physicians and veterinarians.
Risk factors for vector-borne CD are related to the likelihood of infectious bites. Specific risk factors for CD in areas of endemicity based on large-scale seroprevalence and triatome surveillance surveys were the presence of the vector, nearby cropland and grassland, disarray of the domiciliary environment, and mud and thatch or tarred cardboard homes and outbuildings (190–192). Other factors, including evidence of triatome infestation of the home and the presence of a companion animal sharing a room with an individual, were significantly associated with CD in Peru (193). Studies demonstrated preferential vector feeding on mammals, such as caged guinea pigs and dogs over birds or cats, with a higher percentage of triatome bugs feeding and reaching engorgement on preferred species (194). This indicates the importance of T. cruzi domestic reservoirs for transmission to humans.
(ii) Dogs as reservoir species and risk.
Dogs are considered the predominant domestic reservoir for CD in many areas of endemicity. In Texas, there were 537 confirmed canine CD cases between 1993 and 2007 (195–197). Dogs develop acute and chronic disease, similar to human infection. Acute infection in young dogs presents typically as myocarditis with arrhythmia (8). In chronic disease, dogs have chronic progressive cardiac failure due to electrocardiogram (ECG) abnormalities and/or congestive bilateral or right-sided heart failure (8). Dogs in areas of high endemicity in Argentina had seropositivity rates from 25% in young dogs to 92% in dogs 8 years of age or older (197). In Venezuela, one estimate placed the seroprevalence in dogs at 6.9%, almost identical to the human seroprevalence in this region (190). A study in Panama determined an overall T. cruzi infection index of 16.2% in dogs (198). In Campeche, Mexico, the prevalence of T. cruzi seropositivity was higher in dogs than in people, with 9.5% positivity in stray dogs and 5.3% positivity in owned dogs (199). Household cats play a much less definitive role in the domestic transmission of CD, with a lower estimated seroprevalence rate than that for dogs (197). Although triatomes do feed upon cats (38% in one study), they are much less likely to be engorged (194, 197). Therefore, household dogs are estimated to be at a 3-fold-higher risk for transmission than cats in regions of endemicity (197). The presence of domestic companion mammals within the home is a definitive risk factor for human infection with T. cruzi. The nondiscriminatory nature of both triatome species and T. cruzi allows for the persistence of CD in sylvatic, rural, and domestic environments and creates a significant challenge to disease prevention.
Congenital Chagas' disease.
Congenital infection with T. cruzi causes approximately 14,000 cases of CD per year, resulting in a spectrum of clinical signs (200). A majority of congenital infections occur in asymptomatic mothers. Congenital transmission closely follows the serologic prevalence in female populations in areas of endemicity (200). Studies indicated that maternal seropositivity rates ranged from less than 1% to up to 64.5% in areas of Bolivia (200–204). The congenital transmission rate, measured as the number of infected infants born to infected mothers, was 1% to 7% (200–205). A recent comprehensive surveillance of congenital CD in immigrants from areas of endemicity to Spain demonstrated an infection rate of 11.4% in mothers from regions of endemicity, with higher seropositivity rates in mothers from Bolivia (34.1%) (206). Mothers from areas of endemicity had a congenital transmission rate of 3.4% in Spain (206). Based on a recent case of congenital CD in Virginia in an infant born to a Bolivian mother, this may also be true for the United States (207). Based on expected rates of congenital transmission and known seroprevalences in the United States, there may be approximately 58 to 502 congenital cases per year (208). Vector control has made an impact on maternal seroprevalence rates in areas of endemicity, with increasing maternal age being significantly associated with congenital transmission and seropositivity. However, a large number of young infected women remain. Congenital CD will continue to be a long-term maternal and neonatal health challenge both in countries of endemicity and in countries where the disease is not endemic.
Transfusion-associated Chagas' disease.
Transfusion-associated CD occurs when a parasitemic donor donates either blood or organs for transplant, causing acute or chronic disease in the blood or organ recipient. All blood and organ components are infective, and T. cruzi remained viable for at least 18 days at 4°C (209). The likelihood of infection due to transfusion is dependent upon the level of donor parasitemia, the amount of transfused blood, conditions of blood storage and processing, and screening methods in place in the region of residence of the donor (209). In the United States, there have been five published cases of CD associated with blood transfusion and five cases associated with organ transplantation (210–215, 348). Many of these patients suffered from concurrent conditions necessitating a transplant and developed severe acute CD posttransplantation or posttransfusion (188). The American Red Cross and Blood Systems Inc. began screening blood for T. cruzi on 1 January 2007 (188). From 1 January 2007 to 28 June 2012, there were 1,668 confirmed seropositive donations (216). In addition to immigrant infections, recent research suggests the occurrence of vector-borne transmission in the United States (189). This has resulted in T. cruzi-positive blood donations, indicating that all blood donations should be evaluated for the presence of T. cruzi (189). The estimated seroprevalence of blood donors in Latin America is approximately 1.3% (209). Continued screening efforts and long-term vector control in areas of endemicity will be required, due to extensive immigration and the rate of asymptomatic AT.
Oral transmission of Chagas' disease.
Sylvatic T. cruzi infection of opossums, skunks, and raccoons is dependent upon oral transmission, due to the insectivorous nature of these animals (188, 217, 218). Dogs and cats are also insectivorous. In humans, epidemics due to transmission via contaminated fruit and vegetable materials from regions of endemicity have caused increased concern for transmission via food. Some reports suggested that oral transmission is the primary route of T. cruzi dissemination between animals and vectors and the predominant cause of acute human disease in Amazonia (219). The clinical form of oral CD has a clinical presentation similar to that of acute CD, with some differences. Oral exposure results in an acute febrile syndrome 3 to 22 days after exposure, with myalgia, cholangiohepatitis, and gastritis with epistaxis, hematemesis, and, potentially, shock (219).
Between 1980 and 2001, 28 small family-focused outbreaks occurred in Brazil, due to the contamination of juice, water, or food with triatomes, their feces, or secretions from the anal glands of infected mammals (219, 220). Acai and sugar cane juices have been implicated in outbreaks, due to the nature of the preparation of the juice and the grinding of triatome insects into the juice (220). One of the most recent and largest outbreaks of orally acquired CD occurred in a school community in Venezuela (221). There were 103 confirmed cases during this outbreak, with 75% symptomatic cases, and of those, 20.3% required hospitalization (221). This outbreak was significantly associated with the consumption of guava juice, which was prepared the previous night and left to cool in an open container outside (221). The oral transmission of CD, which maybe the predominant means of companion animal infection, highlights the importance of keeping pets inside when possible, food safety and general hygienic practices, and the maintenance of quality control and vigilance during food, particularly juice, preparation.
Prevention
The prevention of CD relies heavily on vector control, the screening of the blood supply and organ donations, and standard food safety practices. Vector control practices in many South American countries have resulted in significant decreases in rates of human seropositivity (222, 223). In these regions, mammalian reservoirs have generally remained, and the number of asymptomatic seropositive people of middle age and older remains high. Therefore, CD will likely continue to be a threat to the blood supply and a risk for congenital infection. Surveillance of blood donors, in conjunction with screening questions about CD for blood donors, has likely reduced the number of acute transfusion-associated CD cases. For pregnant women already infected with T. cruzi, there is no viable prevention of congenital transmission, due to the potential side effects of the current toxic therapy on the fetus (224). For women in general, the prevention of future congenital transmission depends upon recognition and therapy prior to conception (224). Ongoing efforts against CD have greatly reduced the burden of this zoonotic disease in the last 2 decades. However, given the worldwide nature of immigration, the wide variety of competent vectors and reservoirs, and the asymptomatic nature of the disease, Chagas' disease will continue to be a worldwide public health challenge for the foreseeable future.
LEISHMANIASIS
Life Cycle and Mechanisms of Virulence
Leishmaniasis is a vector-borne disease caused by Leishmania species of the family Kinetoplastidae. Infection with Leishmania spp. can result in a spectrum of clinical diseases dependent upon the infecting species. Visceral leishmaniasis (VL) is caused by Leishmania infantum in the Americas and the Mediterranean basin and by L. donovani in India, sub-Saharan Africa, and Asia. Occasionally, cases of VL will arise from cutaneous disease-causing species and has occurred in members of the U.S. military due to infection by L. tropica (225). VL arises from the parasitic infection of phagocytic cells within secondary lymphatic organs (spleen and lymph nodes), liver, and bone marrow. Cutaneous leishmaniasis (CL) (Table 2) arises from an infection of epidermal tissue after promastigote host inoculation. In susceptible hosts and immunocompromised persons, disseminated cutaneous or diffuse cutaneous leishmaniasis may occur as a rare but severe manifestation of CL. A third form of the disease, mucocutaneous leishmaniasis (MCL), arises from a small percentage of cutaneous cases who cleared the disease months or years prior to the onset of MCL. MCL often begins with an involvement of the nasal mucosa, including generalized inflammation and ulceration. Ulceration and necrosis of these areas may be severe, resulting in disfigurement and, occasionally, death. Mechanisms of mucocutaneous lesion formation are poorly understood, but a Leishmania RNA virus (LRV1) was associated with severe mucocutaneous lesions through a TLR3-dependent inflammatory response (296).
Table 2.
Geographic locations, hosts, and companion animal seroprevalences of Leishmania spp.
| Disease entity | Species | Area(s) of endemicity | Predominant reservoir(s) | Canine seroprevalence | Reference(s) |
|---|---|---|---|---|---|
| Cutaneous leishmaniasis | Leishmania major | Middle East, northwestern China, northwestern India, Pakistan, Africa | Gerbil species, jird, fat sand rat | Egypt, 3 cases; Saudi Arabia, 3 cases | 226–233 |
| L. aethiopica | Ethiopia, Kenya, Somalia | Rock hyrax | 234, 235 | ||
| L. mexicana | Central America, Mexico, TX | Yucatan deer mouse, tree rat, other rodents | Mexico, 30.2%, 10.5%a; TX, 8 casesa | 236–241 | |
| L. amazonensis | Brazil | Various forest rodents (grass, pygmy mice) | Brazil, 1 case | 242, 243 | |
| L. tropica | Mediterranean, Middle East, western Asia, Indian subcontinent | Human, foxes, golden jackals, hyrax, dogs | Morocco, 8 cases | 244–248 | |
| L. braziliensis | Central, South America | Forest mammals, marsupial species, opossum | Mexico, 8.2%, 11.57%a; Brazil, 2–3% | 240, 241, 249–252 | |
| L. guyanensis | Guyana, Suriname, northern Amazon basin | Two-toed sloth, forest mammals, marsupial species, opossum | Colombia, 2.2% | 253, 254 | |
| L. peruviana | Peru, Argentinean highlands | Dog? | Peru, 1.8%b | 255 | |
| L. shawi | Brazil | Cebus monkeys, sloths, procyonids | 256 | ||
| L. lainsoni | Brazil, Bolivia, Peru | Lowland paca, rodents | 257 | ||
| L. naiffi | Brazil, French Guyana, Ecuador, Peru | Armadillos | 258 | ||
| L. venezuelensis | Venezuela | Unknown, cat? | Venezuela, 6 casesa | 259 | |
| L. panamensis | Panama, Costa Rica, Colombia | Sloths, kinkajous, marsupial species, opossum | Colombia, 12 cases; Panama, 3.3%; Ecuador, 2 cases | 260–264 | |
| Visceral leishmaniasis | L. donovani | Indian subcontinent, northern and eastern China, Pakistan, Nepal, eastern Africa, Sudan, Kenya | Human, dogs, goats | Sudan, 2 cases; Ethiopia, 14.8%; India, 6.5% | 265–268 |
| L. infantum (syn., L. chagasi) | Middle East, Mediterranean basin, northern and northwestern China, northern and sub-Saharan Africa, Central and South America | Dogs, foxes, jackals, wolves | Mexico, 11.9%, 22.10%a; Brazil, 7.14–57%; Portugal, 4.3–25.2%; Spain, 8.1–13%; Italy, 2.6%; France, 4–8%; Greece, 2–30%; Uzbekistan, 32.1%; Turkey, 20.7%; China, 23.5–28.2%; Iran, 14.2%, 10%a,b; Jerusalem, 6.7%a; Senegal, >40% | 240, 241, 248, 250, 269–295 |
Feline seroprevalence.
Prevalence was measured as positivity on skin or splenic biopsy specimens and culture.
The life cycles of Leishmania spp. are relatively simple, involving a mammalian host and a vector stage (Fig. 10). Phlebotomine sandflies of the genus Lutzomyia in the Americas and Phlebotomus in other regions of endemicity serve as vectors for Leishmania. The sandfly injects infective promastigotes into a susceptible mammal during feeding. Promastigotes (Fig. 11A) are quickly phagocytized by resident phagocytes, transformed into tissue-stage amastigotes, and divide through simple division in a parasitophorous vacuole. Depending upon host and parasite factors, the parasite infects further phagocytic cells either at the site of cutaneous infection or in secondary lymphoid organs, with eventual parasitemia. Sandflies become infected through feeding on a host either with an active skin lesion in CL or with parasitemia in VL. Parasites convert to promastigotes within the sandfly midgut and reproduce to high numbers in 4 to 14 days. These promastigotes migrate to the salivary glands, transform into infectious metacyclic promastigotes, and await the initiation of feeding.
Fig 10.

The life cycle of Leishmania species. Sandflies inject infective promastigotes into a susceptible mammal during feeding. Promastigotes are phagocytosed by resident phagocytes, transform into tissue-stage amastigotes, and multiply within these cells through simple division. The parasite continues to infect phagocytic cells either at the site of cutaneous infection or in secondary lymphoid organs, with eventual parasitemia. Sandflies become infected through feeding on a host either with an active skin lesion in CL or with parasitemia in VL. Parasites convert to promastigotes within the sandfly midgut. Promastigotes migrate from the midgut and transform into highly infectious metacyclic promastigotes.
Fig 11.
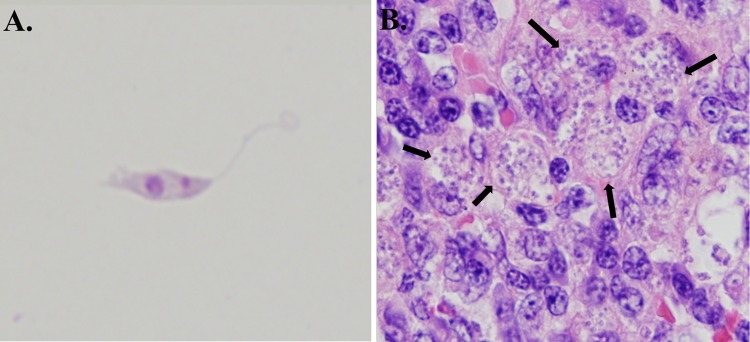
Leishmania parasites in culture and in a tissue section. (A) Leishmania amazonensis promastigote from culture with a visible kinetoplast. (Photo by Pedro Martinez.) (B) Zoonotic visceral leishmaniasis in canine spleen. The spleen was enlarged and infiltrated by large numbers of foamy macrophages containing numerous intracellular Leishmania infantum amastigotes (arrows), confirmed by immunohistochemistry (magnification, ×100).
Leishmania spp. have unique virulence mechanisms, maintaining persistence within host phagocytes to establish long-term chronic infection. After a sandfly bites the host, salivary chemoattractants promote an influx of both neutrophils and macrophages to the feeding site (297). Parasites inhibit phagosome acidification, allowing them to survive within neutrophils, but have not been shown to transform into amastigotes or proliferate within neutrophils (298). At the time of neutrophil apoptosis, surviving parasites are phagocytized by resident and infiltrating macrophages. Dendritic cells also become infected, becoming mature and migrating to the lymph node. L. amazonensis specifically inhibited dendritic cell maturation through enhanced extracellular signal-regulated kinase (ERK) activation from the phagosome, resulting in the decreased production of interleukin 12, a key proinflammatory mediator (299). Leishmania phagocytosis is mediated through complement receptors 1 and 3 and mannose scavenger receptors, indicating both opsonization-dependent and -independent mechanisms of invasion (300). Uptake results in the reorganization of F-actin and delayed phagolysosomal fusion (300, 301). Leishmania spp. are resistant to acidification as amastigotes and persist in late-endosome-associated LAMP1- and Rab7-positive vacuoles (302). Amastigotes replicate within the phagolysosome until eventual host cell lysis. The ability of the parasite to direct phagosome trafficking and delay phagolysosome fusion is dependent upon surface lipophosphoglycans with differing side chains (302–304). Leishmania spp. are also able to acquire nutrients needed for survival through the expression of LIT-1 to acquire the Fe2+ needed for growth and survival (305).
The immune response to all Leishmania species as an intracellular pathogen is dependent upon a timely and appropriate T helper 1 response, including IL-12 production by dendritic cells and macrophages, efficient MHC-II presentation, and subsequent IFN-γ production from T cell populations. The clearance of Leishmania infection by the innate immune system is dependent primarily upon intracellular killing via superoxide and nitric oxide within phagolysosomes of infected macrophages, which is enhanced by IFN-γ stimulation from NK cells early in infection and T cells at later stages. The mechanisms of immunity to various species of Leishmania, and the specific evasion mechanisms utilized by Leishmania species, are beyond the scope of this review and have been thoroughly discussed elsewhere (306, 307).
Epidemiology and Transmission Dynamics
There are 98 countries and 3 territories where Leishmania is endemic, with the majority of cases occurring in developing nations (308). The distribution of competent vector species and leishmaniasis has expanded over the last decade, possibly due to an increasingly amenable environment of vector species due to shifts in climate (309). This results in approximately 200,000 to 400,000 cases of VL and 700,000 to 1.2 million cases of CL each year and an estimated 20,000 to 40,000 deaths (308). More than 90% of VL cases occur in India, Bangladesh, Sudan, South Sudan, Ethiopia, and Brazil (308). Among the 15 disease-causing species of Leishmania, 13 are thought to have some degree of zoonotic transmission, and in the case of L. infantum, dogs are the main reservoir, being largely responsible for the transition from sylvatic to domestic transmission cycles (9, 269). The clinical forms of leishmaniasis are discussed briefly, primarily based on the degree of zoonotic transmission from companion animal species.
Cutaneous leishmaniasis.
CL is caused by a number of Leishmania spp. with widespread distributions and a variety of location-specific reservoir species (Table 2). In the Old World, Leishmania major is a predominant cause of CL. Reservoirs for L. major vary by location but include many rodent species. Reservoir and vector population densities are significantly correlated with the seasonality of infection. Increases in reservoir rodent populations have been associated with numerous zoonotic cutaneous leishmaniasis (ZCL) outbreaks (269, 310). L. aethiopica causes ZCL and appears to be isolated to the highlands of Ethiopia. Reports identified this species at lower altitudes as well, indicating that the distribution is perhaps more widespread or that the area of endemicity is expanding (234). CL caused by L. tropica is considered anthroponotic. However, animal species can be infected and have been suggested to be potential reservoirs, including dogs (Table 2).
In the New World, numerous species of CL-causing Leishmania (Viannia) spp. have been identified in multiple mammalian species. L. mexicana is found from Central America to the Yucatan peninsula in Mexico, and cases have been reported in Texas (236, 311–313). There have also been a number of non-travel-associated reports of CL in companion animals in Texas (236, 311–313). Notably, many of these cases of zoonotic CL were in cats (236, 311, 313), perhaps due to their more outdoor life-style in the United States. There have been 30 autochthonous cases of human CL in Texas through 2008, where CL is considered endemic (237). While companion animal infection and transmission occur, the predominant sylvatic reservoir in Texas is the Southern Plains woodrat, Neotoma micropus (238). As there are vectors throughout the southern United States, it is likely that rates of disease due to L. mexicana will increase in the United States (309).
South American species of Leishmania causing CL, including L. amazonensis, L. braziliensis, L. guyanensis, and L. panamensis, have sylvatic reservoirs. Two CL-causing species in South America have domestic animal reservoirs. L. venezuelensis has been identified in several urban and periurban areas, with a suspected domestic cat reservoir host (259). L. peruviana, a species once limited to altitudes of 1,200 to 3,000 m in the Peruvian Andes, uses the dog as a reservoir, although limited evidence of transmission to sandflies exists (255). The zoonotic transmission of cutaneous leishmaniasis from companion animals, most notably the dog, has been occurring. However, while there have been numerous reports of canine and feline infection and clinical disease with ZCL, their roles as reservoirs have not been firmly established.
Human risk factors for ZCL are dependent upon exposure to vector species and the presence of reservoir species. In all cases, urbanization and wilderness encroachment have resulted in increased interactions between humans and reservoir and vector species and the establishment of (peri)urban domestic life cycles rather than sylvatic ones. The establishment of urban domestic transmission holds the potential for larger outbreaks of ZCL due to a more frequent exposure of naïve human hosts during everyday life versus occasional infection due to human introduction into the sylvatic cycle. Deforestation and agricultural development, including the damming of waterways and irrigation, also create new environments optimal for the survival of rodent reservoirs. An outbreak in Mazar-e Sharif, Afghanistan, was traced to exploding populations of Rhombomys opimus in an area of irrigation canal construction (310). L. guyanensis evolved urbanized life cycles in Brazil, leading to an increased risk of infection dependent upon household distance from the forest (314, 315). Interestingly, long-term surveillance in Bahia, Brazil, suggests that the number of cases in agricultural workers has decreased in the last 20 years, with an overall increase in the rate of disseminated disease in coastal areas (316).
Recent studies have identified younger age (5–19), sleeping without bed nets, ownership of dogs and cattle, and the presence of organic animal material as risk factors for infection (317). Additionally, social factors, including low education and income, increase the likelihood of exposure due to poor housing/environments conducive to the presence of vectors. These factors include mud-walled housing (not brick or concrete), dirt streets and/or floors, the cleanliness of the domestic environment, and working in forested areas (318–320).
Zoonotic visceral leishmaniasis.
Visceral leishmaniasis is caused by Leishmania donovani in India, areas of Asia, Sudan, South Sudan, Kenya, and Ethiopia and by L. infantum (syn., L. chagasi) throughout South America and in areas of Central America and Mexico, the Mediterranean basin, the Middle East, central and southwestern Asia, northwestern China, and northern and sub-Saharan Africa. The transmission of L. donovani is considered solely anthroponotic, although animal infections have been reported, and a reservoir status has been suggested for dogs in Sudan and for goats and possibly other species in Nepal (265, 266).
Zoonotic infection by L. infantum is responsible for the majority of zoonosis-based human cases of VL. As with other species of this genus, vectors for Leishmania in the New World are Lutzomyia spp., predominantly Lutzomyia longipalpis, and those in the Old World are species of the genus Phlebotomus. Canids are considered the primary sylvatic and domestic reservoirs, with foxes, jackals, and wolves filling the role of sylvatic reservoirs and the domestic dog serving as a domestic reservoir (9, 269). Wild felids can be susceptible to infection, although a definitive role in transmission is unclear (321). Similarly, domestic cats in areas of Brazil where the disease is endemic have been infected and could be secondary reservoirs (270). Humans are relatively resistant to L. infantum infection and have high rates of asymptomatic infection. The WHO estimated 200,000 to 400,000 clinical VL cases to be a gross underestimate of the burden of human L. infantum infection worldwide (322). An asymptomatic, immunologically competent patient with low-level or absent parasitemia and a compartmentalization of the parasite within the secondary lymphoid organs and bone marrow may have reduced transmission to sandflies. When zoonotic visceral leishmaniasis (ZVL) occurs in immunocompromised persons or animals, the parasite load increases, and transmission, as well as clinical disease, is likely to occur.
There is a clear role for domestic dogs in the maintenance and transmission of L. infantum. In Brazil, infected dogs in urban and periurban areas of endemicity are common, accompanied by a sufficient environment for L. longipalpis (271). Vertical transmission has been documented for dogs, suggesting a vector-independent means of transmission in these areas as well (323). In northeastern Brazil, there was a reported seroprevalence rate of 32.5%, with parasitemia in almost 47% of seropositive dogs (271). In southeastern Brazil, seroprevalence rates in dogs ranged from 15.9% in urban areas up to 57% in rural areas of endemicity (272, 273). In Mediterranean areas of endemicity, the reported canine seropositivity rates were approximately 8.1% in central Spain, 13% in southern Spain, and between 4 and 25% in Portugal, France, Italy, Greece, Cyprus, and Turkey (274, 324, 325). The presence of infected animals was significantly correlated with human risk, but the incidences of human disease varied by country and region (271, 324). In Europe, the estimated incidence of human ZVL ranges from 0.02 to 0.47 cases per 100,000 people, except in Turkey, where a higher incidence, 1.6 to 8.53 cases/100,000 people, was reported (324, 326). In areas of Brazil where the disease is endemic, the incidence of VL was much higher, with twice the number of cases for the entire Mediterranean region between 2004 and 2008 (326).
Risk factors for ZVL account for differences in human incidences in Brazil versus Europe. Numerous studies have demonstrated a risk for ZVL based on the presence of dogs within the household, housing types (mud-walled housing compared to concrete or brick housing), education level, income, and disease knowledge (271, 272, 327, 328). Clinical ZVL has also been associated with poor nutrition (328). Many of the same factors associated with human ZVL also apply to owner characteristics associated with risks for canine VL (271, 272, 329). Vertical transmission has been characterized for dogs and people, causing an increased risk for infants born to parasitemic mothers (330). The treatment of pregnant women with liposomal amphotericin B appears to be successful in reducing the occurrence of congenital VL (330). Additional risk factors for humans are related to their immunologic status and their ability to clear infection or maintain an asymptomatic state. These factors include concurrent infection with HIV, coinfections with helminth parasites, drug abuse, and other immune suppressions. These comorbidities, especially HIV coinfection, confer a higher risk for the development of ZVL (331). The risks for dogs also include coinfection with other parasites, rickettsial diseases, heartworm disease, or immune suppression (332, 333). Genetic susceptibility may also be a factor for the development of clinical disease. Large-scale studies conducted in numerous countries indicated a role of genetic susceptibility, including polymorphisms of a number of metabolic genes, iron metabolism genes, chemokines, cytokines, and HLA alleles (334). This suggests a complex evolutionary interplay of parasite and host factors, which are likely associated with disease susceptibility.
Prevention
The prevention of leishmaniasis requires the blocking of a step in the parasite's life cycle. The interruption of sandfly transmission is of primary importance for individual and community protection from CL and VL. Avoiding being outside during times of sandfly feeding, typically from dusk until dawn, can greatly reduce transmission. Wearing topical insect repellents and utilizing permethrin-treated bed nets or clothes are also effective in repelling sandflies (335–337). In domestic areas, residual household sprays have been utilized to reduce the presence of vectors, but inconsistent compliance with periodic spraying, cost, and concerns over insecticide resistance limit the efficacy of this type of intervention on a widespread, long-term basis.
Approaches to address reservoir populations have also been implemented in an attempt to reduce ZVL. Brazil has implemented public health policies utilizing voluntary surveillance and the culling of positive dogs to reduce the burden of VL (338). While studies have shown that vigilant surveillance and culling can reduce the canine prevalence of VL to a degree, impacts on human infection are more difficult to ascertain (339). Limitations in diagnostic sensitivity likely lead to false-negative diagnoses for a large number of asymptomatic dogs, delays between testing and dog removal increase the likelihood of transmission, and the financial and emotional costs of the policy implemented in Brazil are high (329, 339, 340). The use of permethrin or deltamethrin collars or topical applications has shown efficacy in reducing sandfly feeding and transmission in areas of endemicity (341, 342). However, the cost and necessity for reapplication make these interventions more difficult to utilize in many regions of endemicity. Limiting the degree of human infection is also important for the control and prevention of severe clinical disease. Public health efforts have resulted in a reduction in the number of severe cases in Brazil. There is currently no vaccine for human leishmaniasis. Continued research may result in a vaccine with long-term, efficacious protection.
CONCLUSIONS
Zoonotic diseases are an important challenge to the health of the public worldwide. Rudolf Virchow, a 19th-century German physician, politician, and luminary promoter of public health and social medicine, once stated, “There is no scientific barrier, nor should there be, between veterinary medicine and human medicine” (343). In fact, more than 60% of recently emergent human pathogens are zoonotic and include more than 200 different bacteria, viruses, protozoa, and other parasites (344). However, in many regions of the world, human and veterinary medicine remain segregated. Companion animals, such as dogs, cats, and others, have a significant role in companionship in the United States, with an estimated 62% of homes owning a pet in 2008, equaling 72.9 million pet-owning homes in the United States alone (1). Companion animals benefit human health in many ways, including social interactions and support and the documented health benefits of reduced blood pressure, reduced heart disease, and a reduction in health care costs (345, 346). Recent studies indicated that infants in pet-owning homes are healthier, with lower rates of otitis, reduced numbers of respiratory infections, and reductions in the use of necessary antibiotics (347). However, when interactions between species are increased, the transmission of pathogens common to both species becomes a greater risk to humans and pets alike. Here we discuss the mechanisms of zoonotic protozoal disease virulence, human and companion animal epidemiology, means of transmission, and subsequent ways to prevent zoonotic disease. In all instances, good hygiene, including proper hand washing, is critical for halting zoonotic transmission. To prevent the vector-borne spread of Babesia and Leishmania spp., the use of topical insecticides on pets and their owners, as well as other means to prevent infectious insect bites, is indicated. The transmission of Toxoplasma gondii, a classical zoonotic disease, can additionally be prevented by proper food preparation and water sanitation. Although the zoonotic role of Giardia is debated, irrespective of how the protists arrived in water, proper water sanitation methods will hinder Giardia transmission. Surprisingly, Trypanosoma cruzi has emerged as a food- and, particularly, juice-borne disease, requiring the adaptation of food sanitation methods for the prevention of T. cruzi transmission. As dogs and cats often consume insects, keeping them inside, where the chances that they can be a meal for the vector or vice versa are reduced, can prevent T. cruzi or other vector-borne infections. An understanding of zoonotic protozoa and their transmission and epidemiology is a necessary step toward the prevention of these zoonotic diseases. We cannot completely eliminate our exposure to these pathogens, adapted for long-term survival within multiple host species over millennia, and we do not want to eliminate our exposure to our pets. Instead, understanding what factors increase zoonotic protozoal disease risk and utilizing and promoting effective means for their prevention can reduce the incidence and global spread of zoonotic protozoal pathogens in the future.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI50803 and the Pfizer Animal Health/Morris Animal Foundation Fellowship for Advanced Study.
Biographies

Kevin J. Esch, D.V.M., M.P.H., is a Ph.D. candidate in immunobiology and an anatomic pathology resident in the Department of Veterinary Pathology at the Iowa State University College of Veterinary Medicine. Dr. Esch's primary research focus is on T cell immunopathology during visceral leishmaniasis and the epidemiology and ecology of canine and human Leishmania infantum chagasi infections in northeastern Brazil. He is interested in the exhaustion of T cell function during symptomatic visceral leishmaniasis and mechanisms of T cell suppression mediated through coinhibitory receptors. He has an additional interest in the pathophysiology of immune-mediated glomerulonephropathies and glomerular podocyte and mesangial cell responses to immune complexes and complement. Dr. Esch received his D.V.M. from the Iowa State University of Medicine and his M.P.H. from the University of Iowa.

Christine A. Petersen graduated with her D.V.M. from Cornell University in 1998 and then completed her Ph.D. in Immunology and Infectious Diseases at the Harvard School of Public Health in 2004. With the first seeds planted as a veterinary student doing a summer research project on African trypanosomiasis in Nairobi, Kenya, Dr. Petersen's scientific career has focused on the prevention of zoonotic vector-borne diseases, which primarily includes leishmaniasis. As an Associate Professor at Iowa State University, she also teaches veterinary and graduate coursework and conducts outreach related to the diagnosis, treatment, and prevention of zoonotic diseases within companion animal and human populations. Additionally, she codirects the animal shelter medicine program and supervises the student-run trap-neuter-return program for feral cats, two populations ripe for the spread of zoonotic infectious diseases. Her interest in “One Health” was started while a veterinary student by a cadre of veterinarians working in global developing areas.
REFERENCES
- 1. APPA 2012. APPA national pet owners survey 2011-2012. APPA, Washington, DC: http://www.americanpetproducts.org/press_industrytrends.asp [Google Scholar]
- 2. Krause-Parello CA. 2012. Pet ownership and older women: the relationships among loneliness, pet attachment support, human social support, and depressed mood. Geriatr. Nurs. 33:194–203 [DOI] [PubMed] [Google Scholar]
- 3. McConnell AR, Brown CM, Shoda TM, Stayton LE, Martin CE. 2011. Friends with benefits: on the positive consequences of pet ownership. J. Pers. Soc. Psychol. 101:1239–1252 [DOI] [PubMed] [Google Scholar]
- 4. Larsen BA, Lingaas F. 1997. Dogs and health. A review of documented connections. Tidsskr. Nor. Laegeforen. 117:4375–4379 (In Norwegian.) [PubMed] [Google Scholar]
- 5. Raina P, Waltner-Toews D, Bonnett B, Woodward C, Abernathy T. 1999. Influence of companion animals on the physical and psychological health of older people: an analysis of a one-year longitudinal study. J. Am. Geriatr. Soc. 47:323–329 [DOI] [PubMed] [Google Scholar]
- 6. Ballweber LR, Xiao L, Bowman DD, Kahn G, Cama VA. 2010. Giardiasis in dogs and cats: update on epidemiology and public health significance. Trends Parasitol. 26:180–189 [DOI] [PubMed] [Google Scholar]
- 7. Jones JL, Dargelas V, Roberts J, Press C, Remington JS, Montoya JG. 2009. Risk factors for Toxoplasma gondii infection in the United States. Clin. Infect. Dis. 49:878–884 [DOI] [PubMed] [Google Scholar]
- 8. Barr SC. 2009. Canine Chagas' disease (American trypanosomiasis) in North America. Vet. Clin. North Am. Small Anim. Pract. 39:1055–1064 [DOI] [PubMed] [Google Scholar]
- 9. Petersen CA. 2009. Leishmaniasis, an emerging disease found in companion animals in the United States. Top. Companion Anim. Med. 24:182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chuang YM, Ho YC, Chang HT, Yu CJ, Yang PC, Hsueh PR. 2008. Disseminated cryptococcosis in HIV-uninfected patients. Eur. J. Clin. Microbiol. 27:307–310 [DOI] [PubMed] [Google Scholar]
- 11. Cribier BJ, Bakshi R. 2004. Terbinafine in the treatment of onychomycosis: a review of its efficacy in high-risk populations and in patients with nondermatophyte infections. Br. J. Dermatol. 150:414–420 [DOI] [PubMed] [Google Scholar]
- 12. Dubey JP, Lindsay DS, Lappin MR. 2009. Toxoplasmosis and other intestinal coccidial infections in cats and dogs. Vet. Clin. North Am. Small Anim. Pract 39:1009–1034 [DOI] [PubMed] [Google Scholar]
- 13. Dubey JP. 2009. History of the discovery of the life cycle of Toxoplasma gondii. Int. J. Parasitol. 39:877–882 [DOI] [PubMed] [Google Scholar]
- 14. Elmore SA, Jones JL, Conrad PA, Patton S, Lindsay DS, Dubey JP. 2010. Toxoplasma gondii: epidemiology, feline clinical aspects, and prevention. Trends Parasitol. 26:190–196 [DOI] [PubMed] [Google Scholar]
- 15. Malmasi A, Mosallanejad B, Mohebali M, Sharifian Fard M, Taheri M. 2009. Prevention of shedding and re-shedding of Toxoplasma gondii oocysts in experimentally infected cats treated with oral clindamycin: a preliminary study. Zoonoses Public Health 56:102–104 [DOI] [PubMed] [Google Scholar]
- 16. Dubey JP, Frenkel JK. 1976. Feline toxoplasmosis from acutely infected mice and the development of Toxoplasma cysts. J. Protozool. 23:537–546 [DOI] [PubMed] [Google Scholar]
- 17. Yoshida N, Tyler KM, Llewellyn MS. 2011. Invasion mechanisms among emerging food-borne protozoan parasites. Trends Parasitol. 27:459–466 [DOI] [PubMed] [Google Scholar]
- 18. Carruthers V, Boothroyd JC. 2007. Pulling together: an integrated model of Toxoplasma cell invasion. Curr. Opin. Microbiol. 10:83–89 [DOI] [PubMed] [Google Scholar]
- 19. Dubey JP. 2006. Comparative infectivity of oocysts and bradyzoites of Toxoplasma gondii for intermediate (mice) and definitive (cats) hosts. Vet. Parasitol. 140:69–75 [DOI] [PubMed] [Google Scholar]
- 20. Jones JL, Kruszon-Moran D, Wilson M, McQuillan G, Navin T, McAuley JB. 2001. Toxoplasma gondii infection in the United States: seroprevalence and risk factors. Am. J. Epidemiol. 154:357–365 [DOI] [PubMed] [Google Scholar]
- 21. Weiss LM, Dubey JP. 2009. Toxoplasmosis: a history of clinical observations. Int. J. Parasitol. 39:895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lang C, Gross U, Luder CG. 2007. Subversion of innate and adaptive immune responses by Toxoplasma gondii. Parasitol. Res. 100:191–203 [DOI] [PubMed] [Google Scholar]
- 23. Munoz M, Liesenfeld O, Heimesaat MM. 2011. Immunology of Toxoplasma gondii. Immunol. Rev. 240:269–285 [DOI] [PubMed] [Google Scholar]
- 24. Saeij JP, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, Boothroyd JC. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314:1780–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Butcher BA, Kim L, Panopoulos AD, Watowich SS, Murray PJ, Denkers EY. 2005. IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-alpha in host macrophages. J. Immunol. 174:3148–3152 [DOI] [PubMed] [Google Scholar]
- 27. Lang C, Algner M, Beinert N, Gross U, Luder CG. 2006. Diverse mechanisms employed by Toxoplasma gondii to inhibit IFN-gamma-induced major histocompatibility complex class II gene expression. Microbes Infect. 8:1994–2005 [DOI] [PubMed] [Google Scholar]
- 28. Khan IA, Matsuura T, Kasper LH. 1995. IL-10 mediates immunosuppression following primary infection with Toxoplasma gondii in mice. Parasite Immunol. 17:185–195 [DOI] [PubMed] [Google Scholar]
- 29. Santos TA, Portes JA, Damasceno-Sa JC, Caldas LA, Souza W, Damatta RA, Seabra SH. 2011. Phosphatidylserine exposure by Toxoplasma gondii is fundamental to balance the immune response granting survival of the parasite and of the host. PLoS One 6:e27867 doi:10.1371/journal.pone.0027867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vutova P, Wirth M, Hippe D, Gross U, Schulze-Osthoff K, Schmitz I, Luder CG. 2007. Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant processing and degradation of caspase 8. Cell. Microbiol. 9:1556–1570 [DOI] [PubMed] [Google Scholar]
- 31. Khan IA, Matsuura T, Kasper LH. 1996. Activation-mediated CD4+ T cell unresponsiveness during acute Toxoplasma gondii infection in mice. Int. Immunol. 8:887–896 [DOI] [PubMed] [Google Scholar]
- 32. Nash PB, Purner MB, Leon RP, Clarke P, Duke RC, Curiel TJ. 1998. Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J. Immunol. 160:1824–1830 [PubMed] [Google Scholar]
- 33. House PK, Vyas A, Sapolsky R. 2011. Predator cat odors activate sexual arousal pathways in brains of Toxoplasma gondii infected rats. PLoS One 6:e23277 doi:10.1371/journal.pone.0023277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lindsay DS, Dubey JP, Butler JM, Blagburn BL. 1997. Mechanical transmission of Toxoplasma gondii oocysts by dogs. Vet. Parasitol. 73:27–33 [DOI] [PubMed] [Google Scholar]
- 35. Vollaire MR, Radecki SV, Lappin MR. 2005. Seroprevalence of Toxoplasma gondii antibodies in clinically ill cats in the United States. Am. J. Vet. Res. 66:874–877 [DOI] [PubMed] [Google Scholar]
- 36. Jones JL, Kruszon-Moran D, Sanders-Lewis K, Wilson M. 2007. Toxoplasma gondii infection in the United States, 1999-2004, decline from the prior decade. Am. J. Trop. Med. Hyg. 77:405–410 [PubMed] [Google Scholar]
- 37. Robert-Gangneux F, Darde ML. 2012. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin. Microbiol. Rev. 25:264–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pappas G, Roussos N, Falagas ME. 2009. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int. J. Parasitol. 39:1385–1394 [DOI] [PubMed] [Google Scholar]
- 39. Conrad PA, Miller MA, Kreuder C, James ER, Mazet J, Dabritz H, Jessup DA, Gulland F, Grigg ME. 2005. Transmission of Toxoplasma: clues from the study of sea otters as sentinels of Toxoplasma gondii flow into the marine environment. Int. J. Parasitol. 35:1155–1168 [DOI] [PubMed] [Google Scholar]
- 40. Shapiro K, Conrad PA, Mazet JA, Wallender WW, Miller WA, Largier JL. 2010. Effect of estuarine wetland degradation on transport of Toxoplasma gondii surrogates from land to sea. Appl. Environ. Microbiol. 76:6821–6828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dubey JP, Hill DE, Jones JL, Hightower AW, Kirkland E, Roberts JM, Marcet PL, Lehmann T, Vianna MC, Miska K, Sreekumar C, Kwok OC, Shen SK, Gamble HR. 2005. Prevalence of viable Toxoplasma gondii in beef, chicken, and pork from retail meat stores in the United States: risk assessment to consumers. J. Parasitol. 91:1082–1093 [DOI] [PubMed] [Google Scholar]
- 42. Boyer K, Hill D, Mui E, Wroblewski K, Karrison T, Dubey JP, Sautter M, Noble AG, Withers S, Swisher C, Heydemann P, Hosten T, Babiarz J, Lee D, Meier P, McLeod R. 2011. Unrecognized ingestion of Toxoplasma gondii oocysts leads to congenital toxoplasmosis and causes epidemics in North America. Clin. Infect. Dis. 53:1081–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Holland GN. 2003. Ocular toxoplasmosis: a global reassessment. Part I: epidemiology and course of disease. Am. J. Ophthalmol. 136:973–988 [DOI] [PubMed] [Google Scholar]
- 44. Villena I, Ancelle T, Delmas C, Garcia P, Brezin AP, Thulliez P, Wallon M, King L, Goulet V. 2010. Congenital toxoplasmosis in France in 2007: first results from a national surveillance system. Euro Surveill. 15(25):pii=19600.http://www.eurosurveillance.org/View Article.aspx?ArticleId=19600 [DOI] [PubMed] [Google Scholar]
- 45. Jones JL, Roberts JM. 2012. Toxoplasmosis hospitalizations in the United States, 2008, and trends, 1993-2008. Clin. Infect. Dis. 54:e58–61 [DOI] [PubMed] [Google Scholar]
- 46. Yoder JS, Harral C, Beach MJ. 2010. Giardiasis surveillance—United States, 2006-2008. MMWR Surveill. Summ. 59:15–25 [PubMed] [Google Scholar]
- 47. Yoder JS, Beach MJ. 2007. Giardiasis surveillance—United States, 2003-2005. MMWR Surveill. Summ. 56:11–18 [PubMed] [Google Scholar]
- 48. Feng Y, Xiao L. 2011. Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin. Microbiol. Rev. 24:110–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Covacin C, Aucoin DP, Elliot A, Thompson RC. 2011. Genotypic characterisation of Giardia from domestic dogs in the USA. Vet. Parasitol. 177:28–32 [DOI] [PubMed] [Google Scholar]
- 50. Ankarklev J, Jerlstrom-Hultqvist J, Ringqvist E, Troell K, Svard SG. 2010. Behind the smile: cell biology and disease mechanisms of Giardia species. Nat. Rev. Microbiol. 8:413–422 [DOI] [PubMed] [Google Scholar]
- 51. Rendtorff RC. 1954. The experimental transmission of human intestinal protozoan parasites. II. Giardia lamblia cysts given in capsules. Am. J. Hyg. (Lond.) 59:209–220 [DOI] [PubMed] [Google Scholar]
- 52. Hetsko ML, McCaffery JM, Svard SG, Meng TC, Que X, Gillin FD. 1998. Cellular and transcriptional changes during excystation of Giardia lamblia in vitro. Exp. Parasitol. 88:172–183 [DOI] [PubMed] [Google Scholar]
- 53. Reiner DS, Hetsko ML, Meszaros JG, Sun CH, Morrison HG, Brunton LL, Gillin FD. 2003. Calcium signaling in excystation of the early diverging eukaryote, Giardia lamblia. J. Biol. Chem. 278:2533–2540 [DOI] [PubMed] [Google Scholar]
- 54. Ward W, Alvarado L, Rawlings ND, Engel JC, Franklin C, McKerrow JH. 1997. A primitive enzyme for a primitive cell: the protease required for excystation of Giardia. Cell 89:437–444 [DOI] [PubMed] [Google Scholar]
- 55. Slavin I, Saura A, Carranza PG, Touz MC, Nores MJ, Lujan HD. 2002. Dephosphorylation of cyst wall proteins by a secreted lysosomal acid phosphatase is essential for excystation of Giardia lamblia. Mol. Biochem. Parasitol. 122:95–98 [DOI] [PubMed] [Google Scholar]
- 56. Bernander R, Palm JE, Svard SG. 2001. Genome ploidy in different stages of the Giardia lamblia life cycle. Cell. Microbiol. 3:55–62 [DOI] [PubMed] [Google Scholar]
- 57. Hagen KD, Hirakawa MP, House SA, Schwartz CL, Pham JK, Cipriano MJ, De La Torre MJ, Sek AC, Du G, Forsythe BM, Dawson SC. 2011. Novel structural components of the ventral disc and lateral crest in Giardia intestinalis. PLoS Negl. Trop. Dis. 5:e1442 doi:10.1371/journal.pntd.0001442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jenkins MC, O'Brien CN, Murphy C, Schwarz R, Miska K, Rosenthal B, Trout JM. 2009. Antibodies to the ventral disc protein delta-giardin prevent in vitro binding of Giardia lamblia trophozoites. J. Parasitol. 95:895–899 [DOI] [PubMed] [Google Scholar]
- 59. Chin AC, Teoh DA, Scott KG, Meddings JB, Macnaughton WK, Buret AG. 2002. Strain-dependent induction of enterocyte apoptosis by Giardia lamblia disrupts epithelial barrier function in a caspase-3-dependent manner. Infect. Immun. 70:3673–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Panaro MA, Cianciulli A, Mitolo V, Mitolo CI, Acquafredda A, Brandonisio O, Cavallo P. 2007. Caspase-dependent apoptosis of the HCT-8 epithelial cell line induced by the parasite Giardia intestinalis. FEMS Immunol. Med. Microbiol. 51:302–309 [DOI] [PubMed] [Google Scholar]
- 61. Scott KG, Meddings JB, Kirk DR, Lees-Miller SP, Buret AG. 2002. Intestinal infection with Giardia spp. reduces epithelial barrier function in a myosin light chain kinase-dependent fashion. Gastroenterology 123:1179–1190 [DOI] [PubMed] [Google Scholar]
- 62. Buret AG, Mitchell K, Muench DG, Scott KG. 2002. Giardia lamblia disrupts tight junctional ZO-1 and increases permeability in non-transformed human small intestinal epithelial monolayers: effects of epidermal growth factor. Parasitology 125:11–19 [DOI] [PubMed] [Google Scholar]
- 63. Troeger H, Epple HJ, Schneider T, Wahnschaffe U, Ullrich R, Burchard GD, Jelinek T, Zeitz M, Fromm M, Schulzke JD. 2007. Effect of chronic Giardia lamblia infection on epithelial transport and barrier function in human duodenum. Gut 56:328–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Prucca CG, Rivero FD, Lujan HD. 2011. Regulation of antigenic variation in Giardia lamblia. Annu. Rev. Microbiol. 65:611–630 [DOI] [PubMed] [Google Scholar]
- 65. Cotton JA, Beatty JK, Buret AG. 2011. Host parasite interactions and pathophysiology in Giardia infections. Int. J. Parasitol. 41:925–933 [DOI] [PubMed] [Google Scholar]
- 66. Roxstrom-Lindquist K, Palm D, Reiner D, Ringqvist E, Svard SG. 2006. Giardia immunity—an update. Trends Parasitol. 22:26–31 [DOI] [PubMed] [Google Scholar]
- 67. Solaymani-Mohammadi S, Singer SM. 2010. Giardia duodenalis: the double-edged sword of immune responses in giardiasis. Exp. Parasitol. 126:292–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yoder JS, Hlavsa MC, Craun GF, Hill V, Roberts V, Yu PA, Hicks LA, Alexander NT, Calderon RL, Roy SL, Beach MJ. 2008. Surveillance for waterborne disease and outbreaks associated with recreational water use and other aquatic facility-associated health events—United States, 2005-2006. MMWR Surveill. Summ. 57:1–29 [PubMed] [Google Scholar]
- 69. Daly ER, Roy SJ, Blaney DD, Manning JS, Hill VR, Xiao L, Stull JW. 2010. Outbreak of giardiasis associated with a community drinking-water source. Epidemiol. Infect. 138:491–500 [DOI] [PubMed] [Google Scholar]
- 70. Yoder J, Roberts V, Craun GF, Hill V, Hicks LA, Alexander NT, Radke V, Calderon RL, Hlavsa MC, Beach MJ, Roy SL. 2008. Surveillance for waterborne disease and outbreaks associated with drinking water and water not intended for drinking—United States, 2005-2006. MMWR Surveill. Summ. 57:39–62 [PubMed] [Google Scholar]
- 71. Brunkard JM, Ailes E, Roberts VA, Hill V, Hilborn ED, Craun GF, Rajasingham A, Kahler A, Garrison L, Hicks L, Carpenter J, Wade TJ, Beach MJ, Yoder JS. 2011. Surveillance for waterborne disease outbreaks associated with drinking water—United States, 2007-2008. MMWR Surveill. Summ. 60:38–68 [PubMed] [Google Scholar]
- 72. Craun GF, Brunkard JM, Yoder JS, Roberts VA, Carpenter J, Wade T, Calderon RL, Roberts JM, Beach MJ, Roy SL. 2010. Causes of outbreaks associated with drinking water in the United States from 1971 to 2006. Clin. Microbiol. Rev. 23:507–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Goncalves AL, Belizario TL, Pimentel JB, Penatti MP, Pedroso RS. 2011. Prevalence of intestinal parasites in preschool children in the region of Uberlandia, State of Minas Gerais, Brazil. Rev. Soc. Bras. Med. Trop. 44:191–193 [DOI] [PubMed] [Google Scholar]
- 74. Matos SM, Assis AM, Prado MS, Strina A, Santos LA, Jesus SR, Barreto ML. 2008. Giardia duodenalis infection and anthropometric status in preschoolers in Salvador, Bahia State, Brazil. Cad. Saude Publica 24:1527–1535 [DOI] [PubMed] [Google Scholar]
- 75. de Souza EA, da Silva-Nunes M, Malafronte RS, Muniz PT, Cardoso MA, Ferreira MU. 2007. Prevalence and spatial distribution of intestinal parasitic infections in a rural Amazonian settlement, Acre State, Brazil. Cad. Saude Publica 23:427–434 [DOI] [PubMed] [Google Scholar]
- 76. Molina N, Minvielle M, Grenovero S, Salomon C, Basualdo J. 2011. High prevalences of infection with Giardia intestinalis genotype B among children in urban and rural areas of Argentina. Ann. Trop. Med. Parasitol. 105:299–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Laishram S, Kang G, Ajjampur SS. 2012. Giardiasis: a review on assemblage distribution and epidemiology in India. Indian J. Gastroenterol. 31:3–12 [DOI] [PubMed] [Google Scholar]
- 78. Dib HH, Lu SQ, Wen SF. 2008. Prevalence of Giardia lamblia with or without diarrhea in South East, South East Asia and the Far East. Parasitol. Res. 103:239–251 [DOI] [PubMed] [Google Scholar]
- 79. Tangtrongsup S, Scorza V. 2010. Update on the diagnosis and management of Giardia spp infections in dogs and cats. Top. Companion Anim. Med. 25:155–162 [DOI] [PubMed] [Google Scholar]
- 80. Mark-Carew MP, Wade SE, Chang YF, Schaaf S, Mohammed HO. 2012. Prevalence of Giardia duodenalis assemblages among dairy herds in the New York City watershed. Vet. Parasitol. 185:151–157 [DOI] [PubMed] [Google Scholar]
- 81. Feng Y, Ortega Y, Cama V, Terrel J, Xiao L. 2008. High intragenotypic diversity of Giardia duodenalis in dairy cattle on three farms. Parasitol. Res. 103:87–92 [DOI] [PubMed] [Google Scholar]
- 82. Geurden T, Thomas P, Casaert S, Vercruysse J, Claerebout E. 2008. Prevalence and molecular characterisation of Cryptosporidium and Giardia in lambs and goat kids in Belgium. Vet. Parasitol. 155:142–145 [DOI] [PubMed] [Google Scholar]
- 83. Santin M, Dargatz D, Fayer R. 2012. Prevalence of Giardia duodenalis assemblages in weaned cattle on cow-calf operations in the United States. Vet. Parasitol. 183:231–236 [DOI] [PubMed] [Google Scholar]
- 84. Stuart JM, Orr HJ, Warburton FG, Jeyakanth S, Pugh C, Morris I, Sarangi J, Nichols G. 2003. Risk factors for sporadic giardiasis: a case-control study in southwestern England. Emerg. Infect. Dis. 9:229–233 [DOI] [PubMed] [Google Scholar]
- 85. Welch TP. 2000. Risk of giardiasis from consumption of wilderness water in North America: a systematic review of epidemiologic data. Int. J. Infect. Dis. 4:100–103 [DOI] [PubMed] [Google Scholar]
- 86. Hoque ME, Hope VT, Kjellstrom T, Scragg R, Lay-Yee R. 2002. Risk of giardiasis in Aucklanders: a case-control study. Int. J. Infect. Dis. 6:191–197 [DOI] [PubMed] [Google Scholar]
- 87. Coles CL, Levy A, Dagan R, Deckelbaum RJ, Fraser D. 2009. Risk factors for the initial symptomatic Giardia infection in a cohort of young Arab-Bedouin children. Ann. Trop. Paediatr. 29:291–300 [DOI] [PubMed] [Google Scholar]
- 88. Novotny TE, Hopkins RS, Shillam P, Janoff EN. 1990. Prevalence of Giardia lamblia and risk factors for infection among children attending day-care facilities in Denver. Public Health Rep. 105:72–75 [PMC free article] [PubMed] [Google Scholar]
- 89. Hlavsa MC, Roberts VA, Anderson AR, Hill VR, Kahler AM, Orr M, Garrison LE, Hicks LA, Newton A, Hilborn ED, Wade TJ, Beach MJ, Yoder JS. 2011. Surveillance for waterborne disease outbreaks and other health events associated with recreational water—United States, 2007-2008. MMWR Surveill. Summ. 60:1–32 [PubMed] [Google Scholar]
- 90. Julio C, Vilares A, Oleastro M, Ferreira I, Gomes S, Monteiro L, Nunes B, Tenreiro R, Angelo H. 2012. Prevalence and risk factors for Giardia duodenalis infection among children: a case study in Portugal. Parasit. Vectors 5:22 doi:10.1186/1756-3305-5-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Moreira ED, Jr, Nassri VB, Santos RS, Matos JF, de Carvalho WA, Silvani CS, Santana e Sant'ana C. 2005. Association of Helicobacter pylori infection and giardiasis: results from a study of surrogate markers for fecal exposure among children. World J. Gastroenterol. 11:2759–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Center for Food Safety and Public Health 1 May 2005, posting date Giardiasis technical fact sheet. Iowa State University, Ames, IA: http://www.cfsph.iastate.edu/Factsheets/pdfs/giardiasis.pdf [Google Scholar]
- 93. Babes V. 1888. Sur l'hémoglobinurie bactérienne duboeuf. C. R. Hebd. Seances Acad. Sci. 107:692–694 [Google Scholar]
- 94. Uilenberg G. 2006. Babesia—a historical overview. Vet. Parasitol. 138:3–10 [DOI] [PubMed] [Google Scholar]
- 95. Gohil S, Kats LM, Sturm A, Cooke BM. 2010. Recent insights into alteration of red blood cells by Babesia bovis: moovin' forward. Trends Parasitol. 26:591–599 [DOI] [PubMed] [Google Scholar]
- 96. Gray J, Zintl A, Hildebrandt A, Hunfeld KP, Weiss L. 2010. Zoonotic babesiosis: overview of the disease and novel aspects of pathogen identity. Ticks Tick Borne Dis. 1:3–10 [DOI] [PubMed] [Google Scholar]
- 97. Johnson ST, Cable RG, Tonnetti L, Spencer B, Rios J, Leiby DA. 2009. Seroprevalence of Babesia microti in blood donors from Babesia-endemic areas of the northeastern United States: 2000 through 2007. Transfusion 49:2574–2582 [DOI] [PubMed] [Google Scholar]
- 98. Magnarelli LA, Stafford KC, III, Ijdo JW, Fikrig E. 2006. Antibodies to whole-cell or recombinant antigens of Borrelia burgdorferi, Anaplasma phagocytophilum, and Babesia microti in white-footed mice. J. Wildl. Dis. 42:732–738 [DOI] [PubMed] [Google Scholar]
- 99. Leiby DA, Chung AP, Gill JE, Houghton RL, Persing DH, Badon S, Cable RG. 2005. Demonstrable parasitemia among Connecticut blood donors with antibodies to Babesia microti. Transfusion 45:1804–1810 [DOI] [PubMed] [Google Scholar]
- 100. Centers for Disease Control and Prevention 2012. Babesiosis surveillance—18 states, 2011. MMWR Morb. Mortal. Wkly. Rep. 61:505–509 [PubMed] [Google Scholar]
- 101. Quick RE, Herwaldt BL, Thomford JW, Garnett ME, Eberhard ML, Wilson M, Spach DH, Dickerson JW, Telford SR, III, Steingart KR, Pollock R, Persing DH, Kobayashi JM, Juranek DD, Conrad PA. 1993. Babesiosis in Washington State: a new species of Babesia? Ann. Intern. Med. 119:284–290 [DOI] [PubMed] [Google Scholar]
- 102. Kjemtrup AM, Lee B, Fritz CL, Evans C, Chervenak M, Conrad PA. 2002. Investigation of transfusion transmission of a WA1-type babesial parasite to a premature infant in California. Transfusion 42:1482–1487 [DOI] [PubMed] [Google Scholar]
- 103. Herwaldt BL, Kjemtrup AM, Conrad PA, Barnes RC, Wilson M, McCarthy MG, Sayers MH, Eberhard ML. 1997. Transfusion-transmitted babesiosis in Washington State: first reported case caused by a WA1-type parasite. J. Infect. Dis. 175:1259–1262 [DOI] [PubMed] [Google Scholar]
- 104. Prince HE, Lape-Nixon M, Patel H, Yeh C. 2010. Comparison of the Babesia duncani (WA1) IgG detection rates among clinical sera submitted to a reference laboratory for WA1 IgG testing and blood donor specimens from diverse geographic areas of the United States. Clin. Vaccine Immunol. 17:1729–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bloch EM, Herwaldt BL, Leiby DA, Shaieb A, Herron RM, Chervenak M, Reed W, Hunter R, Ryals R, Hagar W, Xayavong MV, Slemenda SB, Pieniazek NJ, Wilkins PP, Kjemtrup AM. 2012. The third described case of transfusion-transmitted Babesia duncani. Transfusion 52:1517–1522 [DOI] [PubMed] [Google Scholar]
- 106. Persing DH, Herwaldt BL, Glaser C, Lane RS, Thomford JW, Mathiesen D, Krause PJ, Phillip DF, Conrad PA. 1995. Infection with a Babesia-like organism in northern California. N. Engl. J. Med. 332:298–303 [DOI] [PubMed] [Google Scholar]
- 107. Herwaldt B, Persing DH, Precigout EA, Goff WL, Mathiesen DA, Taylor PW, Eberhard ML, Gorenflot AF. 1996. A fatal case of babesiosis in Missouri: identification of another piroplasm that infects humans. Ann. Intern. Med. 124:643–650 [DOI] [PubMed] [Google Scholar]
- 108. Herwaldt BL, de Bruyn G, Pieniazek NJ, Homer M, Lofy KH, Slemenda SB, Fritsche TR, Persing DH, Limaye AP. 2004. Babesia divergens-like infection, Washington State. Emerg. Infect. Dis. 10:622–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Beattie JF, Michelson ML, Holman PJ. 2002. Acute babesiosis caused by Babesia divergens in a resident of Kentucky. N. Engl. J. Med. 347:697–698 [DOI] [PubMed] [Google Scholar]
- 110. Yabsley MJ, Romines J, Nettles VF. 2006. Detection of Babesia and Anaplasma species in rabbits from Texas and Georgia, USA. Vector Borne Zoonotic Dis. 6:7–13 [DOI] [PubMed] [Google Scholar]
- 111. Yamane I, Gardner IA, Ryan CP, Levy M, Urrico J, Conrad PA. 1994. Serosurvey of Babesia canis, Babesia gibsoni, and Erlichia canis in pound dogs in California, USA. Prev. Vet. Med. 18:293–304 [Google Scholar]
- 112. Hasle G, Bjune GA, Christensson D, Roed KH, Whist AC, Leinaas HP. 2010. Detection of Babesia divergens in southern Norway by using an immunofluorescence antibody test in cow sera. Acta Vet. Scand. 52:55 doi:10.1186/1751-0147-52-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lempereur L, Lebrun M, Cuvelier P, Sepult G, Caron Y, Saegerman C, Shiels B, Losson B. 2012. Longitudinal field study on bovine Babesia spp. and Anaplasma phagocytophilum infections during a grazing season in Belgium. Parasitol. Res. 110:1525–1530 [DOI] [PubMed] [Google Scholar]
- 114. Devos J, Geysen D. 2004. Epidemiological study of the prevalence of Babesia divergens in a veterinary practice in the mid-east of France. Vet. Parasitol. 125:237–249 [DOI] [PubMed] [Google Scholar]
- 115. Tampieri MP, Galuppi R, Bonoli C, Cancrini G, Moretti A, Pietrobelli M. 2008. Wild ungulates as Babesia hosts in northern and central Italy. Vector Borne Zoonotic Dis. 8:667–674 [DOI] [PubMed] [Google Scholar]
- 116. Cassini R, Marcer F, di Regalbono AF, Cancrini G, Gabrielli S, Moretti A, Galuppi R, Tampieri MP, Pietrobelli M. 2012. New insights into the epidemiology of bovine piroplasmoses in Italy. Vet. Parasitol. 184:77–82 [DOI] [PubMed] [Google Scholar]
- 117. Hunfeld KP, Lambert A, Kampen H, Albert S, Epe C, Brade V, Tenter AM. 2002. Seroprevalence of Babesia infections in humans exposed to ticks in midwestern Germany. J. Clin. Microbiol. 40:2431–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Maamun JM, Suleman MA, Akinyi M, Ozwara H, Kariuki T, Carlsson HE. 2011. Prevalence of Babesia microti in free-ranging baboons and African green monkeys. J. Parasitol. 97:63–67 [DOI] [PubMed] [Google Scholar]
- 119. Bonnet S, Jouglin M, L'Hostis M, Chauvin A. 2007. Babesia sp. EU1 from roe deer and transmission within Ixodes ricinus. Emerg. Infect. Dis. 13:1208–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Duh D, Petrovec M, Avsic-Zupanc T. 2005. Molecular characterization of human pathogen Babesia EU1 in Ixodes ricinus ticks from Slovenia. J. Parasitol. 91:463–465 [DOI] [PubMed] [Google Scholar]
- 121. Herwaldt BL, Caccio S, Gherlinzoni F, Aspock H, Slemenda SB, Piccaluga P, Martinelli G, Edelhofer R, Hollenstein U, Poletti G, Pampiglione S, Loschenberger K, Tura S, Pieniazek NJ. 2003. Molecular characterization of a non-Babesia divergens organism causing zoonotic babesiosis in Europe. Emerg. Infect. Dis. 9:942–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Haselbarth K, Tenter AM, Brade V, Krieger G, Hunfeld KP. 2007. First case of human babesiosis in Germany—clinical presentation and molecular characterisation of the pathogen. Int. J. Med. Microbiol. 297:197–204 [DOI] [PubMed] [Google Scholar]
- 123. Centeno-Lima S, do Rosario V, Parreira R, Maia AJ, Freudenthal AM, Nijhof AM, Jongejan F. 2003. A fatal case of human babesiosis in Portugal: molecular and phylogenetic analysis. Trop. Med. Int. Health 8:760–764 [DOI] [PubMed] [Google Scholar]
- 124. Calvo de Mora A, Garcia Castellano JM, Herrera C, Jimenez-Alonso J. 1985. Human babesiosis: report of a case with fatal outcome. Med. Clin. (Barc.) 85:515–516 (In Spanish.) [PubMed] [Google Scholar]
- 125. Gorenflot A, Moubri K, Precigout E, Carcy B, Schetters TP. 1998. Human babesiosis. Ann. Trop. Med. Parasitol. 92:489–501 [DOI] [PubMed] [Google Scholar]
- 126. Terkawi MA, Thekisoe OM, Katsande C, Latif AA, Mans BJ, Matthee O, Mkize N, Mabogoane N, Marais F, Yokoyama N, Xuan X, Igarashi I. 2011. Serological survey of Babesia bovis and Babesia bigemina in cattle in South Africa. Vet. Parasitol. 182:337–342 [DOI] [PubMed] [Google Scholar]
- 127. Iseki H, Zhou L, Kim C, Inpankaew T, Sununta C, Yokoyama N, Xuan X, Jittapalapong S, Igarashi I. 2010. Seroprevalence of Babesia infections of dairy cows in northern Thailand. Vet. Parasitol. 170:193–196 [DOI] [PubMed] [Google Scholar]
- 128. Silva MG, Henriques G, Sanchez C, Marques PX, Suarez CE, Oliva A. 2009. First survey for Babesia bovis and Babesia bigemina infection in cattle from Central and Southern regions of Portugal using serological and DNA detection methods. Vet. Parasitol. 166:66–72 [DOI] [PubMed] [Google Scholar]
- 129. Urdaz-Rodriguez JH, Fosgate GT, Waghela SD, Alleman AR, Rae DO, Donovan GA, Melendez P. 2009. Seroprevalence estimation and management factors associated with high herd seropositivity for Babesia bovis in commercial dairy farms of Puerto Rico. Trop. Anim. Health Prod. 41:1465–1473 [DOI] [PubMed] [Google Scholar]
- 130. Barros SL, Madruga CR, Araujo FR, Menk CF, de Almeida MA, Melo EP, Kessler RH. 2005. Serological survey of Babesia bovis, Babesia bigemina, and Anaplasma marginale antibodies in cattle from the semi-arid region of the state of Bahia, Brazil, by enzyme-linked immunosorbent assays. Mem. Inst. Oswaldo Cruz 100:513–517 [DOI] [PubMed] [Google Scholar]
- 131. Cassini R, Zanutto S, Frangipane di Regalbono A, Gabrielli S, Calderini P, Moretti A, Tampieri MP, Pietrobelli M. 2009. Canine piroplasmosis in Italy: epidemiological aspects in vertebrate and invertebrate hosts. Vet. Parasitol. 165:30–35 [DOI] [PubMed] [Google Scholar]
- 132. Beck R, Vojta L, Mrljak V, Marinculic A, Beck A, Zivicnjak T, Caccio SM. 2009. Diversity of Babesia and Theileria species in symptomatic and asymptomatic dogs in Croatia. Int. J. Parasitol. 39:843–848 [DOI] [PubMed] [Google Scholar]
- 133. Bourdoiseau G. 2006. Canine babesiosis in France. Vet. Parasitol. 138:118–125 [DOI] [PubMed] [Google Scholar]
- 134. Wudhikarn K, Perry EH, Kemperman M, Jensen KA, Kline SE. 2011. Transfusion-transmitted babesiosis in an immunocompromised patient: a case report and review. Am. J. Med. 124:800–805 [DOI] [PubMed] [Google Scholar]
- 135. Jasmer DP, Reduker DW, Hines SA, Perryman LE, McGuire TC. 1992. Surface epitope localization and gene structure of a Babesia bovis 44-kilodalton variable merozoite surface antigen. Mol. Biochem. Parasitol. 55:75–83 [DOI] [PubMed] [Google Scholar]
- 136. Florin-Christensen M, Suarez CE, Hines SA, Palmer GH, Brown WC, McElwain TF. 2002. The Babesia bovis merozoite surface antigen 2 locus contains four tandemly arranged and expressed genes encoding immunologically distinct proteins. Infect. Immun. 70:3566–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Mosqueda J, McElwain TF, Palmer GH. 2002. Babesia bovis merozoite surface antigen 2 proteins are expressed on the merozoite and sporozoite surface, and specific antibodies inhibit attachment and invasion of erythrocytes. Infect. Immun. 70:6448–6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Mosqueda J, McElwain TF, Stiller D, Palmer GH. 2002. Babesia bovis merozoite surface antigen 1 and rhoptry-associated protein 1 are expressed in sporozoites, and specific antibodies inhibit sporozoite attachment to erythrocytes. Infect. Immun. 70:1599–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Moro MH, David CS, Magera JM, Wettstein PJ, Barthold SW, Persing DH. 1998. Differential effects of infection with a Babesia-like piroplasm, WA1, in inbred mice. Infect. Immun. 66:492–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Li Y, Terkawi MA, Nishikawa Y, Aboge GO, Luo Y, Ooka H, Goo YK, Yu L, Cao S, Sun Y, Yamagishi J, Masatani T, Yokoyama N, Igarashi I, Xuan X. 2012. Macrophages are critical for cross-protective immunity conferred by Babesia microti against Babesia rodhaini infection in mice. Infect. Immun. 80:311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Goff WL, Johnson WC, Parish SM, Barrington GM, Tuo W, Valdez RA. 2001. The age-related immunity in cattle to Babesia bovis infection involves the rapid induction of interleukin-12, interferon-gamma and inducible nitric oxide synthase mRNA expression in the spleen. Parasite Immunol. 23:463–471 [DOI] [PubMed] [Google Scholar]
- 142. Leiby DA. 2011. Transfusion-transmitted Babesia spp.: bull's-eye on Babesia microti. Clin. Microbiol. Rev. 24:14–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. M'Fadyean J, Schein E. 1911. A new species of piroplasm found in the blood of British cattle. J. Comp. Pathol. 24:340–354 [Google Scholar]
- 144. Garnham PCC, Bray RS. 1959. The susceptibility of the higher primates to piroplasms. J. Protozool. 6:352–355 [Google Scholar]
- 145. Garnham PCC, Voller A. 1965. Experimental studies on Babesia divergens in rhesus monkeys with special reference to its diagnosis by serologic methods. Acta Protozool. 3:183–187 [Google Scholar]
- 146. Lewis D, Williams H. 1979. Infection of the Mongolian gerbil with the cattle piroplasm Babesia divergens. Nature 278:170–171 [DOI] [PubMed] [Google Scholar]
- 147. Lewis D, Young ER, Baggott DG, Osborn GD. 1981. Babesia divergens infection of the Mongolian gerbil: titration of infective dose and preliminary observations on the disease produced. J. Comp. Pathol. 91:565–572 [DOI] [PubMed] [Google Scholar]
- 148. Homer MJ, Aguilar-Delfin I, Telford SR, III, Krause PJ, Persing DH. 2000. Babesiosis. Clin. Microbiol. Rev. 13:451–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zintl A, Mulcahy G, Skerrett HE, Taylor SM, Gray JS. 2003. Babesia divergens, a bovine blood parasite of veterinary and zoonotic importance. Clin. Microbiol. Rev. 16:622–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Martinot M, Zadeh MM, Hansmann Y, Grawey I, Christmann D, Aguillon S, Jouglin M, Chauvin A, De Briel D. 2011. Babesiosis in immunocompetent patients, Europe. Emerg. Infect. Dis. 17:114–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hunfeld KP, Allwinn R, Peters S, Kraiczy P, Brade V. 1998. Serologic evidence for tick-borne pathogens other than Borrelia burgdorferi (TOBB) in Lyme borreliosis patients from midwestern Germany. Wien. Klin. Wochenschr. 110:901–908 [PubMed] [Google Scholar]
- 152. Bastian S, Jouglin M, Brisseau N, Malandrin L, Klegou G, L'Hostis M, Chauvin A. 2012. Antibody prevalence and molecular identification of Babesia spp. in roe deer in France. J. Wildl. Dis. 48:416–424 [DOI] [PubMed] [Google Scholar]
- 153. Goethert HK, Telford SR., III 2003. Enzootic transmission of Babesia divergens among cottontail rabbits on Nantucket Island, Massachusetts. Am. J. Trop. Med. Hyg. 69:455–460 [PubMed] [Google Scholar]
- 154. Kjemtrup AM, Thomford J, Robinson T, Conrad PA. 2000. Phylogenetic relationships of human and wildlife piroplasm isolates in the western United States inferred from the 18S nuclear small subunit RNA gene. Parasitology 120(Part 5):487–493 [DOI] [PubMed] [Google Scholar]
- 155. Conrad PA, Kjemtrup AM, Carreno RA, Thomford J, Wainwright K, Eberhard M, Quick R, Telford SR, III, Herwaldt BL. 2006. Description of Babesia duncani n. sp. (Apicomplexa: Babesiidae) from humans and its differentiation from other piroplasms. Int. J. Parasitol. 36:779–789 [DOI] [PubMed] [Google Scholar]
- 156. Kjemtrup AM, Wainwright K, Miller M, Penzhorn BL, Carreno RA. 2006. Babesia conradae, sp. nov., a small canine Babesia identified in California. Vet. Parasitol. 138:103–111 [DOI] [PubMed] [Google Scholar]
- 157. Herwaldt BL, Linden JV, Bosserman E, Young C, Olkowska D, Wilson M. 2011. Transfusion-associated babesiosis in the United States: a description of cases. Ann. Intern. Med. 155:509–519 [DOI] [PubMed] [Google Scholar]
- 158. Sethi S, Alcid D, Kesarwala H, Tolan RW., Jr 2009. Probable congenital babesiosis in infant, New Jersey, USA. Emerg. Infect. Dis. 15:788–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Aderinboye O, Syed SS. 2010. Congenital babesiosis in a four-week-old female infant. Pediatr. Infect. Dis. J. 29:188 doi:10.1097/INF.0b013e3181c36eb9 [DOI] [PubMed] [Google Scholar]
- 160. Joseph JT, Purtill K, Wong SJ, Munoz J, Teal A, Madison-Antenucci S, Horowitz HW, Aguero-Rosenfeld ME, Moore JM, Abramowsky C, Wormser GP. 2012. Vertical transmission of Babesia microti, United States. Emerg. Infect. Dis. 18:1318–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. New DL, Quinn JB, Qureshi MZ, Sigler SJ. 1997. Vertically transmitted babesiosis. J. Pediatr. 131:163–164 [DOI] [PubMed] [Google Scholar]
- 162. Esernio-Jenssen D, Scimeca PG, Benach JL, Tenenbaum MJ. 1987. Transplacental/perinatal babesiosis. J. Pediatr. 110:570–572 [DOI] [PubMed] [Google Scholar]
- 163. Grabowski EF, Giardina PJ, Goldberg D, Masur H, Read SE, Hirsch RL, Benach JL. 1982. Babesiosis transmitted by a transfusion of frozen-thawed blood. Ann. Intern. Med. 96:466–467 [DOI] [PubMed] [Google Scholar]
- 164. Yabsley MJ, Davidson WR, Stallknecht DE, Varela AS, Swift PK, Devos JC, Jr, Dubay SA. 2005. Evidence of tick-borne organisms in mule deer (Odocoileus hemionus) from the western United States. Vector Borne Zoonotic Dis. 5:351–362 [DOI] [PubMed] [Google Scholar]
- 165. Chagas C. 1909. Nova tripanosomiaze humana. Estudos sobre morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n.g., n. sp., ajente etiologico de nova entidade morbida do homen. Mem. Inst. Oswaldo Cruz 1:159–218 [Google Scholar]
- 166. World Health Organization 2008. The global burden of disease: 2004 update. WHO Press, Geneva, Switzerland [Google Scholar]
- 167. Teixeira AR, Hecht MM, Guimaro MC, Sousa AO, Nitz N. 2011. Pathogenesis of Chagas' disease: parasite persistence and autoimmunity. Clin. Microbiol. Rev. 24:592–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Padilla AM, Bustamante JM, Tarleton RL. 2009. CD8+ T cells in Trypanosoma cruzi infection. Curr. Opin. Immunol. 21:385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Caradonna KL, Burleigh BA. 2011. Mechanisms of host cell invasion by Trypanosoma cruzi. Adv. Parasitol. 76:33–61 [DOI] [PubMed] [Google Scholar]
- 170. El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, DA Silveira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T, Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E, Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T, Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nelson S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Pettersson U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tarleton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B, Wortman J, White O, Fraser CM, Stuart KD, Andersson B. 2005. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309:409–415 [DOI] [PubMed] [Google Scholar]
- 171. Padilla AM, Simpson LJ, Tarleton RL. 2009. Insufficient TLR activation contributes to the slow development of CD8+ T cell responses in Trypanosoma cruzi infection. J. Immunol. 183:1245–1252 [DOI] [PubMed] [Google Scholar]
- 172. Nagajyothi F, Weiss LM, Silver DL, Desruisseaux MS, Scherer PE, Herz J, Tanowitz HB. 2011. Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl. Trop. Dis. 5:e953 doi:10.1371/journal.pntd.0000953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Woolsey AM, Sunwoo L, Petersen CA, Brachmann SM, Cantley LC, Burleigh BA. 2003. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J. Cell Sci. 116:3611–3622 [DOI] [PubMed] [Google Scholar]
- 174. Ley V, Robbins ES, Nussenzweig V, Andrews NW. 1990. The exit of Trypanosoma cruzi from the phagosome is inhibited by raising the pH of acidic compartments. J. Exp. Med. 171:401–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Andrews NW, Whitlow MB. 1989. Secretion by Trypanosoma cruzi of a hemolysin active at low pH. Mol. Biochem. Parasitol. 33:249–256 [DOI] [PubMed] [Google Scholar]
- 176. Hall BF, Webster P, Ma AK, Joiner KA, Andrews NW. 1992. Desialylation of lysosomal membrane glycoproteins by Trypanosoma cruzi: a role for the surface neuraminidase in facilitating parasite entry into the host cell cytoplasm. J. Exp. Med. 176:313–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Andrade LO, Andrews NW. 2004. Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J. Exp. Med. 200:1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Campos MA, Almeida IC, Takeuchi O, Akira S, Valente EP, Procopio DO, Travassos LR, Smith JA, Golenbock DT, Gazzinelli RT. 2001. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J. Immunol. 167:416–423 [DOI] [PubMed] [Google Scholar]
- 179. Oliveira AC, Peixoto JR, de Arruda LB, Campos MA, Gazzinelli RT, Golenbock DT, Akira S, Previato JO, Mendonca-Previato L, Nobrega A, Bellio M. 2004. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J. Immunol. 173:5688–5696 [DOI] [PubMed] [Google Scholar]
- 180. Bartholomeu DC, Ropert C, Melo MB, Parroche P, Junqueira CF, Teixeira SM, Sirois C, Kasperkovitz P, Knetter CF, Lien E, Latz E, Golenbock DT, Gazzinelli RT. 2008. Recruitment and endo-lysosomal activation of TLR9 in dendritic cells infected with Trypanosoma cruzi. J. Immunol. 181:1333–1344 [DOI] [PubMed] [Google Scholar]
- 181. Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. 2006. TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J. Immunol. 177:3515–3519 [DOI] [PubMed] [Google Scholar]
- 182. Caetano BC, Carmo BB, Melo MB, Cerny A, dos Santos SL, Bartholomeu DC, Golenbock DT, Gazzinelli RT. 2011. Requirement of UNC93B1 reveals a critical role for TLR7 in host resistance to primary infection with Trypanosoma cruzi. J. Immunol. 187:1903–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Rodriguez AM, Santoro F, Afchain D, Bazin H, Capron A. 1981. Trypanosoma cruzi infection in B-cell-deficient rats. Infect. Immun. 31:524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Takehara HA, Perini A, da Silva MH, Mota I. 1981. Trypanosoma cruzi: role of different antibody classes in protection against infection in the mouse. Exp. Parasitol. 52:137–146 [DOI] [PubMed] [Google Scholar]
- 185. Kirchhoff LV. 2011. Epidemiology of American trypanosomiasis (Chagas disease). Adv. Parasitol. 75:1–18 [DOI] [PubMed] [Google Scholar]
- 186. Araujo A, Jansen AM, Reinhard K, Ferreira LF. 2009. Paleoparasitology of Chagas disease—a review. Mem. Inst. Oswaldo Cruz 104(Suppl 1):9–16 [DOI] [PubMed] [Google Scholar]
- 187. Miles MA, Feliciangeli MD, de Arias MR. 2003. American trypanosomiasis (Chagas' disease) and the role of molecular epidemiology in guiding control strategies. BMJ 326:1444–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Bern C, Kjos S, Yabsley MJ, Montgomery SP. 2011. Trypanosoma cruzi and Chagas' disease in the United States. Clin. Microbiol. Rev. 24:655–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Cantey PT, Stramer SL, Townsend RL, Kamel H, Ofafa K, Todd CW, Currier M, Hand S, Varnado W, Dotson E, Hall C, Jett PL, Montgomery SP. 2012. The United States Trypanosoma cruzi Infection Study: evidence for vector-borne transmission of the parasite that causes Chagas disease among United States blood donors. Transfusion 52:1922–1930 [DOI] [PubMed] [Google Scholar]
- 190. Bonfante-Cabarcas R, Rodriguez-Bonfante C, Vielma BO, Garcia D, Saldivia AM, Aldana E, Curvelo JL. 2011. Seroprevalence for Trypanosoma cruzi infection and associated factors in an endemic area of Venezuela. Cad. Saude Publica 27:1917–1929 (In Spanish.) [DOI] [PubMed] [Google Scholar]
- 191. King RJ, Cordon-Rosales C, Cox J, Davies CR, Kitron UD. 2011. Triatoma dimidiata infestation in Chagas disease endemic regions of Guatemala: comparison of random and targeted cross-sectional surveys. PLoS Negl. Trop. Dis. 5:e1035 doi:10.1371/journal.pntd.0001035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Gurevitz JM, Ceballos LA, Gaspe MS, Alvarado-Otegui JA, Enriquez GF, Kitron U, Gurtler RE. 2011. Factors affecting infestation by Triatoma infestans in a rural area of the humid Chaco in Argentina: a multi-model inference approach. PLoS Negl. Trop. Dis. 5:e1349 doi:10.1371/journal.pntd.0001349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Bowman NM, Kawai V, Gilman RH, Bocangel C, Galdos-Cardenas G, Cabrera L, Levy MZ, Cornejo del Carpio JG, Delgado F, Rosenthal L, Pinedo-Cancino VV, Steurer F, Seitz AE, Maguire JH, Bern C. 2011. Autonomic dysfunction and risk factors associated with Trypanosoma cruzi infection among children in Arequipa, Peru. Am. J. Trop. Med. Hyg. 84:85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Gurtler RE, Ceballos LA, Ordonez-Krasnowski P, Lanati LA, Stariolo R, Kitron U. 2009. Strong host-feeding preferences of the vector Triatoma infestans modified by vector density: implications for the epidemiology of Chagas disease. PLoS Negl. Trop. Dis. 3:e447 doi:10.1371/journal.pntd.0000447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Kjos SA, Snowden KF, Craig TM, Lewis B, Ronald N, Olson JK. 2008. Distribution and characterization of canine Chagas disease in Texas. Vet. Parasitol. 152:249–256 [DOI] [PubMed] [Google Scholar]
- 196. Gurtler RE, Cohen JE, Cecere MC, Lauricella MA, Chuit R, Segura EL. 1998. Influence of humans and domestic animals on the household prevalence of Trypanosoma cruzi in Triatoma infestans populations in northwest Argentina. Am. J. Trop. Med. Hyg. 58:748–758 [DOI] [PubMed] [Google Scholar]
- 197. Gurtler RE, Cecere MC, Lauricella MA, Cardinal MV, Kitron U, Cohen JE. 2007. Domestic dogs and cats as sources of Trypanosoma cruzi infection in rural northwestern Argentina. Parasitology 134:69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Pineda V, Saldana A, Monfante I, Santamaria A, Gottdenker NL, Yabsley MJ, Rapoport G, Calzada JE. 2011. Prevalence of trypanosome infections in dogs from Chagas disease endemic regions in Panama, Central America. Vet. Parasitol. 178:360–363 [DOI] [PubMed] [Google Scholar]
- 199. Balan LU, Yerbes IM, Pina MA, Balmes J, Pascual A, Hernandez O, Lopez R, Monteon V. 2011. Higher seroprevalence of Trypanosoma cruzi infection in dogs than in humans in an urban area of Campeche, Mexico. Vector Borne Zoonotic Dis. 11:843–844 [DOI] [PubMed] [Google Scholar]
- 200. Bern C, Martin DL, Gilman RH. 2011. Acute and congenital Chagas disease. Adv. Parasitol. 75:19–47 [DOI] [PubMed] [Google Scholar]
- 201. Romero M, Postigo J, Schneider D, Chippaux JP, Santalla JA, Brutus L. 2011. Door-to-door screening as a strategy for the detection of congenital Chagas disease in rural Bolivia. Trop. Med. Int. Health 16:562–569 [DOI] [PubMed] [Google Scholar]
- 202. Brutus L, Castillo H, Bernal C, Salas NA, Schneider D, Santalla JA, Chippaux JP. 2010. Detectable Trypanosoma cruzi parasitemia during pregnancy and delivery as a risk factor for congenital Chagas disease. Am. J. Trop. Med. Hyg. 83:1044–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Bern C, Verastegui M, Gilman RH, Lafuente C, Galdos-Cardenas G, Calderon M, Pacori J, Del Carmen Abastoflor M, Aparicio H, Brady MF, Ferrufino L, Angulo N, Marcus S, Sterling C, Maguire JH. 2009. Congenital Trypanosoma cruzi transmission in Santa Cruz, Bolivia. Clin. Infect. Dis. 49:1667–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Cardoso EJ, Valdez GC, Campos AC, Sanchez RD, Mendoza CR, Hernandez AP, Ramirez MH, Habana JR, Gonzalez EB, Matzumura PD, Carlier Y. 2012. Maternal fetal transmission of Trypanosoma cruzi: a problem of public health little studied in Mexico. Exp. Parasitol. 131:425–432 [DOI] [PubMed] [Google Scholar]
- 205. Torrico F, Alonso-Vega C, Suarez E, Rodriguez P, Torrico MC, Dramaix M, Truyens C, Carlier Y. 2004. Maternal Trypanosoma cruzi infection, pregnancy outcome, morbidity, and mortality of congenitally infected and non-infected newborns in Bolivia. Am. J. Trop. Med. Hyg. 70:201–209 [PubMed] [Google Scholar]
- 206. Barona-Vilar C, Gimenez-Marti MJ, Fraile T, Gonzalez-Steinbauer C, Parada C, Gil-Brusola A, Bravo D, Gomez MD, Navarro D, Perez-Tamarit A, Fernandez-Silveira L, Fullana-Montoro A, Borras R. 2011. Prevalence of Trypanosoma cruzi infection in pregnant Latin American women and congenital transmission rate in a non-endemic area: the experience of the Valencian Health Programme (Spain). Epidemiol. Infect. 140:1896–1903 [DOI] [PubMed] [Google Scholar]
- 207. Lazarte RA, Litman-Mazo F, Crewalk Keim J-ADE, Baram M, Klassen-Fischer MK, Massif D, Zabielski ML, Sockwell D, Kurkjian K, Moore A, Qvarnstrom Y, Steurer F. 2012. Congenital transmission of Chagas disease—Virginia, 2010. MMWR Morb. Mortal. Wkly. Rep. 61:477–479 [PubMed] [Google Scholar]
- 208. Yadon ZE, Schmunis GA. 2009. Congenital Chagas disease: estimating the potential risk in the United States. Am. J. Trop. Med. Hyg. 81:927–933 [DOI] [PubMed] [Google Scholar]
- 209. Wendel S. 2010. Transfusion transmitted Chagas disease: is it really under control? Acta Trop. 115:28–34 [DOI] [PubMed] [Google Scholar]
- 210. Cimo PL, Luper WE, Scouros MA. 1993. Transfusion-associated Chagas' disease in Texas: report of a case. Tex. Med. 89:48–50 [PubMed] [Google Scholar]
- 211. Centers for Disease Control and Prevention 2002. Chagas disease after organ transplantation—United States, 2001. MMWR Morb. Mortal. Wkly. Rep. 51:210–212 [PubMed] [Google Scholar]
- 212. Kun H, Moore A, Mascola L, Steurer F, Lawrence G, Kubak B, Radhakrishna S, Leiby D, Herron R, Mone T, Hunter R, Kuehnert M. 2009. Transmission of Trypanosoma cruzi by heart transplantation. Clin. Infect. Dis. 48:1534–1540 [DOI] [PubMed] [Google Scholar]
- 213. Grant IH, Gold JW, Wittner M, Tanowitz HB, Nathan C, Mayer K, Reich L, Wollner N, Steinherz L, Ghavimi F, O'Reilly RJ, Armstrong D. 1989. Transfusion-associated acute Chagas disease acquired in the United States. Ann. Intern. Med. 111:849–851 [DOI] [PubMed] [Google Scholar]
- 214. Leiby DA, Lenes BA, Tibbals MA, Tames-Olmedo MT. 1999. Prospective evaluation of a patient with Trypanosoma cruzi infection transmitted by transfusion. N. Engl. J. Med. 341:1237–1239 [DOI] [PubMed] [Google Scholar]
- 215. Young C, Losikoff P, Chawla A, Glasser L, Forman E. 2007. Transfusion-acquired Trypanosoma cruzi infection. Transfusion 47:540–544 [DOI] [PubMed] [Google Scholar]
- 216. Chagas Biovigilance Network 28 June 2012, posting date Chagas biovigilance: RIPA positive map. AABB, Bethesda, MD: http://www.aabb.org/programs/biovigilance/Pages/chagas.aspx#1 [Google Scholar]
- 217. Davis DS, Russell LH, Adams LG, Yaeger RG, Robinson RM. 1980. An experimental infection of Trypanosoma cruzi in striped skunks (Mephitis mephitis). J. Wildl. Dis. 16:403–406 [DOI] [PubMed] [Google Scholar]
- 218. Roellig DM, Ellis AE, Yabsley MJ. 2009. Oral transmission of Trypanosoma cruzi with opposing evidence for the theory of carnivory. J. Parasitol. 95:360–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Shikanai-Yasuda MA, Carvalho NB. 2012. Oral transmission of Chagas disease. Clin. Infect. Dis. 54:845–852 [DOI] [PubMed] [Google Scholar]
- 220. Pereira KS, Schmidt FL, Guaraldo AM, Franco RM, Dias VL, Passos LA. 2009. Chagas' disease as a foodborne illness. J. Food Prot. 72:441–446 [DOI] [PubMed] [Google Scholar]
- 221. Alarcon de Noya B, Diaz-Bello Z, Colmenares C, Ruiz-Guevara R, Mauriello L, Zavala-Jaspe R, Suarez JA, Abate T, Naranjo L, Paiva M, Rivas L, Castro J, Marques J, Mendoza I, Acquatella H, Torres J, Noya O. 2010. Large urban outbreak of orally acquired acute Chagas disease at a school in Caracas, Venezuela. J. Infect. Dis. 201:1308–1315 [DOI] [PubMed] [Google Scholar]
- 222. Borges JD, Assis GF, Gomes LV, Dias JC, Pinto ID, Martins-Filho OA, Torres RM, Vinas PA, Bahia MT, Machado-Coelho GL, Lana M. 2006. Seroprevalence of Chagas disease in schoolchildren from two municipalities of Jequitinhonha Valley, Minas Gerais, Brazil; six years following the onset of epidemiological surveillance. Rev. Inst. Med. Trop. Sao Paulo 48:81–86 [DOI] [PubMed] [Google Scholar]
- 223. Segura EL, Cura EN, Estani SA, Andrade J, Lansetti JC, de Rissio AM, Campanini A, Blanco SB, Gurtler RE, Alvarez M. 2000. Long-term effects of a nationwide control program on the seropositivity for Trypanosoma cruzi infection in young men from Argentina. Am. J. Trop. Med. Hyg. 62:353–362 [DOI] [PubMed] [Google Scholar]
- 224. Carlier Y, Torrico F, Sosa-Estani S, Russomando G, Luquetti A, Freilij H, Albajar Vinas P. 2011. Congenital Chagas disease: recommendations for diagnosis, treatment and control of newborns, siblings and pregnant women. PLoS Negl. Trop. Dis. 5:e1250 doi:10.1371/journal.pntd.0001250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Magill AJ, Grogl M, Gasser RA, Jr, Sun W, Oster CN. 1993. Visceral infection caused by Leishmania tropica in veterans of Operation Desert Storm. N. Engl. J. Med. 328:1383–1387 [DOI] [PubMed] [Google Scholar]
- 226. Morsy TA, Schnur LF, Feinsod FM, Salem AM, Wahba MM, el Said SM. 1987. Natural infections of Leishmania major in domestic dogs from Alexandria, Egypt. Am. J. Trop. Med. Hyg. 37:49–52 [DOI] [PubMed] [Google Scholar]
- 227. Akhavan AA, Yaghoobi-Ershadi MR, Khamesipour A, Mirhendi H, Alimohammadian MH, Rassi Y, Arandian MH, Jafari R, Abdoli H, Shareghi N, Ghanei M, Jalali-zand N. 2010. Dynamics of Leishmania infection rates in Rhombomys opimus (Rodentia: Gerbillinae) population of an endemic focus of zoonotic cutaneous leishmaniasis in Iran. Bull. Soc. Pathol. Exot. 103:84–89 [DOI] [PubMed] [Google Scholar]
- 228. Azizi K, Moemenbellah-Fard MD, Fakoorziba MR, Fekri S. 2011. Gerbillus nanus (Rodentia: Muridae): a new reservoir host of Leishmania major. Ann. Trop. Med. Parasitol. 105:431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Ghawar W, Toumi A, Snoussi MA, Chlif S, Zaatour A, Boukthir A, Hamida NB, Chemkhi J, Diouani MF, Ben-Salah A. 2011. Leishmania major infection among Psammomys obesus and Meriones shawi: reservoirs of zoonotic cutaneous leishmaniasis in Sidi Bouzid (central Tunisia). Vector Borne Zoonotic Dis. 11:1561–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Toumi A, Chlif S, Bettaieb J, Ben Alaya N, Boukthir A, Ahmadi ZE, Ben Salah A. 2012. Temporal dynamics and impact of climate factors on the incidence of zoonotic cutaneous leishmaniasis in central Tunisia. PLoS Negl. Trop. Dis. 6:e1633 doi:10.1371/journal.pntd.0001633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Wasserberg G, Abramsky Z, Anders G, El-Fari M, Schoenian G, Schnur L, Kotler BP, Kabalo I, Warburg A. 2002. The ecology of cutaneous leishmaniasis in Nizzana, Israel: infection patterns in the reservoir host, and epidemiological implications. Int. J. Parasitol. 32:133–143 [DOI] [PubMed] [Google Scholar]
- 232. Yavar R, Abedin S, Reza AM, Ali OM, Sina R, Mehdi M, Reza YE, Fatemeh M, Babak F. 2011. Phlebotomus papatasi and Meriones libycus as the vector and reservoir host of cutaneous leishmaniasis in Qomrood District, Qom Province, central Iran. Asian Pac. J. Trop. Med. 4:97–100 [DOI] [PubMed] [Google Scholar]
- 233. Elbihari S, Cheema AH, el-Hassan AM. 1987. Leishmania infecting man and wild animals in Saudi Arabia. 4. Canine cutaneous leishmaniasis in the Eastern Province. Trans. R. Soc. Trop. Med. Hyg. 81:925–927 [DOI] [PubMed] [Google Scholar]
- 234. Gebre-Michael T, Balkew M, Ali A, Ludovisi A, Gramiccia M. 2004. The isolation of Leishmania tropica and L. aethiopica from Phlebotomus (Paraphlebotomus) species (Diptera: Psychodidae) in the Awash Valley, northeastern Ethiopia. Trans. R. Soc. Trop. Med. Hyg. 98:64–70 [DOI] [PubMed] [Google Scholar]
- 235. Lemma W, Erenso G, Gadisa E, Balkew M, Gebre-Michael T, Hailu A. 2009. A zoonotic focus of cutaneous leishmaniasis in Addis Ababa, Ethiopia. Parasit. Vectors 2:60 doi:10.1186/1756-3305-2-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Trainor KE, Porter BF, Logan KS, Hoffman RJ, Snowden KF. 2010. Eight cases of feline cutaneous leishmaniasis in Texas. Vet. Pathol. 47:1076–1081 [DOI] [PubMed] [Google Scholar]
- 237. Wright NA, Davis LE, Aftergut KS, Parrish CA, Cockerell CJ. 2008. Cutaneous leishmaniasis in Texas: a northern spread of endemic areas. J. Am. Acad. Dermatol. 58:650–652 [DOI] [PubMed] [Google Scholar]
- 238. Kerr SF, McHugh CP, Dronen NO., Jr 1995. Leishmaniasis in Texas: prevalence and seasonal transmission of Leishmania mexicana in Neotoma micropus. Am. J. Trop. Med. Hyg. 53:73–77 [PubMed] [Google Scholar]
- 239. Van Wynsberghe NR, Canto-Lara SB, Sosa-Bibiano EI, Rivero-Cardenas NA, Andrade-Narvaez FJ. 2009. Comparison of small mammal prevalence of Leishmania (Leishmania) mexicana in five foci of cutaneous leishmaniasis in the State of Campeche, Mexico. Rev. Inst. Med. Trop. Sao Paulo 51:87–94 [DOI] [PubMed] [Google Scholar]
- 240. Arjona-Jimenez G, Villegas N, Lopez-Cespedes A, Marin C, Longoni SS, Bolio-Gonzalez ME, Rodriguez-Vivas RI, Sauri-Arceo CH, Sanchez-Moreno M. 2012. Prevalence of antibodies against three species of Leishmania (L. mexicana, L. braziliensis, L. infantum) and possible associated factors in dogs from Merida, Yucatan, Mexico. Trans. R. Soc. Trop. Med. Hyg. 106:252–258 [DOI] [PubMed] [Google Scholar]
- 241. Longoni SS, Lopez-Cespedes A, Sanchez-Moreno M, Bolio-Gonzalez ME, Sauri-Arceo CH, Rodriguez-Vivas RI, Marin C. 2012. Detection of different Leishmania spp. and Trypanosoma cruzi antibodies in cats from the Yucatan Peninsula (Mexico) using an iron superoxide dismutase excreted as antigen. Comp. Immunol. Microbiol. Infect. Dis. 35:469–476 [DOI] [PubMed] [Google Scholar]
- 242. Telleria J, Bosseno MF, Tarifa T, Buitrago R, Martinez E, Torrez M, Le Pont F, Breniere SF. 1999. Putative reservoirs of Leishmania amazonensis in a sub-Andean focus of Bolivia identified by kDNA-polymerase chain reaction. Mem. Inst. Oswaldo Cruz 94:5–6 [DOI] [PubMed] [Google Scholar]
- 243. Tolezano JE, Uliana SR, Taniguchi HH, Araujo MF, Barbosa JA, Barbosa JE, Floeter-Winter LM, Shaw JJ. 2007. The first records of Leishmania (Leishmania) amazonensis in dogs (Canis familiaris) diagnosed clinically as having canine visceral leishmaniasis from Aracatuba County, Sao Paulo State, Brazil. Vet. Parasitol. 149:280–284 [DOI] [PubMed] [Google Scholar]
- 244. Guessous-Idrissi N, Berrag B, Riyad M, Sahibi H, Bichichi M, Rhalem A. 1997. Leishmania tropica: etiologic agent of a case of canine visceral leishmaniasis in northern Morocco. Am. J. Trop. Med. Hyg. 57:172–173 [DOI] [PubMed] [Google Scholar]
- 245. Dereure J, Rioux JA, Gallego M, Perieres J, Pratlong F, Mahjour J, Saddiki H. 1991. Leishmania tropica in Morocco: infection in dogs. Trans. R. Soc. Trop. Med. Hyg. 85:595. [DOI] [PubMed] [Google Scholar]
- 246. Jacobson RL, Eisenberger CL, Svobodova M, Baneth G, Sztern J, Carvalho J, Nasereddin A, El Fari M, Shalom U, Volf P, Votypka J, Dedet JP, Pratlong F, Schonian G, Schnur LF, Jaffe CL, Warburg A. 2003. Outbreak of cutaneous leishmaniasis in northern Israel. J. Infect. Dis. 188:1065–1073 [DOI] [PubMed] [Google Scholar]
- 247. Talmi-Frank D, Kedem-Vaanunu N, King R, Bar-Gal GK, Edery N, Jaffe CL, Baneth G. 2010. Leishmania tropica infection in golden jackals and red foxes, Israel. Emerg. Infect. Dis. 16:1973–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Mohebali M, Hajjaran H, Hamzavi Y, Mobedi I, Arshi S, Zarei Z, Akhoundi B, Naeini KM, Avizeh R, Fakhar M. 2005. Epidemiological aspects of canine visceral leishmaniosis in the Islamic Republic of Iran. Vet. Parasitol. 129:243–251 [DOI] [PubMed] [Google Scholar]
- 249. Schallig HD, da Silva ES, van der Meide WF, Schoone GJ, Gontijo CM. 2007. Didelphis marsupialis (common opossum): a potential reservoir host for zoonotic leishmaniasis in the metropolitan region of Belo Horizonte (Minas Gerais, Brazil). Vector Borne Zoonotic Dis. 7:387–393 [DOI] [PubMed] [Google Scholar]
- 250. Quaresma PF, Rego FD, Botelho HA, da Silva SR, Moura AJ, Junior, Teixeira Neto RG, Madeira FM, Carvalho MB, Paglia AP, Melo MN, Gontijo CM. 2011. Wild, synanthropic and domestic hosts of Leishmania in an endemic area of cutaneous leishmaniasis in Minas Gerais State, Brazil. Trans. R. Soc. Trop. Med. Hyg. 105:579–585 [DOI] [PubMed] [Google Scholar]
- 251. Aguilar CM, Rangel EF, Garcia L, Fernandez E, Momen H, Grimaldi Filho G, De Vargas Z. 1989. Zoonotic cutaneous leishmaniasis due to Leishmania (Viannia) braziliensis associated with domestic animals in Venezuela and Brazil. Mem. Inst. Oswaldo Cruz 84:19–28 [DOI] [PubMed] [Google Scholar]
- 252. Dantas-Torres F. 2007. The role of dogs as reservoirs of Leishmania parasites, with emphasis on Leishmania (Leishmania) infantum and Leishmania (Viannia) braziliensis. Vet. Parasitol. 149:139–146 [DOI] [PubMed] [Google Scholar]
- 253. Christensen HA, Arias JR, de Vasquez AM, de Freitas RA. 1982. Hosts of sandfly vectors of Leishmania braziliensis guyanensis in the central Amazon of Brazil. Am. J. Trop. Med. Hyg. 31:239–242 [DOI] [PubMed] [Google Scholar]
- 254. Santaella J, Ocampo CB, Saravia NG, Mendez F, Gongora R, Gomez MA, Munstermann LE, Quinnell RJ. 2011. Leishmania (Viannia) infection in the domestic dog in Chaparral, Colombia. Am. J. Trop. Med. Hyg. 84:674–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Llanos-Cuentas EA, Roncal N, Villaseca P, Paz L, Ogusuku E, Perez JE, Caceres A, Davies CR. 1999. Natural infections of Leishmania peruviana in animals in the Peruvian Andes. Trans. R. Soc. Trop. Med. Hyg. 93:15–20 [DOI] [PubMed] [Google Scholar]
- 256. Lainson R, Braga RR, De Souza AA, Povoa MM, Ishikawa EA, Silveira FT. 1989. Leishmania (Viannia) shawi sp. n., a parasite of monkeys, sloths and procyonids in Amazonian Brazil. Ann. Parasitol. Hum. Comp. 64:200–207 [DOI] [PubMed] [Google Scholar]
- 257. Lainson R, Shaw JJ, Souza AA, Silveira FT, Falqueto A. 1992. Further observations on Lutzomyia ubiquitalis (Psychodidae: Phlebotominae), the sandfly vector of Leishmania (Viannia) lainsoni. Mem. Inst. Oswaldo Cruz 87:437–439 [DOI] [PubMed] [Google Scholar]
- 258. Lainson R, Shaw JJ. 1989. Leishmania (Viannia) naiffi sp. n., a parasite of the armadillo, Dasypus novemcinctus (L.) in Amazonian Brazil. Ann. Parasitol. Hum. Comp. 64:3–9 [DOI] [PubMed] [Google Scholar]
- 259. Bonfante-Garrido R, Urdaneta I, Urdaneta R, Alvarado J. 1991. Natural infection of cats with Leishmania in Barquisimeto, Venezuela. Trans. R. Soc. Trop. Med. Hyg. 85:53. [DOI] [PubMed] [Google Scholar]
- 260. Dereure J, Espinel I, Barrera C, Guerrini F, Martini A, Echeverria R, Guderian RH, Le Pont F. 1994. Leishmaniasis in Ecuador. 4. Natural infestation of the dog by Leishmania panamensis. Ann. Soc. Belg. Med. Trop. 74:29–33 (In French.) [PubMed] [Google Scholar]
- 261. Herrer A, Christensen HA. 1980. Leishmania braziliensis in the Panamanian two-toed sloth, Choloepus hoffmanni. Am. J. Trop. Med. Hyg. 29:1196–1200 [DOI] [PubMed] [Google Scholar]
- 262. Thatcher VE, Eisenmann C, Hertig M. 1965. A natural infection of Leishmania in the kinkajou, Potos flavus, in Panama. J. Parasitol. 51:1022–1023 [PubMed] [Google Scholar]
- 263. Velez ID, Carrillo LM, Lopez L, Rodriguez E, Robledo SM. 2012. An epidemic outbreak of canine cutaneous leishmaniasis in Colombia caused by Leishmania braziliensis and Leishmania panamensis. Am. J. Trop. Med. Hyg. 86:807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Herrer A, Christensen HA. 1976. Natural cutaneous leishmaniasis among dogs in Panama. Am. J. Trop. Med. Hyg. 25:59–63 [DOI] [PubMed] [Google Scholar]
- 265. Bhattarai NR, Van der Auwera G, Rijal S, Picado A, Speybroeck N, Khanal B, De Doncker S, Das ML, Ostyn B, Davies C, Coosemans M, Berkvens D, Boelaert M, Dujardin JC. 2010. Domestic animals and epidemiology of visceral leishmaniasis, Nepal. Emerg. Infect. Dis. 16:231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Dereure J, El-Safi SH, Bucheton B, Boni M, Kheir MM, Davoust B, Pratlong F, Feugier E, Lambert M, Dessein A, Dedet JP. 2003. Visceral leishmaniasis in eastern Sudan: parasite identification in humans and dogs; host-parasite relationships. Microbes Infect. 5:1103–1108 [DOI] [PubMed] [Google Scholar]
- 267. Kalayou S, Tadelle H, Bsrat A, Abebe N, Haileselassie M, Schallig HD. 2011. Serological evidence of Leishmania donovani infection in apparently healthy dogs using direct agglutination test (DAT) and rk39 dipstick tests in Kafta Humera, north-west Ethiopia. Transbound. Emerg. Dis. 58:255–262 [DOI] [PubMed] [Google Scholar]
- 268. Sharma NL, Mahajan VK, Negi AK, Verma GK. 2009. The rK39 immunochromatic dipstick testing: a study for K39 seroprevalence in dogs and human leishmaniasis patients for possible animal reservoir of cutaneous and visceral leishmaniasis in endemic focus of Satluj river valley of Himachal Pradesh (India). Indian J. Dermatol. Venereol. Leprol. 75:52–55 [DOI] [PubMed] [Google Scholar]
- 269. Gramiccia M, Gradoni L. 2005. The current status of zoonotic leishmaniases and approaches to disease control. Int. J. Parasitol. 35:1169–1180 [DOI] [PubMed] [Google Scholar]
- 270. Vides JP, Schwardt TF, Sobrinho LS, Marinho M, Laurenti MD, Biondo AW, Leutenegger C, Marcondes M. 2011. Leishmania chagasi infection in cats with dermatologic lesions from an endemic area of visceral leishmaniosis in Brazil. Vet. Parasitol. 178:22–28 [DOI] [PubMed] [Google Scholar]
- 271. Lima ID, Queiroz JW, Lacerda HG, Queiroz PV, Pontes NN, Barbosa JD, Martins DR, Weirather JL, Pearson RD, Wilson ME, Jeronimo SM. 2012. Leishmania infantum chagasi in northeastern Brazil: asymptomatic infection at the urban perimeter. Am. J. Trop. Med. Hyg. 86:99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Coura-Vital W, Marques MJ, Veloso VM, Roatt BM, Aguiar-Soares RD, Reis LE, Braga SL, Morais MH, Reis AB, Carneiro M. 2011. Prevalence and factors associated with Leishmania infantum infection of dogs from an urban area of Brazil as identified by molecular methods. PLoS Negl. Trop. Dis. 5:e1291 doi:10.1371/journal.pntd.0001291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Falqueto A, Ferreira AL, dos Santos CB, Porrozzi R, da Costa MV, Teva A, Cupolillo E, Campos-Neto A, Grimaldi G., Jr 2009. Cross-sectional and longitudinal epidemiologic surveys of human and canine Leishmania infantum visceral infections in an endemic rural area of southeast Brazil (Pancas, Espirito Santo). Am. J. Trop. Med. Hyg. 80:559–565 [PubMed] [Google Scholar]
- 274. Martin-Sanchez J, Morales-Yuste M, Acedo-Sanchez C, Baron S, Diaz V, Morillas-Marquez F. 2009. Canine leishmaniasis in southeastern Spain. Emerg. Infect. Dis. 15:795–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Cortes S, Vaz Y, Neves R, Maia C, Cardoso L, Campino L. 2012. Risk factors for canine leishmaniasis in an endemic Mediterranean region. Vet. Parasitol. 189:189–196 [DOI] [PubMed] [Google Scholar]
- 276. Tabar MD, Altet L, Francino O, Sanchez A, Ferrer L, Roura X. 2008. Vector-borne infections in cats: molecular study in Barcelona area (Spain). Vet. Parasitol. 151:332–336 [DOI] [PubMed] [Google Scholar]
- 277. Molina R, Jimenez MI, Cruz I, Iriso A, Martin-Martin I, Sevillano O, Melero S, Bernal J. 2012. The hare (Lepus granatensis) as potential sylvatic reservoir of Leishmania infantum in Spain. Vet. Parasitol. 190:268–271 [DOI] [PubMed] [Google Scholar]
- 278. Tenorio MS, Oliveira e Sousa L, Paixao MS, Alves MF, Paulan SC, Lima FL, Jusi MM, Tasca KI, Machado RZ, Starke-Buzetti WA. 2011. Visceral leishmaniasis in a captive crab-eating fox Cerdocyon thous. J. Zoo Wildl. Med. 42:608–616 [DOI] [PubMed] [Google Scholar]
- 279. Jusi MM, Starke-Buzetti WA, Oliveira TM, Tenorio MS, Sousa LO, Machado RZ. 2011. Molecular and serological detection of Leishmania spp. in captive wild animals from Ilha Solteira, SP, Brazil. Rev. Bras. Parasitol. Vet. 20:219–222 [DOI] [PubMed] [Google Scholar]
- 280. Tzamouranis N, Schnur LF, Garifallou A, Pateraki E, Serie C. 1984. Leishmaniasis in Greece. I. Isolation and identification of the parasite causing human and canine visceral leishmaniasis. Ann. Trop. Med. Parasitol. 78:363–368 [DOI] [PubMed] [Google Scholar]
- 281. Maia C, Gomes J, Cristovao J, Nunes M, Martins A, Rebelo E, Campino L. 2010. Feline Leishmania infection in a canine leishmaniasis endemic region, Portugal. Vet. Parasitol. 174:336–340 [DOI] [PubMed] [Google Scholar]
- 282. Cardoso L, Lopes AP, Sherry K, Schallig H, Solano-Gallego L. 2010. Low seroprevalence of Leishmania infantum infection in cats from northern Portugal based on DAT and ELISA. Vet. Parasitol. 174:37–42 [DOI] [PubMed] [Google Scholar]
- 283. Sherry K, Miro G, Trotta M, Miranda C, Montoya A, Espinosa C, Ribas F, Furlanello T, Solano-Gallego L. 2011. A serological and molecular study of Leishmania infantum infection in cats from the Island of Ibiza (Spain). Vector Borne Zoonotic Dis. 11:239–245 [DOI] [PubMed] [Google Scholar]
- 284. Hatam GR, Adnani SJ, Asgari Q, Fallah E, Motazedian MH, Sadjjadi SM, Sarkari B. 2010. First report of natural infection in cats with Leishmania infantum in Iran. Vector Borne Zoonotic Dis. 10:313–316 [DOI] [PubMed] [Google Scholar]
- 285. Nasereddin A, Salant H, Abdeen Z. 2008. Feline leishmaniasis in Jerusalem: serological investigation. Vet. Parasitol. 158:364–369 [DOI] [PubMed] [Google Scholar]
- 286. Coelho WM, do Amarante AF, Apolinario JC, Coelho NM, de Lima VM, Perri SH, Bresciani KD. 2011. Seroepidemiology of Toxoplasma gondii, Neospora caninum, and Leishmania spp. infections and risk factors for cats from Brazil. Parasitol. Res. 109:1009–1013 [DOI] [PubMed] [Google Scholar]
- 287. Cardoso L, Mendao C, Madeira de Carvalho L. 2012. Prevalence of Dirofilaria immitis, Ehrlichia canis, Borrelia burgdorferi sensu lato, Anaplasma spp. and Leishmania infantum in apparently healthy and CVBD-suspect dogs in Portugal—a national serological study. Parasit. Vectors 5:62 doi:10.1186/1756-3305-5-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Athanasiou LV, Kontos VI, Saridomichelakis MN, Rallis TS, Diakou A. 2012. A cross-sectional sero-epidemiological study of canine leishmaniasis in Greek mainland. Acta Trop. 122:291–295 [DOI] [PubMed] [Google Scholar]
- 289. Kovalenko DA, Nasyrova RM, Ponomareva VI, Fatullaeva AA, Razakov SA, Ponirovskii EN, Strelkova MV, Zhirenkina EN, Morozov EN, Dzhaf C, Banet G, Shnur L, Varburg A, Shonian G. 2011. Human and canine visceral leishmaniasis in the Papsky District, Namangan Region, Uzbekistan: seroepidemiological and seroepizootological surveys. Med. Parazitol. (Mosk.) 2011:32–37 (In Russian.) [PubMed] [Google Scholar]
- 290. Shang LM, Peng WP, Jin HT, Xu D, Zhong NN, Wang WL, Wu YX, Liu Q. 2011. The prevalence of canine Leishmania infantum infection in Sichuan Province, southwestern China detected by real time PCR. Parasit. Vectors 4:173 doi:10.1186/1756-3305-4-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Sousa S, Lopes AP, Cardoso L, Silvestre R, Schallig H, Reed SG, Cordeiro da Silva A. 2011. Seroepidemiological survey of Leishmania infantum infection in dogs from northeastern Portugal. Acta Trop. 120:82–87 [DOI] [PubMed] [Google Scholar]
- 292. Faye B, Banuls AL, Bucheton B, Dione MM, Bassanganam O, Hide M, Dereure J, Choisy M, Ndiaye JL, Konate O, Claire M, Senghor MW, Faye MN, Sy I, Niang AA, Molez JF, Victoir K, Marty P, Delaunay P, Knecht R, Mellul S, Diedhiou S, Gaye O. 2010. Canine visceral leishmaniasis caused by Leishmania infantum in Senegal: risk of emergence in humans? Microbes Infect. 12:1219–1225 [DOI] [PubMed] [Google Scholar]
- 293. Baldelli R, Battelli G, Maroli M, Mollicone E, Gudi A, Stegagno G, Tasini G. 2001. A new stable focus of canine leishmaniasis in northern Italy. Parassitologia 43:151–153 [PubMed] [Google Scholar]
- 294. Ozensoy Toz S, Sakru N, Ertabaklar H, Demir S, Sengul M, Ozbel Y. 2009. Serological and entomological survey of zoonotic visceral leishmaniasis in Denizli Province, Aegean Region, Turkey. New Microbiol. 32:93–100 [PubMed] [Google Scholar]
- 295. Lemesre JL, Holzmuller P, Goncalves RB, Bourdoiseau G, Hugnet C, Cavaleyra M, Papierok G. 2007. Long-lasting protection against canine visceral leishmaniasis using the LiESAp-MDP vaccine in endemic areas of France: double-blind randomised efficacy field trial. Vaccine 25:4223–4234 [DOI] [PubMed] [Google Scholar]
- 296. Ives A, Ronet C, Prevel F, Ruzzante G, Fuertes-Marraco S, Schutz F, Zangger H, Revaz-Breton M, Lye LF, Hickerson SM, Beverley SM, Acha-Orbea H, Launois P, Fasel N, Masina S. 2011. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 331:775–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. 2008. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321:970–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Laufs H, Muller K, Fleischer J, Reiling N, Jahnke N, Jensenius JC, Solbach W, Laskay T. 2002. Intracellular survival of Leishmania major in neutrophil granulocytes after uptake in the absence of heat-labile serum factors. Infect. Immun. 70:826–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Boggiatto PM, Jie F, Ghosh M, Gibson-Corley KN, Ramer-Tait AE, Jones DE, Petersen CA. 2009. Altered dendritic cell phenotype in response to Leishmania amazonensis amastigote infection is mediated by MAP kinase, ERK. Am. J. Pathol. 174:1818–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Sibley LD. 2011. Invasion and intracellular survival by protozoan parasites. Immunol. Rev. 240:72–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Ueno N, Bratt CL, Rodriguez NE, Wilson ME. 2009. Differences in human macrophage receptor usage, lysosomal fusion kinetics and survival between logarithmic and metacyclic Leishmania infantum chagasi promastigotes. Cell. Microbiol. 11:1827–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Lodge R, Descoteaux A. 2008. Leishmania invasion and phagosome biogenesis. Subcell. Biochem. 47:174–181 [DOI] [PubMed] [Google Scholar]
- 303. Sacks DL. 2001. Leishmania-sand fly interactions controlling species-specific vector competence. Cell. Microbiol. 3:189–196 [DOI] [PubMed] [Google Scholar]
- 304. Desjardins M, Descoteaux A. 1997. Inhibition of phagolysosomal biogenesis by the Leishmania lipophosphoglycan. J. Exp. Med. 185:2061–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Huynh C, Sacks DL, Andrews NW. 2006. A Leishmania amazonensis ZIP family iron transporter is essential for parasite replication within macrophage phagolysosomes. J. Exp. Med. 203:2363–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Lambertz U, Silverman JM, Nandan D, McMaster WR, Clos J, Foster LJ, Reiner NE. 2012. Secreted virulence factors and immune evasion in visceral leishmaniasis. J. Leukoc. Biol. 91:887–899 [DOI] [PubMed] [Google Scholar]
- 307. Olivier M, Gregory DJ, Forget G. 2005. Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin. Microbiol. Rev. 18:293–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. WHO 2012. Leishmaniasis: epidemiology and access to medicines. WHO, Geneva, Switzerland [Google Scholar]
- 309. Gonzalez C, Wang O, Strutz SE, Gonzalez-Salazar C, Sanchez-Cordero V, Sarkar S. 2010. Climate change and risk of leishmaniasis in North America: predictions from ecological niche models of vector and reservoir species. PLoS Negl. Trop. Dis. 4:e585 doi:10.1371/journal.pntd.0000585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Faulde M, Schrader J, Heyl G, Amirih M, Hoerauf A. 2008. Zoonotic cutaneous leishmaniasis outbreak in Mazar-e Sharif, northern Afghanistan: an epidemiological evaluation. Int. J. Med. Microbiol. 298:543–550 [DOI] [PubMed] [Google Scholar]
- 311. Barnes JC, Stanley O, Craig TM. 1993. Diffuse cutaneous leishmaniasis in a cat. J. Am. Vet. Med. Assoc. 202:416–418 [PubMed] [Google Scholar]
- 312. Sellon RK, Menard MM, Meuten DJ, Lengerich EJ, Steurer FJ, Breitschwerdt EB. 1993. Endemic visceral leishmaniasis in a dog from Texas. J. Vet. Intern. Med. 7:16–19 [DOI] [PubMed] [Google Scholar]
- 313. Craig TM, Barton CL, Mercer SH, Droleskey BE, Jones LP. 1986. Dermal leishmaniasis in a Texas cat. Am. J. Trop. Med. Hyg. 35:1100–1102 [DOI] [PubMed] [Google Scholar]
- 314. Barrett TV, Senra MS. 1989. Leishmaniasis in Manaus, Brazil. Parasitol. Today 5:255–257 [DOI] [PubMed] [Google Scholar]
- 315. Desjeux P. 2001. Worldwide increasing risk factors for leishmaniasis. Med. Microbiol. Immunol. 190:77–79 [DOI] [PubMed] [Google Scholar]
- 316. Jirmanus L, Glesby MJ, Guimaraes LH, Lago E, Rosa ME, Machado PR, Carvalho EM. 2012. Epidemiological and clinical changes in American tegumentary leishmaniasis in an area of Leishmania (Viannia) braziliensis transmission over a 20-year period. Am. J. Trop. Med. Hyg. 86:426–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Votypka J, Kasap OE, Volf P, Kodym P, Alten B. 2012. Risk factors for cutaneous leishmaniasis in Cukurova region, Turkey. Trans. R. Soc. Trop. Med. Hyg. 106:186–190 [DOI] [PubMed] [Google Scholar]
- 318. Morrone A, Pitidis A, Pajno MC, Dassoni F, Latini O, Barnabas GA, Padovese V. 2011. Epidemiological and geographical aspects of leishmaniasis in Tigray, northern Ethiopia: a retrospective analysis of medical records, 2005-2008. Trans. R. Soc. Trop. Med. Hyg. 105:273–280 [DOI] [PubMed] [Google Scholar]
- 319. Reithinger R, Mohsen M, Leslie T. 2010. Risk factors for anthroponotic cutaneous leishmaniasis at the household level in Kabul, Afghanistan. PLoS Negl. Trop. Dis. 4:e639 doi:10.1371/journal.pntd.0000639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Pedrosa FA, Ximenes RA. 2009. Sociodemographic and environmental risk factors for American cutaneous leishmaniasis (ACL) in the State of Alagoas, Brazil. Am. J. Trop. Med. Hyg. 81:195–201 [PubMed] [Google Scholar]
- 321. Dahroug MA, Almeida AB, Sousa VR, Dutra V, Turbino NC, Nakazato L, de Souza RL. 2010. Leishmania (Leishmania) chagasi in captive wild felids in Brazil. Trans. R. Soc. Trop. Med. Hyg. 104:73–74 [DOI] [PubMed] [Google Scholar]
- 322. Michel G, Pomares C, Ferrua B, Marty P. 2011. Importance of worldwide asymptomatic carriers of Leishmania infantum (L. chagasi) in human. Acta Trop. 119:69–75 [DOI] [PubMed] [Google Scholar]
- 323. Boggiatto PM, Gibson-Corley KN, Metz K, Gallup JM, Hostetter JM, Mullin K, Petersen CA. 2011. Transplacental transmission of Leishmania infantum as a means for continued disease incidence in North America. PLoS Negl. Trop. Dis. 5:e1019 doi:10.1371/journal.pntd.0001019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Dujardin JC, Campino L, Canavate C, Dedet JP, Gradoni L, Soteriadou K, Mazeris A, Ozbel Y, Boelaert M. 2008. Spread of vector-borne diseases and neglect of leishmaniasis, Europe. Emerg. Infect. Dis. 14:1013–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Galvez R, Miro G, Descalzo MA, Nieto J, Dado D, Martin O, Cubero E, Molina R. 2010. Emerging trends in the seroprevalence of canine leishmaniasis in the Madrid region (central Spain). Vet. Parasitol. 169:327–334 [DOI] [PubMed] [Google Scholar]
- 326. Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M. 2012. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7:e35671 doi:10.1371/journal.pone.0035671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. de Almeida AS, Medronho RA, Werneck GL. 2011. Identification of risk areas for visceral leishmaniasis in Teresina, Piaui State, Brazil. Am. J. Trop. Med. Hyg. 84:681–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Harhay MO, Olliaro PL, Vaillant M, Chappuis F, Lima MA, Ritmeijer K, Costa CH, Costa DL, Rijal S, Sundar S, Balasegaram M. 2011. Who is a typical patient with visceral leishmaniasis? Characterizing the demographic and nutritional profile of patients in Brazil, East Africa, and South Asia. Am. J. Trop. Med. Hyg. 84:543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Esch KJ, Pontes NN, Arruda P, O'Connor A, Morais L, Jeronimo SM, Petersen CA. 2012. Preventing zoonotic canine leishmaniasis in northeastern Brazil: pet attachment and adoption of community leishmania prevention. Am. J. Trop. Med. Hyg. 87:822–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Pagliano P, Carannante N, Rossi M, Gramiccia M, Gradoni L, Faella FS, Gaeta GB. 2005. Visceral leishmaniasis in pregnancy: a case series and a systematic review of the literature. J. Antimicrob. Chemother. 55:229–233 [DOI] [PubMed] [Google Scholar]
- 331. Nascimento ET, Moura ML, Queiroz JW, Barroso AW, Araujo AF, Rego EF, Wilson ME, Pearson RD, Jeronimo SM. 2011. The emergence of concurrent HIV-1/AIDS and visceral leishmaniasis in Northeast Brazil. Trans. R. Soc. Trop. Med. Hyg. 105:298–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Cortese L, Terrazzano G, Piantedosi D, Sica M, Prisco M, Ruggiero G, Ciaramella P. 2011. Prevalence of anti-platelet antibodies in dogs naturally co-infected by Leishmania infantum and Ehrlichia canis. Vet. J. 188:118–121 [DOI] [PubMed] [Google Scholar]
- 333. Aresu L, Valenza F, Ferroglio E, Pregel P, Uslenghi F, Tarducci A, Zanatta R. 2007. Membranoproliferative glomerulonephritis type III in a simultaneous infection of Leishmania infantum and Dirofilaria immitis in a dog. J. Vet. Diagn. Invest. 19:569–572 [DOI] [PubMed] [Google Scholar]
- 334. Blackwell JM, Fakiola M, Ibrahim ME, Jamieson SE, Jeronimo SB, Miller EN, Mishra A, Mohamed HS, Peacock CS, Raju M, Sundar S, Wilson ME. 2009. Genetics and visceral leishmaniasis: of mice and man. Parasite Immunol. 31:254–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Soto J, Medina F, Dember N, Berman J. 1995. Efficacy of permethrin-impregnated uniforms in the prevention of malaria and leishmaniasis in Colombian soldiers. Clin. Infect. Dis. 21:599–602 [DOI] [PubMed] [Google Scholar]
- 336. Rojas CA, Weigle KA, Tovar R, Morales AL, Alexander B. 2006. A multifaceted intervention to prevent American cutaneous leishmaniasis in Colombia: results of a group-randomized trial. Biomedica 26(Suppl 1):152–166 [PubMed] [Google Scholar]
- 337. Mutinga MJ, Renapurkar DM, Wachira DW, Basimike M, Mutero CM. 1993. A bioassay to evaluate the efficacy of permethrin-impregnated screens used against phlebotomine sandflies (Diptera: Psychodidae) in Baringo District of Kenya. East Afr. Med. J. 70:168–170 [PubMed] [Google Scholar]
- 338. Tesh RB. 1995. Control of zoonotic visceral leishmaniasis: is it time to change strategies? Am. J. Trop. Med. Hyg. 52:287–292 [DOI] [PubMed] [Google Scholar]
- 339. Costa CH. 2011. How effective is dog culling in controlling zoonotic visceral leishmaniasis? A critical evaluation of the science, politics and ethics behind this public health policy. Rev. Soc. Bras. Med. Trop. 44:232–242 [DOI] [PubMed] [Google Scholar]
- 340. Harhay MO, Olliaro PL, Costa DL, Costa CH. 2011. Urban parasitology: visceral leishmaniasis in Brazil. Trends Parasitol. 27:403–409 [DOI] [PubMed] [Google Scholar]
- 341. Courtenay O, Kovacic V, Gomes PA, Garcez LM, Quinnell RJ. 2009. A long-lasting topical deltamethrin treatment to protect dogs against visceral leishmaniasis. Med. Vet. Entomol. 23:245–256 [DOI] [PubMed] [Google Scholar]
- 342. Quinnell RJ, Courtenay O. 2009. Transmission, reservoir hosts and control of zoonotic visceral leishmaniasis. Parasitology 136:1915–1934 [DOI] [PubMed] [Google Scholar]
- 343. Saunders LZ. 2000. Virchow's contributions to veterinary medicine: celebrated then, forgotten now. Vet. Pathol. 37:199–207 [DOI] [PubMed] [Google Scholar]
- 344. WHO 2011. Managing zoonotic public health risks at the human-animal-ecosystem interface. World Health Organization, Geneva, Switzerland [Google Scholar]
- 345. Allen K, Blascovich J, Mendes WB. 2002. Cardiovascular reactivity and the presence of pets, friends, and spouses: the truth about cats and dogs. Psychosom. Med. 64:727–739 [DOI] [PubMed] [Google Scholar]
- 346. Arhant-Sudhir K, Arhant-Sudhir R, Sudhir K. 2011. Pet ownership and cardiovascular risk reduction: supporting evidence, conflicting data and underlying mechanisms. Clin. Exp. Pharmacol. Physiol. 38:734–738 [DOI] [PubMed] [Google Scholar]
- 347. Bergroth E, Remes S, Pekkanen J, Kauppila T, Buchele G, Keski-Nisula L. 2012. Respiratory tract illnesses during the first year of life: effect of dog and cat contacts. Pediatrics 130:211–220 [DOI] [PubMed] [Google Scholar]
- 348. Gerseler PJ, Ito JI, Tegtmeier BR, Kerndt PR, Krance R. 1988. Fulminant Chagas disease in bone marrow transplantation, abstr 418. Abstr. 27th Intersci. Conf. Antimicrob. Agents Chemother., Washington, DC [Google Scholar]
- 349. Kirchoff LV. 2011. American trypanosomiasis (Chagas disease), chapter 99. In Guerrant RL, Walker DH, Weller PF. (ed), Tropical infectious diseases: principles, pathogens, and practice. Elsevier, Amsterdam, Netherlands [Google Scholar]
- 350. Jeronimo SMB, Sousa AQ, Pearson RD. 2011. Leishmaniasis, chapter 100. In Guerrant RL, Walker DH, Weller PF. (ed), Tropical infectious diseases: principles, pathogens, and practice. Elsevier, Amsterdam, Netherlands [Google Scholar]