

Abstract
In pathogenic simian immunodeficiency virus (SIV) and human immunodeficiency virus (HIV) infections, the translocation of microbial products from the gastrointestinal (GI) tract to portal and systemic circulation has been proposed as a major driver of the chronic immune activation that is associated with disease progression. Consistently, microbial translocation is not present in nonpathogenic SIV infections of natural host species. In vivo studies demonstrated that HIV/SIV-associated microbial translocation results from a series of immunopathological events occurring at the GI mucosa: (i) early and severe mucosal CD4+ depletion, (ii) mucosal immune hyperactivation/persistent inflammation; (iii) damage to the integrity of the intestinal epithelium with enterocyte apoptosis and tight junction disruption; and (iv) subverted the gut microbiome, with a predominance of opportunistic bacteria. Direct in situ evidence of microbial translocation has been provided for SIV-infected rhesus macaques showing translocated microbial products in the intestinal lamina propria and distant sites. While the mechanisms by which microbial translocation causes immune activation remain controversial, a key pathogenic event appears to be innate immunity activation via Toll-like receptors and other pathogen recognition receptors. Accumulating clinical observations suggest that microbial translocation might affect HIV disease progression, response to therapy, and non-AIDS comorbidities. Given its detrimental effect on overall immunity, several interventions to prevent/block microbial translocation are currently under investigation as novel therapeutic agents for HIV/AIDS.
INTRODUCTION
The distinctive feature of untreated human immunodeficiency virus (HIV) infection is the progressive depletion of CD4+ T lymphocytes from blood, lymphoid organs, and mucosal tissues. While HIV infects and kills CD4+ T lymphocytes, virus cytopathicity alone is unlikely to provide a satisfying explanation for the course of the disease, which seems to result from complex virus-host interactions ultimately leading to chronic immune system activation and a substantial disruption of T-cell homeostasis (1). Studies conducted using both simian immunodeficiency virus (SIV)-infected macaques and HIV-infected individuals have clearly demonstrated that this immune dysfunction arises within a few weeks from virus transmission (acute infection), when viral dissemination at mucosal tissues results in the massive depletion of mucosal CCR5+ CD4+ T cells from direct and indirect virus cytopathicity (2, 3) as well as CD8+ T-cell-mediated killing of infected CD4+ T cells (4). CD4+ T-cell depletion is particularly rapid within the gastrointestinal (GI) tract, where a significant fraction of T lymphocytes reside, and is relatively slower in peripheral blood.
During the transition from acute to chronic infection, HIV replication is reduced but not fully suppressed by the host adaptive immune response, and during the long chronic phase of the infection, the CD4+ T-cell loss continues to progress. Importantly, the tempo of HIV- and SIV-associated CD4+ T-cell depletion is related to both the set-point level of viremia and the magnitude of virus-associated chronic immune activation (1, 5). HIV/SIV-associated chronic immune activation is characterized by polyclonal B-cell activation (6), high T-cell turnover of both CD4+ and CD8+ T cells with an enhanced cell surface expression of molecules indicative of cell activation (7), and high levels of circulating proinflammatory cytokines and chemokines (8).
HIV/SIV-associated chronic immune activation is not just a consequence of continuous viral replication but rather represents a complex and multifactorial phenomenon that ultimately plays a major role in disrupting T-lymphocyte homeostasis, affecting normal immune function, and even compromising the response to antiviral therapy (reviewed in reference9). In addition, newly generated activated T cells are themselves targets of HIV infection, in turn sustaining viral replication (10) and therefore creating a self-perpetuating vicious circle between HIV replication and immune activation. For these reasons, it is not surprising that the magnitude of this chronic immune activation has been long identified as a major determinant of disease progression independent of the level of virus replication (5, 11, 12). Of note, antiretroviral therapy (ART), which results in the complete suppression of HIV replication, is not sufficient to fully turn off immune activation, and indeed, HIV-infected individuals with poor CD4+ recovery on virologically suppressive ART often exhibit higher levels of immune activation (13, 14). In support of an independent pathogenic role of chronic immune activation is the classical observation that nonpathogenic SIV infection in natural nonhuman primates such as sooty mangabeys and African green monkeys is associated with high levels of viral replication and low levels of immune activation (15).
The mechanisms causing HIV/SIV-associated immune activation remain incompletely understood, and it is possible that specific factors contribute differently to this phenotype in different subsets of patients. The mechanisms that have been proposed to cause HIV-associated immune activation include (i) the direct effect of specific virus gene products (Env, Nef, and Tat, etc.), (ii) the innate and adaptive immune responses to the virus, (iii) an ineffective regulation of normally generated antiviral immune responses, (iv) bystander activation of T and B lymphocytes caused by an increased level of production of proinflammatory cytokines (e.g., tumor necrosis factor alpha [TNF-α], interleukin-1 [IL-1], IL-6, and several others), (v) the presence of clinical or subclinical coinfections, and, more recently, (vi) the preferential infection of central memory CD4+ T cells (as opposed to effector memory CD4+ T cells) as a factor responsible for concentrating the bulk of the antigenic load in central lymphoid tissues (16, 17). In this rather complex immunopathogenic context, the translocation of microbial products from the intestinal lumen to the systemic circulation appears to be a central factor that determines the severity of HIV/SIV-associated chronic immune activation.
The specific purpose of this article is to comprehensively review the literature on the pathogenic role of microbial translocation in the setting of SIV and HIV disease, its possible causes, and the consequences in terms of the loss of immune function, HIV disease progression, and response to therapy. An understanding of the mechanisms responsible for HIV-associated immune activation and microbial translocation will set the basis for the development of therapeutic strategies to prevent or reduce this phenomenon and thus improve the prognosis of HIV infection.
DEFINITION OF MICROBIAL TRANSLOCATION
Gut “translocation” of bacteria or other microbes is defined as the nonphysiological passage of the gastrointestinal microflora through the intestinal epithelial barrier and the lamina propria and eventually to local mesenteric lymph nodes and, from there, to extranodal sites (18–20). Under normal circumstances, translocating microbes and microbial products are phagocytosed within the lamina propria and the mesenteric lymph nodes (21). However, if the host mucosal immune system is compromised, these defense mechanisms may fail, thus permitting the egress and survival of bacteria at distant, extraintestinal sites (21–23).
Lipopolysaccharide (LPS), a component of Gram-negative bacterial cell walls and a known agonist of Toll-like receptor 4 (TLR-4) (24), is considered a major marker of microbial translocation (25–27). In addition to local defense against microbial translocation at the level of the GI mucosa and within the liver, several lines of protection are active in the systemic circulation to neutralize translocating LPS and its downstream effects as a potent immune-activating molecule, thus limiting the detrimental effects of microbial translocation. These protective factors include IgM, IgG, and IgA specific for the LPS core antigen and endotoxin core antibodies (EndoCAb), and indeed, clinical conditions featuring an acute excess of circulating endotoxin result in the consumption of EndoCAb (e.g., sepsis) as they bind and neutralize LPS (28). In other clinical settings (e.g., inflammatory bowel disease [IBD]), ongoing chronic microbial translocation results in elevated LPS levels that are associated with high EndoCAb titers. Importantly, LPS induce several responses in the innate immune system, with the interaction of LPS with LPS binding protein (LBP), which catalytically transfers LPS onto membrane or soluble CD14 (sCD14), leading to NF-κB activation and cytokine production (29), thus determining a state of aberrant immune activation.
Microbial translocation has been described for various diseases, with immunocytochemical evidence for a role of bacteria such as Listeria, Escherichia coli, and Streptococcus in the pathogenesis of Crohn's disease (30). Similarly, in the mouse model of graft-versus-host disease following stem cell transplantation, microbial translocation negatively affects the expression of intestinal α-defensins, leading to the outgrowth of E. coli, which causes septicemia (31). High levels of endotoxemia have also been described for subjects who have undergone laparotomic cholecystectomy, suggesting that intestinal manipulation may impair gut mucosal barrier function (27). The idea that microbial translocation is a key mechanism of mucosal immune dysfunction, chronic systemic immune activation, and, ultimately, disease progression during HIV infection was originally proposed by Brenchley et al. in a landmark hypothesis article (32). Several months later, those authors reported significantly increased levels of circulating LPS in both chronically HIV-infected individuals and SIV-infected rhesus macaques (RM) that positively correlated with measures of innate and adaptive immune activation (33). After that initial report, numerous studies have confirmed that HIV infection is associated with a substantial loss of CD4+ T cells residing in the gut, in turn leading to microbial translocation (34, 35).
Translocating bacteria and microbial components may stimulate innate immune cells through the TLR pathways as well as other innate immune receptors, thus contributing to the proinflammatory cytokine milieu and systemic immune activation associated with chronic HIV infection (33). Consistent with the above-mentioned findings on several biological and pathological models, acute HIV infection is associated with relatively normal LPS levels, with increased LBP, sCD14, and EndoCAb levels compared to levels in uninfected individuals, thus suggesting that the ongoing translocation of LPS is rapidly counteracted by the host Ig response. However, EndoCAb titers decrease progressively as HIV infection enters its chronic phase, which becomes characterized by higher plasma levels of LPS.
MEASUREMENT OF MICROBIAL TRANSLOCATION
The extent of microbial translocation can be assessed either directly through the measurement of bacterial by-products in plasma, such as LPS and bacterial DNA or RNA fragments, or indirectly by sCD14, LBP, EndoCAb, and antiflagellin antibodies. Recently, plasma levels of intestinal fatty acid binding protein (I-FABP), a marker of enterocyte damage (36), have also been used to correlate intestinal impairment and microbial translocation (37, 38). Table 1 summarizes the characteristics of the principal microbial translocation markers measured in plasma/serum of HIV-infected patients. In the majority of published studies, sCD14, LBP, and EndoCAb are measured in the plasma or serum by a solid-phase sandwich enzyme-linked immunosorbent assay (ELISA), which generally produces reliable and reproducible results (47). In particular, sCD14 has been widely used as a marker of microbial translocation, given the relatively easy standardization of its measurement between different laboratories. It must be noted, however, that sCD14 serves as a biomarker of monocyte activation (29), and although it correlates with LPS, it is not a direct and specific marker of microbial translocation per se. Conversely, the commercial Limulus amebocyte lysate (LAL) assay allows for the quantitative determination of LPS in reference to known endotoxin concentrations and is therefore a direct measure of endotoxemia. However, the assay presents technical difficulties due to possible reagent contamination and the inhibitory nature of some plasma components which have rendered the assay difficult to reproduce. The pretreatment of samples by heating has been shown to provide more reliable results and is thus mandatory for plasma LPS measurements using the LAL technique (48). Finally, it should be remembered that as LPS is a specific product of the cell wall of Gram-negative bacteria, the LPS assay fails to detect translocating Gram-positive bacteria.
Table 1.
Microbial translocation markers measured in plasma/serum of HIV-infected patients
| Marker | Description | Method | Limit(s) | References |
|---|---|---|---|---|
| LPS | Component of outer membrane of Gram-negative bacteria | LAL test | Reagent contamination | 33, 37, 39–44 |
| Plasma inhibitors | ||||
| Specific only for Gram-negative bacteria | ||||
| sCD14 | Protein expressed by monocytes/macrophages; the soluble form appears after shedding or is directly secreted from intracellular vesicles | ELISA | Proinflammatory cytokines such as IFN-α and IFN-β can increase plasma levels | 33, 37, 40–43 |
| LBP | Acute-phase protein that binds LPS and presents it to CD14 and TLR-4 | ELISA | Produced by hepatocytes, influenced by HCV/HBV coinfection | 33, 43 |
| EndoCAb | IgM, IgG, and IgA antibodies directed against the LPS core antigen that neutralize LPS activity | ELISA | Under normal conditions, when LPS gain access to circulation, these antibodies bind to and clear LPS, and their titers decrease | 33, 37, 41–43 |
| When microbial products are found in the systemic circulation chronically, EndoCAb levels are increased as part of the normal humoral response to antigenic stimulation | ||||
| Bacterial DNA fragments | DNA fragments derived from bacterial degradation | 16S rRNA gene PCR | Reagent contamination (i.e., exogenous DNA in bacterially expressed Taq polymerase) | 37, 39, 40, 45, 46 |
| Sample contamination | ||||
| Amount of circulating bacterial DNA |
An alternative method to assess microbial translocation is the detection and quantification of the universally conserved microbial 16S rRNA gene in plasma (49). The majority of 16S rRNA gene PCR assays are designed for the qualitative detection of amplicons, which allows for the identification of different bacterial species (39, 40, 50). Novel and highly sensitive real-time PCRs have recently been developed, focusing on the accurate quantification of 16S rRNA gene sequences (45, 46, 51, 52). However, exogenous DNA contamination during workflow or the presence of DNA traces in PCR reagents, including Taq DNA polymerase from the production strain, may account for false-positive results (53, 54). Given these obstacles, diverse approaches have been reported to optimize such techniques and include the use of shrimp nuclease (SNuc) (46), the removal of human background DNA, and bacterial DNA enrichment (55). Despite the attempts to implement sensitive techniques to detect traces of microbial translocation in peripheral blood of HIV-infected patients, concerns remain regarding the risk of contamination. Bacterial flagellin is known as a microbial compound with strong immune-modulatory properties that has an essential role under conditions of intestinal damage, such as inflammatory bowel disease. Recently, elevated levels of flagellin-specific IgG were described for three cohorts of HIV-1-infected patients with a significantly elevated ratio of flagellin-specific IgG to total IgG (56) and were subsequently demonstrated to be reduced following highly active ART (HAART) despite not reaching the levels in HIV-negative controls (57). Combined, these data suggest that flagellin should be considered an important microbial product that plays an important role in immune activation due to translocation from the gut.
MICROBIAL TRANSLOCATION IN THE PATHOGENESIS OF HIV AND SIV INFECTIONS
A seminal study by Brenchley et al. demonstrated for the first time that both chronically HIV-infected individuals and SIV-infected rhesus macaques exhibit significantly increased levels of circulating LPS. Particularly, HIV-infected patients in an advanced stage of disease (<200 CD4+ T cells/ml) had significantly higher plasma LPS levels than uninfected individuals, thus suggesting the presence of increased microbial translocation in this study population. Interestingly, a positive correlation was found between LPS levels in plasma and measures of innate and adaptive immune activation (33, 39), thus supporting the hypothesis that microbial translocation may result in T-cell activation, which is known to influence HIV disease progression (5, 11, 12, 58). Table 2 shows an overview of studies suggesting that microbial translocation through the gastrointestinal tract is a cause of immune activation and immune dysfunction in HIV/SIV infections.
Table 2.
Overview of studies suggesting that microbial translocation through the gastrointestinal tract is a cause of immune activation and immune dysfunction in HIV/SIV infectionsa
| Reference | Study design | Study population(s) | Main findings |
|---|---|---|---|
| 33 | Cross-sectional | HIV-infected patients | Higher levels of LPS in HIV-infected patients |
| SIV-infected rhesus macaques | LPS level increases following SIV infection in RM | ||
| Positive correlation between plasma LPS levels and circulating CD8+ CD38+ HLA-DR+ T cells | |||
| Inverse correlation between plasma LPS levels and TNF+ IL-1+ monocytes upon LPS stimulation | |||
| Inverse correlation between plasma LPS levels and plasma EndoCAb titers | |||
| 39 | Cross-sectional | INRs on HAART | Higher LPS levels in INRs and advanced naïve patients |
| Full-responder patients on HAART | Positive correlation between LPS and proliferating Ki67+ CD8+ T cells in INRs | ||
| Advanced naïve patients | Higher plasma enterobacterial DNA levels in INRs | ||
| 45 | Cross-sectional and longitudinal | Case Western Reserve University Cohort, with (i) HIV-negative patients, (ii) HIV-positive naïve patients, (iii) HIV-positive treated patients with undetectable viremia, and (iv) HIV-positive treated patients with detectable viremia | Increased bacterial 16S rRNA levels in HIV-infected patients |
| UCSF SCOPE Cohort | Correlation of bacterial 16S rRNA with LPS in HAART-treated patients | ||
| ACTG study 5014 | Correlation of 16S rRNA levels (i) directly with the frequency of CD38+ and HLA-DR+ CD8+ T cells and (ii) inversely with the degree of CD4+ restoration on HAART | ||
| Decrease in 16S rRNA levels following HAART | |||
| 59 | Cross-sectional | HIV-infected patients, HAART naïve | Correlation between Ki67-expressing T cells in the gut and plasma LPS levels |
| HIV-infected patients on HAART | |||
| HIV-negative controls | |||
| 60 | Cross-sectional | HIV-1-infected patients, HAART naïve | Higher LPS levels in patients with AIDS than in controls |
| HIV-1-infected patients with AIDS | Correlation between LPS levels, CD4+ T-cell lymphopenia, and HIV RNA load | ||
| HIV-2-infected patients, HAART naïve | Inverse correlation between plasma LPS levels and expression of IL-12 and IFN-α following TLR stimulation | ||
| HIV-2-infected patients with AIDS | No difference between HIV-1- and HIV-2-infected individuals within the same disease stage, CD4+ T-cell count, or viral load intervals | ||
| HIV-negative controls | |||
| 61 | Cross-sectional | SIV-uninfected pigtail macaques | Increased plasma LPS levels and intestinal permeability in uninfected pigtail macaques |
| SIV-uninfected rhesus macaques | Tight-epithelial-barrier damage and increased lamina propria LPS levels in uninfected pigtail macaques | ||
| HIV-negative controls | Correlation between breaches in the colon, microbial translocation, and local immune activation | ||
| Microbial translocation and immune activation in the colon result in systemic microbial translocation and immune activation | |||
| 62 | Cross-sectional | HIV-infected patients, HAART naïve | Decrease in activated CD14+ CD16+ monocytes, correlating with HIV RNA, in course of HAART |
| HIV-infected patients on HAART | Elevated sCD14 and TNF-α levels persist in course of HAART | ||
| HIV-negative controls | |||
| 63 | Cross-sectional | SIV-uninfected sooty mangabeys | Highest LPS levels in lamina propria of chronically infected/AIDS animals |
| SIV-infected sooty mangabeys | Damage of epithelial barrier associated with infiltration of microbial products into the lamina propria | ||
| SIV-uninfected rhesus macaques | Correlation between damage of the epithelial barrier and microbial translocation (highest in chronically infected/AIDS animals) | ||
| SIV-infected rhesus macaques | |||
| 40 | Longitudinal | HIV-infected patients starting HAART with CD4+ counts of <200 cells/μl | Persistence of high LPS and sCD14 levels despite suppressive HAART |
| Lack of probiotic Lactobacillaceae DNA in plasma both prior and after therapy in patients with CD4+ counts of <200 cells/μl following HAART | |||
| 64 | Cross-sectional | HIV-infected patients, HAART naïve | Negative correlation between total bacterial load and duodenal activated CD4+ and CD8+ T cells |
| HIV-infected patients on HAART | Positive correlation between fecal Bacteroidales (rRNA) and activated, peripheral CD8+ T cells | ||
| HIV-negative controls | Negative correlation between fecal Enterobacteriales (rRNA) and duodenal CD4+ T cells | ||
| 65 | Cross-sectional | HIV-infected patients, HAART naïve | Correlation between gut HIV DNA, plasma LPS, and peripheral CD8+ CD38+ T cells in HAART-treated patients |
| HIV-infected patients on HAART | |||
| HIV-negative controls | |||
| 66 | Cross-sectional | HIV-infected patients, HAART naïve | Depletion of gut Th17 cells, which is normalized by HAART |
| HIV-infected patients on HAART | Persistence of high LPS levels despite HAART | ||
| HIV-negative controls | Inverse correlation was observed between plasma LPS levels and gut Th17 frequencies | ||
| Correlation between plasma LPS levels and gut HIV proviral reservoir | |||
| 38 | Cross-sectional | HIV-infected patients on HAART | Positive correlation between LPS and sCD14 and gut-homing CD4+ T cells in the blood |
| HIV-negative controls | Inverse correlation between LPS and sCD14 and gut-homing CD4+ T cells in the gut | ||
| Positive correlation between peripheral CD4+ Ki67+ cells and circulating gut-homing CD4+ T cells |
Studies are listed in chronological order.
The possibility that microbial TLR ligands might affect T-lymphocyte activation in HIV-uninfected individuals was elegantly investigated by Funderburg et al. (67). In that study, in vitro exposure to microbial TLR ligands promoted the cell surface expression of lymphocyte-homing and activation/apoptosis molecules on CD4+ and CD8+ T cells (67). Consistent with those observations, in vitro stimulation by microbial and viral TLR ligands was shown to induce T-cell activation in both antiretroviral-naïve and ART-treated, HIV-infected patients (68, 69). By extension, those in vitro findings suggested that systemic exposure to TLR ligands in HIV-infected patients results in heightened immune activation, effector T-cell sequestration in lymphoid tissues, and T-cell turnover, thus reinforcing the hypothesis that microbial translocation is a crucial determinant of immune activation.
Pathogenesis of HIV/SIV-Associated Microbial Translocation
From the early phase of acute HIV or SIV infection, and throughout the course of chronic disease, the gastrointestinal tract suffers from a substantial immunological and structural disruption, which includes the depletion of CD4+ T cells, with a preferential loss of the IL-17-producing subpopulation (Th17 cells), the establishment of local mucosal hyperactivation/inflammation, the exhaustion of intestinal macrophage phagocytic function, and structural epithelial damage (apoptosis of enterocytes and disruption of tight junctions, etc.). Collectively, these alterations may result in the increased passage of gut microflora and microbial products into the systemic circulation. Interestingly, several aspects of HIV-associated mucosal immune dysfunction were described prior to the original formulation of the microbial translocation hypothesis, including the presence of a rapid and severe loss of CD4+ T cells in the gastrointestinal tract of SIV-infected macaques and HIV-infected humans (34, 35, 70–72).
During acute HIV/SIV infection, CD4+ T-cell depletion is more prominent in the gastrointestinal tract than in peripheral blood and lymph nodes. Such severe CD4+ T-cell impairment has been shown to preferentially involve gut-residing memory-activated CD4+ CCR5+ T cells, reflecting the well-known prevalence of CCR5-tropic viruses in the early phases of infection. Almost concomitantly, Veazey et al. and Kewenig et al. showed that more than 70% of intestinal CD4+ cells were depleted as early as 21 days post-SIVmac239 infection (72, 73), and several subsequent studies then confirmed these findings and shed further light onto the mechanistic pathways underlying CD4+ T-cell depletion (34, 74–76). Among those studies, Mattapallil et al. were able to demonstrate the presence of SIV DNA in up to 60% of intestinal CD4+ T cells as early as 10 days after experimental SIVmac251 infection, therefore emphasizing the role of direct virus-mediated killing of CD4+ T cells as a mechanism causing the observed depletion (70). However, Li et al., using in situ hybridization to detect SIV RNA, concluded that only ∼7% of mucosal CD4+ cells were productively infected by SIV during acute infection, thus supporting the hypothesis that apoptosis-mediated death is a pivotal mechanism sustaining mucosal T-lymphocyte depletion (77). Similar data have also been obtained by serial gastrointestinal tract sampling in HIV-infected individuals during early stages of infection, although the depletion of intestinal CD4+ CCR5+ T cells appeared to be somewhat less dramatic than in the SIVmac infection model (34, 35, 71). Following the acute phase of infection, CD4+ T-cell depletion in the intestinal mucosa has been shown to continue throughout chronic disease to a much greater extent than in the peripheral blood and lymph nodes. Taken together, these data indicate that the depletion of gut CD4+ T cells is a multifactorial phenomenon, which involves both direct virus-mediated killing and bystander death of uninfected cells, with a subsequent impairment of the structure and function of the intestinal immune system and mucosal barrier, which ultimately leads to sustained microbial translocation.
Interestingly, studies of nonpathogenic SIV infection in the natural hosts of SIV, e.g., sooty mangabeys (SM) and African green monkeys (AGM), have also shown severe CD4+ T-cell depletion in mucosal tissues during acute infection (78, 79). However, this depletion is not accompanied by sustained microbial translocation and chronic mucosal immune activation, and in fact, in contrast to pathogenic SIV and HIV infections, the early mucosal CD4+ T-cell loss does not seem to progress further in SM and is even followed by a partial recovery in AGM (80). The latter findings notwithstanding, it remains intriguing that natural SIV hosts show no evidence of microbial translocation despite significant intestinal CD4+ T-cell loss, with this finding suggesting that mucosal CD4+ T-cell depletion is necessary but not sufficient for microbial translocation to ensue. In addition to the evolutionary hypothesis that natural SIV hosts have progressively adapted to maintain mucosal immunity despite reduced local CD4+ T-cell levels, several non-mutually exclusive mechanisms might be hypothesized to explain differences in microbial translocation between pathogenic SIV/HIV infections and infections of natural SIV hosts. First and foremost, it is possible that a different “quality” of CD4+ T-cell depletion in natural versus nonnatural SIV hosts at the intestinal mucosal level plays a major role. Indeed, both SIV-infected RM and HIV-infected persons experience a significant and preferential reduction of the IL-17-producing CD4+ (Th17) and CD8+ (Tc17) T-cell subsets throughout the gastrointestinal tract (81–84). Similarly, the depletion of IL-17-producing innate lymphocytes in the GI tracts of SIV-infected Asian macaques has been demonstrated (85, 86).
In marked contrast, natural SIV hosts have been shown to maintain frequencies of functional gut-based Th17 and Th1 CD4+ T cells at levels comparable to those in noninfected animals (87). As intestinal Th17 cells have an important role in antimicrobial immunity, particularly at mucosal sites (88–90), as well as in sustaining enterocyte homeostasis (89, 90), the preferential depletion of Th17 cells in pathogenic HIV/SIV infections may be involved in the damage to the structural and functional integrity of the intestinal barrier and in the loss of control over microbial translocation. Indeed, Favre et al. elegantly demonstrated that the early loss of GI Th17 in pathogenic SIV infection is associated with overt signs of disease progression that include enhanced immune activation and T-cell apoptosis, increased levels of circulating alpha interferon (IFN-α), enhanced transcription of IFN-α-induced genes, and rapid CD4+ T-lymphocyte depletion (87). In contrast, SIV infection in the natural AGM host is characterized by a transient IFN-α inflammatory response which decreases once an SIV viremia plateau is reached (87). Second, both SIV-infected AGM and SM exhibit significantly reduced levels of activated/proliferating T lymphocytes in the intestinal mucosa compared to those in SIV-infected macaques (78), thus suggesting a central role for local immune activation in dictating generalized mucosal immune dysfunction and microbial translocation, possibly by reducing the functionality of other arms of the immune system. Importantly, the exogenous administration of LPS to SIV-infected AGM results in increased levels of systemic immune activation, thus indicating that an effective preservation of mucosal barrier function is likely to be key to the ability of AGM to maintain low-level immune activation despite high-level viremia (91).
MICROBIAL TRANSLOCATION AND IMMUNE ACTIVATION: WHAT IS THE CAUSE AND WHAT IS THE EFFECT? LESSONS LEARNED FROM THE ANIMAL MODEL
Following multiple literature reports that strongly suggested an association between immune activation and microbial translocation parameters in HIV-infected patients (Table 2), two studies demonstrated that the direct administration of LPS in a natural host of SIV (which lacks chronic immune activation and microbial translocation) results in increased immune activation and viral replication (91, 92).
However, while it is commonly accepted that the structural and functional damage of the intestinal mucosa barrier and immune function results in an increased permeability of the mucosal epithelium (93–96) and a consequent unchecked systemic translocation of bioactive microbial products that can trigger immune activation, it remains unclear to what extent this phenomenon contributes to HIV/SIV-associated chronic immune activation. For instance, one could also hypothesize a scenario in which chronic immune activation is the initial driver of mucosal immune dysfunction and that microbial translocation is a secondary amplification of a loop that further aggravates immune activation (9, 80).
While reciprocal interactions between immune activation and microbial translocation are most likely to occur during pathogenic HIV/SIV infections, evidence of microbial translocation as a factor driving pathological immune activation in HIV-infected humans is quite strong although not necessarily conclusive. In the SIV model, Estes et al. provided elegant in vivo evidence of the presence of microbial products, LPS and E. coli, in the colon lamina propria of SIV-infected RM but not of SIV-infected sooty mangabeys (63). In particular, using immunohistochemistry and confocal fluorescence microscopy, those authors were able to show the presence of LPS/E. coli in the colonic mucosa since the earliest phases of infection, with a progressive increase throughout chronic disease that is associated with epithelial barrier dysfunction, as documented by the multifocal disruption of the epithelial GI barrier via staining of the tight junction protein claudin-3. In that same study, LPS was also identified within systemic lymph nodes and liver, the amount of which positively correlated with colonic LPS, therefore confirming the systemic passage of gut-derived microbic elements (63). In addition, the magnitude of epithelial breakdown was correlated with the extent of microbial translocation and intestinal immune activation, as shown by the colocalization of bacterial products and proinflammatory cytokines (IFN-α and IL-18) in the colonic lamina propria. Those authors took a further step by investigating the timing and dynamics of macrophage phagocytosis and concluded that intestinal macrophages appear free from bacterial cells during chronic infection, whose cell-free component is, on the contrary, increased (97). The latter findings suggest that the progressive exhaustion of intestinal macrophage phagocytic function, with the extracellular accumulation of microbial components, is also involved in the pathogenesis of HIV/SIV-associated microbial translocation. Overall, the available data strongly suggest that in SIV infection of RM and, by extension, in HIV infection of humans, a complex series of events leads to mucosal immune dysfunction, damage to the intestinal epithelial barrier, microbial translocation, and chronic systemic immune activation, all ultimately contributing to disease progression. However, the reciprocal contribution of these events to the immunopathogenesis of AIDS remains incompletely defined, and further studies of both humans and nonhuman primates will be needed to better elucidate the chain of events that leads to the progressive demise of the host immune system during pathogenic HIV/SIV infections.
CHARACTERIZATION OF STOOL MICROBES AND TRANSLOCATING FLORA IN HIV-INFECTED PATIENTS
Numerous studies have demonstrated a major influence of the gut microbiome in the maintenance of immunity and health by exerting many immunologic, metabolic, and structural functions. Germfree animals are in fact characterized by a dramatic disruption of gut integrity and the intestinal immune system, with increased susceptibility to infections (98). In these experimental models, the repopulation of the gut microbiome results in a reconstitution of the different components of mucosal immunity (99). Indeed, the specific composition of the gut microbiome is essential to maintain both local and systemic immunity, as recently reviewed by Paiardini et al. (80) and Brenchley and Douek (100). While several human diseases have been associated with a disruption of the gut microbiome (i.e., dysbiosis), it has somehow been difficult to establish with certainty whether and to what extent such a dysbiosis is the cause or the effect of the disease, given the influence of a multitude of potential pathogenic factors. In the macaque model, microbial communities residing in the gut have been described to differ substantially in both quantity and composition between healthy and diseased animal (101).
Interestingly, Gori et al. demonstrated that HIV-infected individuals in early stages of disease are characterized by an impaired fecal flora, with a predominance of opportunistic pathogens (Pseudomonas aeruginosa and Candida albicans) and low levels of protective bacteria (bifidobacteria and lactobacilli) compared with historical data on healthy HIV-uninfected persons (102). Altered stool composition has also been reported, with increased levels of fecal calprotectin, which is secreted by recruited neutrophils in the intestinal lining and has been proposed to be a marker of intestinal inflammation (103). Taken together, these data demonstrate a substantial subversion of physiological gut microbiome and a correlation between pathogenic intestinal flora and gut inflammation in HIV disease (102). In a recent study, Ellis et al. (64) explored the intriguing hypothesis that the gut microflora composition affects both local and systemic immunity, in analogy with what was described in several clinical settings (104, 105). Those authors observed a greater representation of proinflammatory/inflammation-thriving order-level bacteria, such as Enterobacteriales and Bacteroidales (both Gram-negative, LPS-containing pathogens associated with several clinical conditions) in the gut of HIV-infected subjects than in healthy controls. Those authors next investigated correlations between the stool bacterial populations and both T-lymphocyte loss and immune activation at the intestinal and peripheral blood levels. Most interestingly, the total bacterial load in the stools negatively correlated with duodenal T-cell activation, and levels of Enterobacteriales and Bacteroidales fecal DNA were significantly associated with CD4+ T-cell loss in the duodenum and peripheral CD8+ T-cell activation, respectively (64). Therefore, by correlating specific features of the gut microbiome and markers of HIV disease progression, these data provide evidence for a direct role of the gut microbiome in driving local and systemic immune activation in HIV-infected patients (39, 45).
Given the relationship between microbial translocation and CD4+ T-cell homeostasis, based on our previous report on higher levels of microbial rRNA genes specific to Enterobacteriaceae in patients with incomplete immune reconstitution (39), our group investigated whether the composition of microflora translocating in peripheral blood may influence the degree of immunological recovery during the course of ART. In a longitudinal study aimed at assessing the composition of microbial products translocating in peripheral blood prior to and during ART, we reported that patients with a poor immunological response to therapy exhibited a translocating bacterial microflora enriched in Enterobacteriaceae, with no evidence of probiotic Lactobacillus spp. (40), which are known to possess immunomodulatory and anti-inflammatory properties (106), including the suppression of proinflammatory cytokine production from E. coli LPS-activated monocytes (83). This change in the composition of the gut microbiome might result in a disproportionate antigenic challenge that is not contained by counterregulatory bacterially derived factors, overall exacerbating immune activation in this population of HIV-infected individuals. Collectively, these data suggest that early changes in the composition of the intestinal microflora may affect local and systemic immune activation and, by extension, HIV disease progression and the response to therapy.
While intriguing, these data need to be corroborated by further research specifically designed to investigate (i) whether and to what extent the bacterial load and composition in the GI tract correlate with the microbiome in peripheral circulation by extensive genetic sequencing in both compartments and (ii) possible direct correlations between the microbiome at both sites and markers of T-cell depletion and immune activation. By defining putative pathological mechanisms that link the gut/systemic microbiome with immune activation in HIV disease, these studies might provide a rational basis for the investigation of therapeutic approaches aimed at recovering the symbiotic host-microorganism relationships as a way to reduce HIV-associated chronic immune activation and to improve the response to antiretroviral therapy.
CLINICAL IMPLICATIONS OF MICROBIAL TRANSLOCATION IN HIV DISEASE
Microbial Translocation Predicts HIV Disease Progression and Poor Response to ART
The association of mucosal immune dysfunction and microbial translocation with systemic immune activation and disease progression in the course of HIV infection prompts a number of questions of major clinical relevance. A first crucial issue is whether microbial translocation is a predictor of HIV disease progression independently of other established markers such as CD4+ T-cell counts and viral load. If such an association is established, the next questions would be, (i) should markers of microbial translocation be measured as part of the routine clinical and laboratory monitoring of HIV-infected individuals (107); (ii) should the level of markers of microbial translocation be used to make management and therapeutic decisions, e.g., on when to start/modify antiretroviral treatment, and (iii) how reversible is microbial translocation once HIV replication is suppressed by ART?
The role of microbial translocation in predicting disease progression in the absence of antiretroviral therapy as well as the effects of HAART have recently been investigated in several studies. For a cohort of HIV-infected individuals from Rakai, Uganda, who were monitored longitudinally from preinfection to death for levels of proinflammatory cytokines and microbial translocation, Redd et al. failed to find significant associations between levels of LPS, sCD14, and endotoxin antibody and HIV disease progression (108). Moreover, circulating immunoreactive cytokine levels either decreased or remained virtually unchanged throughout the progression of the disease. However, subsequent data by Redd et al. found longitudinal increases in plasma levels of C-reactive protein, which correlated with levels of sCD14 (109).
We obtained different results by investigating markers of microbial translocation (LPS, sCD14, and EndoCAb) in a cohort of 379 HIV-infected, ART-naïve patients with early HIV infection (approximately 3 years after seroconversion) and CD4+ T-cell counts of >200 cells/μl. In our study, circulating levels of LPS were a strong predictor of disease progression, measured through a combined endpoint of AIDS, death, CD4+ T-cell counts of <200 cells/μl, or the start of ART, independent of CD4+ T-cell counts and plasma viremia. Indeed, patients with heightened LPS levels showed substantially accelerated disease progression, with a median time to clinical event of 1.5 years, versus 4 years for patients with lower levels (41). As a possible complement to these findings, previous data strongly suggested a correlation between intestinal immune dysfunction and intestinal functionality, as suggested both by gene expression profiling that associated gut CD4+ T-cell depletion with downregulated expressions of genes involved in mucosal function (110, 111) and by earlier clinical data on malabsorption and nutritional complications in SIV-infected RM (111) and HIV-infected humans (112). Along the same lines, a nested-control study from the Strategies for Management of Anti-Retroviral Therapy (SMART) trial by Sandler et al. reported that plasma levels of sCD14 are independent predictors of overall mortality in HIV disease (37). Taken together, these data strongly suggest a negative effect of mucosal immune dysfunction and microbial translocation on HIV disease progression.
Circulating levels of LPS have been consistently reported to decrease after the initiation of ART, even though they did not return to the levels observed for healthy HIV-uninfected individuals (33, 39, 45, 62, 65). Since persistent immune activation has long been implicated as a factor impairing the immunological response to ART (13, 14), the question as to whether microbial translocation might affect immune recovery after ART has been investigated by different groups in the last few years. Following the original observation by Brenchley et al. of an inverse correlation between CD4+ T-cell reconstitution and microbial translocation (33), this finding was confirmed by several groups. Our group reported, in a cross-sectional study, increased plasma levels of LPS in immunological nonresponder (INR) ART-treated HIV-infected individuals (defined by CD4+ T-cell counts of ≤200 cells/μl despite long-term HIV RNA levels of ≤50 copies/ml while on HAART) compared to levels in subjects with full immunological recovery after ART (39). Analogously, Jiang et al. showed that higher levels of DNA sequences encoding bacterial 16S rRNA genes are associated with greater T-cell activation and impaired CD4+ T-cell restoration after ART (45). Similar data were also reported for a South African cohort, confirming persistently elevated LPS and sCD14 levels in INR despite virologically suppressive ART (62). While these findings provide important insights into the mechanisms determining the immunological response to ART, they still fail to formally prove that the level of microbial translocation is a key factor regulating CD4+ T-cell reconstitution upon virologically suppressive ART. Given the well-described increased risk of AIDS- and non-AIDS-related morbidity and mortality in HIV-infected patients who do not experience full immunological recovery on ART (113, 114), further ad hoc-designed observational and randomized studies are warranted to investigate this crucial aspect of SIV/HIV pathogenesis.
Role of Microbial Translocation in Non-AIDS Comorbidities
Over the past few years, strong evidence has accumulated indicating that long-term-ART-treated HIV-infected individuals exhibit a higher-than-expected risk of degenerative diseases, including cardiovascular disease, cancer, osteoporosis, liver disease, and other end-organ illnesses, therefore resulting in a shortened life expectancy and also depicting a pattern of early and accelerated aging (115). Given the burden of these non-AIDS comorbidities in the clinical management of HIV infection, great scientific effort is now being put into the identification of possible immunopathogenic factors. Among these, some evidence suggests that microbial translocation may play an important role in liver and cardiovascular diseases as well as, possibly, neurocognitive decline. Since LPS induces monocyte activation and enhances the trafficking of these cells to the brain, Ancuta et al. hypothesized a possible association between microbial translocation and HIV-associated neurocognitive dementia (HAND). For a cohort of HIV-infected individuals with CD4+ T-cell counts of <300 cells/μl and with various degrees of neurocognitive impairment, those authors demonstrated higher LPS and sCD14 levels in patients with HAND and described a significant and independent association between circulating LPS and HAND development (116). Kamat et al. most recently expanded these data by associating sCD14 levels in cerebrospinal fluid (CSF) with neurocognitive impairment. Combined, these data suggest the possibility of exploiting plasma and CSF sCD14 measurements in the clinical management of HAND (117). Ellis et al. investigated whether HIV-associated sensor neuropathy (SN) in HIV-infected individuals with good CD4+ recovery on virologically suppressive ART could be associated with monocyte activation and found an association between SN and sCD14 levels in the patients' cerebrospinal fluid (118). Furthermore, Forsyth et al. described a link between endotoxin exposure markers and early Parkinson's disease (119).
Microbial Translocation in Liver Disease
The liver has a crucial, physiological role in LPS detoxification from the circulation, as bacterially derived products from the intestine translocate into the portal system, where they are sensed and cleared by Kupffer cells through interactions with cell surface CD14 (120). However, in the setting of liver disease, bacterial overgrowth, intestinal edema, and altered hepatic architecture result in the increased entry of LPS into the peripheral circulation, in turn activating Kupffer cells. Indeed, LPS has been shown to accelerate liver fibrosis through TLR-4 signaling on Kupffer cells following membrane binding via LPS binding protein (LBP) and CD14 (121). These events lead to the generation of superoxide and the release of proinflammatory and profibrogenic cytokines such as TNF-α, IL-1, IL-6, and IL-12, all of which induce liver damage (122, 123). Consistent with these findings, microbial translocation has been shown to contribute to liver disease in several clinical settings, such as alcoholic liver disease (124, 125) and other enteric processes (126–128). HIV coinfection with hepatitis viruses accelerates liver disease progression; however, the precise mechanisms by which this phenomenon occurs are not fully understood. Given the heightened circulating LPS levels in HIV infection, microbial translocation has been proposed to exert a major role in the worsened liver disease in HIV-infected individuals.
Balagopal et al. demonstrated that in HIV-infected individuals, measures of microbial translocation are strongly associated with markers of hepatitis C virus (HCV)-related liver disease progression (e.g., cirrhosis) (123). Similarly, for 98 HIV/HCV coinfected patients on virologically suppressive ART, we reported increased sCD14 levels in subjects either harboring aggressive HCV genotypes (i.e., genotypes 1 to 4) or presenting with cirrhosis (129). Furthermore, Sandler et al. reported that higher sCD14 levels distinguish patients with severe liver fibrosis from those with minimal fibrosis in a cohort of hepatitis B virus (HBV)/HCV-monoinfected subjects (130). These results suggest a pathogenic role of the host response to LPS in severe HIV/HCV-related liver disease. However, an alternative pathway linking microbial translocation and liver disease progression must be acknowledged, as hepatic disease might in fact substantially affect the ability of the liver to metabolize circulating LPS. This hepatic dysfunction would in turn result in increased LPS plasma levels (which, in the context of HIV coinfection, would sum up to the already increased levels of LPS translocation from damaged gastrointestinal mucosa) to further contribute to liver damage.
The apparent association between markers of microbial translocation and advanced liver disease has led different groups to investigate whether microbial translocation (either per se or through the host response) may influence the short- and long-term virological outcomes of anti-HCV therapy. As predicted by this hypothesis, the rate of sustained virological response (SVR) to standard anti-HCV treatment with pegylated alpha interferon (peg-IFN-α) plus ribavirin is lower in HIV/HCV-coinfected subjects than in HCV-monoinfected patients (131, 132). Although several factors have been associated with the response to anti-HCV therapy, the determinants of successful full-course peg-IFN-α–ribavirin therapy are still poorly defined (133–137). Interestingly, increased plasma sCD14 levels have been proven to be independently associated with poor responses to anti-HCV treatment (129, 130), thus suggesting an independent role of microbial translocation in the outcome of anti-HCV therapy. Contrasting data, however, were obtained for a cohort of ART-naïve HIV/HCV-coinfected patients with high CD4+ T-cell counts, for whom higher sCD14 levels were independently associated with a decreased risk of liver disease progression, defined as a time to Fibrosis 4 (Fib-4) score of >1.45 or liver-related death. Since sCD14 prevents the interaction of LPS with membrane-bound CD14 on the surface of phagocytes, thus hampering the inflammatory response (138), those authors proposed a model in which high sCD14 levels preserve liver function through the downregulation of the LPS-induced downstream inflammatory cascade (139).
Microbial Translocation in Cardiovascular Disease
HIV-infected patients are at an increased risk of cardiovascular events compared to age- and sex-matched healthy controls. The reasons for this phenomenon are likely complex, although recent studies suggested that chronic inflammation and immune activation/senescence may contribute to the initiation and progression of atherosclerosis in HIV infection (37, 140–142). LPS represents an important source of inflammatory stimuli (143) and has been investigated in the setting of atherosclerosis (144). The Bruneck study by Wiedermann et al. in 1999 provided the first epidemiological evidence in support of a clinical association between levels of LPS and cardiovascular risk (145, 146). Consistent with these findings, the role of bacterial endotoxin as a proinflammatory mediator of atherosclerosis has been widely reported for the HIV-uninfected population (143). Studies on the role of bacterial bioproducts (i.e., LPS) and macrophage activators (sCD14) in the pathogenesis of atherosclerosis in HIV disease are under way, and discordant results have been generated so far. Merlini et al. reported higher levels of sCD14 in ART-treated HIV-infected individuals with increased carotid intima media thickness (cIMT) in an unadjusted analysis; however, this effect was lost after adjusting for classical cardiovascular predictors (147). Similarly, Kelesidis et al. recently showed that in HIV-infected subjects with low cardiovascular risk, sCD14 and LPS levels are independently associated with increases in the yearly rate of change in cIMT (148). Intriguing data were recently presented by Pandrea et al., who identified a nonhuman primate model for the role of microbial translocation in cardiovascular comorbidity (92). In that study, SIVagm-infected pigtail macaques showed elevated circulating levels of LPS in all stages of infection that correlated well with increased levels of coagulation markers, i.e., D-dimer and the thrombin-antithrombin complex. Most interestingly, and in analogy with what was observed for HIV-infected patients, these animals showed histopathological signs indicative of cardiovascular comorbidity, such as renal thrombotic microangiopathy. Furthermore, the reduction of microbial translocation-driven immune activation also resulted in the reduction of coagulation markers, thus pointing to a role of microbial translocation in the onset of cardiovascular disease (92).
Collectively, these data seem to support the hypothesis that microbial translocation, by inducing substantial immune activation, might contribute to the pathogenesis of several age-associated degenerative clinical conditions described for ART-treated, HIV-infected individuals. However, although interesting, the studies presented above have to be considered hypothesis-generating studies, and the possible associations between microbial translocation and non-AIDS comorbidities need further investigation within larger studies. The demonstration that microbial translocation is indeed a major determinant of non-AIDS comorbidities would provide a strong rationale for therapeutic interventions aimed at reducing the overall morbidity and mortality of ART-treated, HIV-infected patients.
TREATMENT OF HIV/SIV-ASSOCIATED MICROBIAL TRANSLOCATION
The detrimental effects of microbial translocation on HIV disease progression, the response to ART, and the development of non-AIDS comorbidities provide a strong rationale for the exploration of novel therapeutic interventions aimed specifically at preventing and reducing microbial translocation, its determinants, and its downstream effects. Several strategies are being investigated, including (i) modification of the intestinal microbiome, (ii) enhancement of the enterocyte barrier, (iii) reduction of local inflammation, and (iv) clearance of microbial bioproducts translocated from the gut.
Modification of GI Microbiome
Strategies aimed at restoring the normal composition of the intestinal microbiome include antibiotics and probiotics. Ideally, the most suitable antibiotics in such a context would be broad-spectrum, nonabsorbed antibiotics available for oral administration. Rifaximin is a semisynthetic, rifamycin-based, nonsystemic, broad-spectrum antibiotic, meaning that very little of the drug will pass the gastrointestinal wall into the circulation. Given its unique properties, it is widely used for the treatment of hepatic encephalopathy and as a gut-sterilizing treatment prior to abdominal surgery, while contrasting results have been obtained thus far on its efficacy in the treatment of inflammatory diseases (100). Rifaximin is now being intensively tested in HIV/SIV infection. Most recently, the efficacy of 3 months of rifaximin in association with the anti-inflammatory agent sulfasalazine was tested by Pandrea et al. in the acute phase of SIVagm infection of pigtail macaques. In that study, the rifaximin-sulfasalazine combination resulted in significant reductions in levels of circulating sCD14 cells, activated CD38+ HLA-DR+ T cells, D-dimer, proinflammatory cytokines, and chemokines in treated animals compared to untreated controls. A slight reduction in the magnitude of the CD4+ T-cell loss at the GI mucosa was observed for rifaximin-sulfasalazine-treated macaques versus controls, with no changes in circulating CD4+ T-cell counts (92). In HIV-infected humans, rifaximin is under investigation within the ACTG A5286 trial, which is a randomized, open-label, two-arm study that will test 4 weeks of treatment with rifaximin in HIV-infected patients on virologically suppressive ART and with low-level CD4+ T-cell recovery (<350 cells/μl). Study endpoints include rifaximin safety and efficacy in preventing persistent HIV-driven inflammation (https://actgnetwork.org/). While awaiting the completion of ongoing trials, it should be acknowledged that antibiotic treatment holds limited promise as an antimicrobial translocation treatment for HIV infection. Indeed, long-term antibiotic therapies would in fact result in the spread of resistant microbes, metabolic disadvantages, and further damage to the GI barrier.
An alternative approach is represented by the use of probiotics, which have been shown to modulate the gut microflora by inhibiting proinflammatory cytokines, decreasing gut permeability, and stimulating mucosal immunity. Previous studies demonstrated the usefulness of probiotics in gastrointestinal diseases such as IBD (100). In an observational, retrospective, 3-year study carried out on a cohort of HIV-infected individuals in Tanzania, Irvine et al. reported that probiotic-yogurt consumption was associated with an increase in CD4+ T-cell counts compared with a control group of participants not consuming the yogurt (149). More recently, Klatt et al. assessed the use of probiotics in addition to ART in SIVmac239-infected macaques for about 5 months. Interestingly, those authors observed an increase in antigen-presenting cell frequency and function; enhanced T-cell immunity in the intestinal mucosa, with Th17 cells becoming more polyfunctional; and reduced T-cell activation in animals treated with probiotics (150).
Interventions To Improve the Enterocyte Barrier
Given the dramatic structural and functional changes in the intestinal epithelial barrier during HIV infection, the possibility of positively supporting enterocyte homeostasis by means of immunomodulatory molecules is quite intriguing. In this context, two possible candidates are IL-22 and IL-17, due to their role in mucosal immunity and the association between the loss of IL-17- and IL-22-secreting lymphocytes and mucosal immune dysfunction during SIV infection (86).
Of note, encouraging results are provided by data on a murine model of ulcerative colitis, where the administration of IL-22 reduced intestinal inflammation (151). More recently, data have been presented to support a possible role of recombinant human IL-7 (rhIL-7) in the restoration of gut mucosal T-cell homeostasis in HIV-infected individuals. Indeed, the administration of rhIL-7 to a cohort of subjects with low-level CD4+ T-cell recovery on ART was efficacious in expanding both circulating and gut-residing naïve and memory CD4+ T cells, with parallel reductions in plasma levels of sCD14 and D-dimer (152).
Clearance of Microbial Bioproducts Translocated from the Intestinal Lumen
An intriguing therapeutic option is to neutralize microbial bioproducts translocated in peripheral blood as shown in Fig. 1 and 2. One such option is currently being investigated within the ACTG A5296 trial that is specifically aimed at testing the efficacy of oral sevelamer carbonate (which is effective in reducing inflammation and endotoxemia in the context of renal failure) to decrease endotoxemia in HIV-infected individuals naïve to antiretrovirals with CD4+ T-cell counts of ≥400 cells/μl (https://actgnetwork.org/) (153).
Fig 1.

Pathogenesis of microbial translocation in HIV-infected patients. (A) In healthy, HIV-negative subjects, the anatomical and functional integrity of the gastrointestinal (GI) mucosal barrier contains the passage of gut bacteria from the intestinal lumen to the lamina propria, thus resulting in physiological levels of mucosal immune activation and very limited if any microbial translocation in peripheral blood. (B) On the contrary, HIV accounts for a breach in the intestinal epithelial barrier with a loss of tight junctions, enterocyte apoptosis, local immune activation, and the depletion of CD4+ Th17 cells. This results in the passage of pathogenic bacteria and microbial products from the gut lumen to the lamina propria and from the lamina propria to the systemic circulation. At these sites, microbes and bacterial fragments further exacerbate local immune activation. Microbial translocation may be antagonized through different strategies: (i) modification of the GI microbiome by antibiotics, probiotics, and prebiotics; (ii) clearance of microbial bioproducts translocated from the gut (e.g., sevelamer); and (iii) interventions to reduce mucosal immune activation, such as IL-7, IL-17, and IL-22. IL, interleukin; HAART, highly active antiretroviral therapy.
Fig 2.
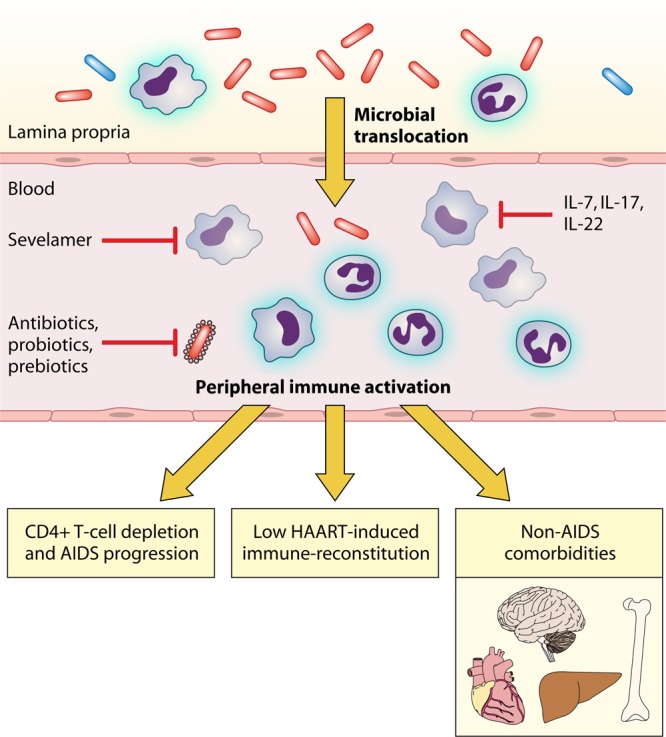
Immunological and clinical consequences of microbial translocation in HIV-infected patients. Gut-derived bacteria translocate from the lamina propria to the systemic circulation, leading to peripheral immune activation, which may be an underlying cause of HIV disease progression, poor immunological responses to HAART, and noninfectious comorbidities, such as neurological impairment and cardiovascular, liver, and bone diseases. Microbial translocation may be antagonized through different strategies: (i) modification of the GI microbiome by antibiotics, probiotics, and prebiotics; (ii) clearance of microbial bioproducts translocated from the gut (e.g., sevelamer); and (iii) interventions to reduce mucosal immune activation, such as IL-7, IL-17, and IL-22. IL, interleukin; HAART, highly active antiretroviral therapy.
CONCLUSIONS
Chronic microbial translocation is a feature of progressive HIV and SIV infections. It is now widely accepted that microbial translocation arises from a substantial immunological and structural disruption at the level of the gastrointestinal mucosa, which starts during acute infection and continues through chronic disease. This mucosal dysfunction includes the depletion of CD4+ T cells, with a preferential loss of Th17 cells; the establishment of local mucosal hyperactivation/inflammation; the exhaustion of intestinal macrophage phagocytic function; and structural epithelial damage (apoptosis of enterocytes and disruption of tight junctions, etc.). Both in vivo and in vitro data have documented that the unchecked continuous passage of immunostimulatory microbial molecules such as LPS from the intestinal lumen to the systemic circulation is able to sustain a substantial activation of innate and adaptive immunity, leading to the paradigm that microbial translocation is a crucial determinant of systemic immune activation in HIV/AIDS. Most interestingly, microbial translocation has been shown to be a clinically significant event, as it is associated with HIV clinical progression, suboptimal CD4+ T-cell recovery on ART, and the early onset of non-AIDS comorbidities such as liver disease progression, atherosclerosis and cardiovascular disease, and neurocognitive impairment. These findings have emphasized the need for therapeutic approaches that would mitigate microbial translocation and its effects on immune homeostasis, possibly contributing to improving HIV prognoses and life expectancy. These interventions could include modification of the intestinal microbiome, enhancement of the enterocyte barrier, reduction of local inflammation, and clearance of microbial bioproducts translocated from the gut.
ACKNOWLEDGMENTS
We are indebted to Alessandro Gottardo for excellent assistance with manuscript figures.
This work was supported by grant number GR 2000-1592029 from the Ministero della Salute-Regione Lombardia to G.M.
Biographies

Giulia Marchetti received her M.D. in 1995 at the University of Milan, Italy. She completed Residency in Infectious Diseases in 2000 (Milan), she took her Ph.D. in HIV/AIDS Clinic and Pathogenesis in 2003 (Milan). She was visiting fellow at the RIVM (Bilthoven, the Netherlands), Rush University (Chicago, IL), and the NIH (Bethesda, MD) between 1996 and 2002. She is currently Assistant Professor at the University of Milan. Dr. Marchetti has been involved in studies of HIV pathogenesis, therapies, and HIV-related comorbidities since 1996 and has authored or coauthored 68 original peer-reviewed publications in this field. In 2011, she obtained a grant from the Italian Health Ministry for the study of Microbial Translocation in HIV-Infected Patients with Poor Immunological Response on HAART. Her work has been presented at national and international conferences; in June 2011, she served as Rapporteur (Basic Science) of the 6th International AIDS Conference in Rome, Italy.

Camilla Tincati, M.D., received her medical training at the University of Milan, Italy. She is specialized in Infectious Diseases and currently works at the Department of Health Sciences, San Paolo Hospital, University of Milan, Italy. As a clinician, she focuses on the management of HIV-infected patients. Her research work aims at the understanding of the pathogenic mechanisms underlying HIV disease progression, immunologic recovery on HAART, and noninfectious comorbidities in HIV-infected patients. In particular, her field of expertise is the study of the homeostatic T-cell imbalances in the pathogenesis of HIV infection and response to antiretroviral treatment. Her work has been published in peer-reviewed journals and presented at international conferences.

Guido Silvestri received his M.D. in Ancona, Italy, in 1987. He completed Residency in Internal Medicine and Clinical Immunology (Florence, Italy, 1990) and Pathology (University of Pennsylvania, 2001) and postdoctoral training between 1993 and 1999 at the IRCM (Montreal, Canada), the NIH (Bethesda, MD), and Emory University. He is currently a Georgia Research Alliance Eminent Scholar in Comparative Pathology and Professor of Pathology and Laboratory Medicine at the Emory University School of Medicine, where he also serves as Chair of the Division of Microbiology and Immunology at the Yerkes National Primate Research Center. Dr. Silvestri has been involved in studies of HIV pathogenesis and vaccines using nonhuman primates since 1993 and has authored or coauthored 148 original peer-reviewed publications in this field, including the highest-impact journals (Nature, Science, and Cell, etc.). He is an Editor of the Journal of Immunology, PLoS Pathogens, Journal of Virology, Clinical Microbiology Reviews, and Journal of Infectious Diseases; he is a member of the IAS, of the CROI organizing committee, and of the ANRS scientific committee. His work has been presented at international meetings. In June 2011, he was Cochair of the 6th International AIDS Conference in Rome, Italy.
REFERENCES
- 1. Douek DC. 2003. Disrupting T-cell homeostasis: how HIV-1 infection causes disease. AIDS Rev. 5:172–177 [PubMed] [Google Scholar]
- 2. Cao J, Park IW, Cooper A, Sodroski J. 1996. Molecular determinants of acute single-cell lysis by human immunodeficiency virus type 1. J. Virol. 70:1340–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lenardo MJ, Angleman SB, Bounkeua V, Dimas J, Duvall MG, Graubard MB, Hornung F, Selkirk MC, Speirs CK, Trageser C, Orenstein JO, Bolton DL. 2002. Cytopathic killing of peripheral blood CD4(+) T lymphocytes by human immunodeficiency virus type 1 appears necrotic rather than apoptotic and does not require env. J. Virol. 76:5082–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McMichael AJ, Rowland-Jones SL. 2001. Cellular immune responses to HIV. Nature 410:980–987 [DOI] [PubMed] [Google Scholar]
- 5. Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narváez AB, Hunt P, Martin JN, Kahn JO, Levy J, McGrath MS, Hecht FM. 2004. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 104:942–947 [DOI] [PubMed] [Google Scholar]
- 6. Lane HC, Masur H, Edgar LC, Whalen G, Rook AH, Fauci AS. 1983. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N. Engl. J. Med. 309:453–458 [DOI] [PubMed] [Google Scholar]
- 7. Hellerstein M, Hanley MB, Cesar D, Siler S, Papageorgopoulos C, Wieder E, Schmidt D, Hoh R, Neese R, Macallan D, Deeks S, McCune JM. 1999. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat. Med. 5:83–89 [DOI] [PubMed] [Google Scholar]
- 8. Valdez H, Lederman MM. 1997. Cytokines and cytokine therapies in HIV infection. AIDS Clin. Rev. 1997–1998:187–228 [PubMed] [Google Scholar]
- 9. Douek DC, Roederer M, Koup RA. 2009. Emerging concepts in the immunopathogenesis of AIDS. Annu. Rev. Med. 60:471–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grossman Z, Feinberg MB, Paul WE. 1998. Multiple modes of cellular activation and virus transmission in HIV infection: a role for chronically and latently infected cells in sustaining viral replication. Proc. Natl. Acad. Sci. U. S. A. 95:6314–6319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, Shih R, Lewis J, Wiley DJ, Phair JP, Wolinsky SM, Detels R. 1999. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J. Infect. Dis. 179:859–870 [DOI] [PubMed] [Google Scholar]
- 12. Hunt PW, Brenchley J, Sinclair E, McCune JM, Roland M, Page-Shafer K, Hsue P, Emu B, Krone M, Lampiris H, Douek D, Martin JN, Deeks SG. 2008. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J. Infect. Dis. 197:126–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, Deeks SG. 2003. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J. Infect. Dis. 187:1534–1543 [DOI] [PubMed] [Google Scholar]
- 14. Marchetti G, Gori A, Casabianca A, Magnani M, Franzetti F, Clerici M, Perno CF, Monforte A, Galli M, Meroni L. 2006. Comparative analysis of T-cell turnover and homeostatic parameters in HIV-infected patients with discordant immune-virological responses to HAART. AIDS 20:1727–1736 [DOI] [PubMed] [Google Scholar]
- 15. Paiardini M, Pandrea I, Apetrei C, Silvestri G. 2009. Lessons learned from the natural hosts of HIV-related viruses. Annu. Rev. Med. 60:485–495 [DOI] [PubMed] [Google Scholar]
- 16. Sodora DL, Silvestri G. 2008. Immune activation and AIDS pathogenesis. AIDS 22:439–446 [DOI] [PubMed] [Google Scholar]
- 17. Paiardini M, Cervasi B, Reyes-Aviles E, Micci L, Ortiz AM, Chahroudi A, Vinton C, Gordon SN, Bosinger SE, Francella N, Hallberg PL, Cramer E, Schlub T, Chan ML, Riddick NE, Collman RG, Apetrei C, Pandrea I, Else J, Munch J, Kirchhoff F, Davenport MP, Brenchley JM, Silvestri G. 2011. Low levels of SIV infection in sooty mangabey central memory CD4+ T cells are associated with limited ccr5 expression. Nat. Med. 17:830–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wolochow H, Hildebrand GJ, Lamanna C. 1966. Translocation of microorganisms across the intestinal wall of the rat: effect of microbial size and concentration. J. Infect. Dis. 116:523–528 [DOI] [PubMed] [Google Scholar]
- 19. Fuller R, Jayne-Williams DJ. 1970. Resistance of the fowl (Gallus domesticus) to invasion by its intestinal flora. I. The effect of hormonal bursectomy on the invasiveness of the intestinal microflora of the fowl. Res. Vet. Sci. 11:363–367 [PubMed] [Google Scholar]
- 20. Fuller R, Jayne-Williams DJ. 1970. Resistance of the fowl (Gallus domesticus) to invasion by its intestinal flora. II. Clearance of translocated intestinal bacteria. Res. Vet. Sci. 11:368–374 [PubMed] [Google Scholar]
- 21. Berg RD. 1995. Bacterial translocation from the gastrointestinal tract. Trends Microbiol. 3:149–154 [DOI] [PubMed] [Google Scholar]
- 22. Alexander JW, Gianotti L, Pyles T, Carey MA, Babcock GF. 1991. Distribution and survival of Escherichia coli translocating from the intestine after thermal injury. Ann. Surg. 213:558–566; discussion 566–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tancrède CH, Andremont AO. 1985. Bacterial translocation and gram-negative bacteremia in patients with hematological malignancies. J. Infect. Dis. 152:99–103 [DOI] [PubMed] [Google Scholar]
- 24. Beutler B. 2000. Tlr4: central component of the sole mammalian LPS sensor. Curr. Opin. Immunol. 12:20–26 [DOI] [PubMed] [Google Scholar]
- 25. Caradonna L, Amati L, Magrone T, Pellegrino NM, Jirillo E, Caccavo D. 2000. Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: biological and clinical significance. J. Endotoxin Res. 6:205–214 [PubMed] [Google Scholar]
- 26. Cooke KR, Gerbitz A, Crawford JM, Teshima T, Hill GR, Tesolin A, Rossignol DP, Ferrara JL. 2001. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J. Clin. Invest. 107:1581–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schietroma M, Carlei F, Cappelli S, Amicucci G. 2006. Intestinal permeability and systemic endotoxemia after laparotomic or laparoscopic cholecystectomy. Ann. Surg. 243:359–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barclay GR, Scott BB, Wright IH, Rogers PN, Smith DG, Poxton IR. 1989. Changes in anti-endotoxin-IgG antibody and endotoxaemia in three cases of gram-negative septic shock. Circ. Shock 29:93–106 [PubMed] [Google Scholar]
- 29. Anderson KV. 2000. Toll signaling pathways in the innate immune response. Curr. Opin. Immunol. 12:13–19 [DOI] [PubMed] [Google Scholar]
- 30. Liu Y, van Kruiningen HJ, West AB, Cartun RW, Cortot A, Colombel JF. 1995. Immunocytochemical evidence of Listeria, Escherichia coli, and Streptococcus antigens in Crohn's disease. Gastroenterology 108:1396–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eriguchi Y, Takashima S, Oka H, Shimoji S, Nakamura K, Uryu H, Shimoda S, Iwasaki H, Shimono N, Ayabe T, Akashi K, Teshima T. 2012. Graft-versus-host disease disrupts intestinal microbial ecology by inhibiting paneth cell production of α-defensins. Blood 120:223–231 [DOI] [PubMed] [Google Scholar]
- 32. Brenchley JM, Price DA, Douek DC. 2006. HIV disease: fallout from a mucosal catastrophe? Nat. Immunol. 7:235–239 [DOI] [PubMed] [Google Scholar]
- 33. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12:1365–1371 [DOI] [PubMed] [Google Scholar]
- 34. Guadalupe M, Reay E, Sankaran S, Prindiville T, Flamm J, McNeil A, Dandekar S. 2003. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 77:11708–11717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M. 2004. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 200:761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pelsers MM, Namiot Z, Kisielewski W, Namiot A, Januszkiewicz M, Hermens WT, Glatz JF. 2003. Intestinal-type and liver-type fatty acid-binding protein in the intestine. Tissue distribution and clinical utility. Clin. Biochem. 36:529–535 [DOI] [PubMed] [Google Scholar]
- 37. Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, Pedersen C, Ruxrungtham K, Lewin SR, Emery S, Neaton JD, Brenchley JM, Deeks SG, Sereti I, Douek DC, INSIGHT SMART Study Group 2011. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J. Infect. Dis. 203:780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mavigner M, Cazabat M, Dubois M, L'Faqihi FE, Requena M, Pasquier C, Klopp P, Amar J, Alric L, Barange K, Vinel JP, Marchou B, Massip P, Izopet J, Delobel P. 2012. Altered CD4+ T cell homing to the gut impairs mucosal immune reconstitution in treated HIV-infected individuals. J. Clin. Invest. 122:62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marchetti G, Bellistrì GM, Borghi E, Tincati C, Ferramosca S, La Francesca M, Morace G, Gori A, Monforte AD. 2008. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. AIDS 22:2035–2038 [DOI] [PubMed] [Google Scholar]
- 40. Merlini E, Bai F, Bellistrì GM, Tincati C, d'Arminio Monforte A, Marchetti G. 2011. Evidence for polymicrobic flora translocating in peripheral blood of HIV-infected patients with poor immune response to antiretroviral therapy. PLoS One 6:e18580 doi:10.1371/journal.pone.0018580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marchetti G, Cozzi-Lepri A, Merlini E, Bellistrì GM, Castagna A, Galli M, Verucchi G, Antinori A, Costantini A, Giacometti A, di Caro A, D'Arminio Monforte A, ICONA Foundation Study Group 2011. Microbial translocation predicts disease progression of HIV-infected antiretroviral-naive patients with high CD4+ cell count. AIDS 25:1385–1394 [DOI] [PubMed] [Google Scholar]
- 42. Bukh AR, Melchjorsen J, Offersen R, Jensen JM, Toft L, Støvring H, Ostergaard L, Tolstrup M, Søgaard OS. 2011. Endotoxemia is associated with altered innate and adaptive immune responses in untreated HIV-1 infected individuals. PLoS One 6:e21275 doi:10.1371/journal.pone.0021275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Papasavvas E, Pistilli M, Reynolds G, Bucki R, Azzoni L, Chehimi J, Janmey PA, DiNubile MJ, Ondercin J, Kostman JR, Mounzer KC, Montaner LJ. 2009. Delayed loss of control of plasma lipopolysaccharide levels after therapy interruption in chronically HIV-1-infected patients. AIDS 23:369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baroncelli S, Galluzzo CM, Pirillo MF, Mancini MG, Weimer LE, Andreotti M, Amici R, Vella S, Giuliano M, Palmisano L. 2009. Microbial translocation is associated with residual viral replication in HAART-treated HIV+ subjects with <50copies/ml HIV-1 RNA. J. Clin. Virol. 46:367–370 [DOI] [PubMed] [Google Scholar]
- 45. Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, Landay A, Martin J, Sinclair E, Asher AI, Deeks SG, Douek DC, Brenchley JM. 2009. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J. Infect. Dis. 199:1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kramski M, Gaeguta AJ, Lichtfuss GF, Rajasuriar R, Crowe SM, French MA, Lewin SR, Center RJ, Purcell DF. 2011. Novel sensitive real-time PCR for quantification of bacterial 16S rRNA genes in plasma of HIV-infected patients as a marker for microbial translocation. J. Clin. Microbiol. 49:3691–3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nixon DE, Landay AL. 2010. Biomarkers of immune dysfunction in HIV. Curr. Opin. HIV AIDS 5:498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fukui H, Brauner B, Bode JC, Bode C. 1989. Chromogenic endotoxin assay in plasma. Selection of plasma pretreatment and production of standard curves. J. Clin. Chem. Clin. Biochem. 27:941–946 [DOI] [PubMed] [Google Scholar]
- 49. Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral JP, Raoult D. 2000. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J. Clin. Microbiol. 38:3623–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Janda JM, Abbott SL. 2007. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45:2761–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Amar J, Fauvel J, Drouet L, Ruidavets JB, Perret B, Chamontin B, Boccalon H, Ferrieres J. 2006. Interleukin 6 is associated with subclinical atherosclerosis: a link with soluble intercellular adhesion molecule 1. J. Hypertens. 24:1083–1088 [DOI] [PubMed] [Google Scholar]
- 52. Ferri E, Novati S, Casiraghi M, Sambri V, Genco F, Gulminetti R, Bandi C. 2010. Plasma levels of bacterial DNA in HIV infection: the limits of quantitative polymerase chain reaction. J. Infect. Dis. 202:176–177; author reply 178 [DOI] [PubMed] [Google Scholar]
- 53. Böttger EC. 1990. Frequent contamination of Taq polymerase with DNA. Clin. Chem. 36:1258–1259 [PubMed] [Google Scholar]
- 54. Corless CE, Guiver M, Borrow R, Edwards-Jones V, Kaczmarski EB, Fox AJ. 2000. Contamination and sensitivity issues with a real-time universal 16S rRNA PCR. J. Clin. Microbiol. 38:1747–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Handschur M, Karlic H, Hertel C, Pfeilstöcker M, Haslberger AG. 2009. Preanalytic removal of human DNA eliminates false signals in general 16s rDNA PCR monitoring of bacterial pathogens in blood. Comp. Immunol. Microbiol. Infect. Dis. 32:207–219 [DOI] [PubMed] [Google Scholar]
- 56. Abdurahman A, Barqasho B, Nowak P, Cuong DD, Amogne W, Troseid M, Vesterbacka J, Sönnerborg A. 2011. Elevated flagellin-specific antibodies during HIV-1 infection, abstr 313. Abstr. 18th Conf. Retroviruses Opportun. Infect., Boston, MA [Google Scholar]
- 57. Barqasho B, Abdurahman A, Nowak P, Vesterbacka J, Andersson LM, Svärd J, Trosseid M, Gisslén M, Sönnerborg A. 2012. Longitudinal analysis of microbial translocation markers in patients on efavirenz or lopinavir/r based antiretroviral therapy, abstr 836. Abstr. 19th Conf. Retroviruses Opportun. Infect., Seattle, WA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Giorgi JV, Lyles RH, Matud JL, Yamashita TE, Mellors JW, Hultin LE, Jamieson BD, Margolick JB, Rinaldo CR, Phair JP, Detels R, Multicenter AIDS Cohort Study 2002. Predictive value of immunologic and virologic markers after long or short duration of HIV-1 infection. J. Acquir. Immune Defic. Syndr. 29:346–355 [DOI] [PubMed] [Google Scholar]
- 59. Ciccone EJ, Read SW, Mannon PJ, Yao MD, Hodge JN, Dewar R, Chairez CL, Proschan MA, Kovacs JA, Sereti I. 2010. Cycling of gut mucosal CD4+ T cells decreases after prolonged anti-retroviral therapy and is associated with plasma LPS levels. Mucosal Immunol. 3:172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nowroozalizadeh S, Månsson F, da Silva Z, Repits J, Dabo B, Pereira C, Biague A, Albert J, Nielsen J, Aaby P, Fenyö EM, Norrgren H, Holmgren B, Jansson M. 2010. Microbial translocation correlates with the severity of both HIV-1 and HIV-2 infections. J. Infect. Dis. 201:1150–1154 [DOI] [PubMed] [Google Scholar]
- 61. Klatt NR, Harris LD, Vinton CL, Sung H, Briant JA, Tabb B, Morcock D, McGinty JW, Lifson JD, Lafont BA, Martin MA, Levine AD, Estes JD, Brenchley JM. 2010. Compromised gastrointestinal integrity in pigtail macaques is associated with increased microbial translocation, immune activation, and IL-17 production in the absence of SIV infection. Mucosal Immunol. 3:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cassol E, Malfeld S, Mahasha P, van der Merwe S, Cassol S, Seebregts C, Alfano M, Poli G, Rossouw T. 2010. Persistent microbial translocation and immune activation in HIV-1-infected South Africans receiving combination antiretroviral therapy. J. Infect. Dis. 202:723–733 [DOI] [PubMed] [Google Scholar]
- 63. Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, Barclay GR, Smedley J, Pung R, Oliveira KM, Hirsch VM, Silvestri G, Douek DC, Miller CJ, Haase AT, Lifson J, Brenchley JM. 2010. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 6:e1001052 doi:10.1371/journal.ppat.1001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ellis CL, Ma ZM, Mann SK, Li CS, Wu J, Knight TH, Yotter T, Hayes TL, Maniar AH, Troia-Cancio PV, Overman HA, Torok NJ, Albanese A, Rutledge JC, Miller CJ, Pollard RB, Asmuth DM. 2011. Molecular characterization of stool microbiota in HIV-infected subjects by panbacterial and order-level 16S ribosomal DNA (rDNA) quantification and correlations with immune activation. J. Acquir. Immune Defic. Syndr. 57:363–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. d'Ettorre G, Paiardini M, Zaffiri L, Andreotti M, Ceccarelli G, Rizza C, Indinnimeo M, Vella S, Mastroianni CM, Silvestri G, Vullo V. 2011. HIV persistence in the gut mucosa of HIV-infected subjects undergoing antiretroviral therapy correlates with immune activation and increased levels of LPS. Curr. HIV Res. 9:148–153 [DOI] [PubMed] [Google Scholar]
- 66. Chege D, Sheth PM, Kain T, Kim CJ, Kovacs C, Loutfy M, Halpenny R, Kandel G, Chun TW, Ostrowski M, Kaul R, Toronto Mucosal Immunology Group 2011. Sigmoid Th17 populations, the HIV latent reservoir, and microbial translocation in men on long-term antiretroviral therapy. AIDS 25:741–749 [DOI] [PubMed] [Google Scholar]
- 67. Funderburg N, Luciano AA, Jiang W, Rodriguez B, Sieg SF, Lederman MM. 2008. Toll-like receptor ligands induce human T cell activation and death, a model for HIV pathogenesis. PLoS One 3:e1915 doi:10.1371/journal.pone.0001915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tincati C, Bellistrì GM, Ancona G, Merlini E, d'Arminio Monforte A, Marchetti G. 2012. Role of in vitro stimulation with lipopolysaccharide on T-cell activation in HIV-infected antiretroviral-treated patients. Clin. Dev. Immunol. 2012:935425 doi:10.1155/2012/935425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Merlini E, Tincati C, Bellistri GM, Bai F, d'Arminio Monforte A, Marchetti G. 2012. Stimulation with microbial components accounts for different levels of activation and HIV production in T cells and macrophages according to the severity of CD4 impairment, abstr 5. Abstr. 8th Int. Workshop HIV Cells Macrophage/Dendritic Lineage Other Reservoirs Pathogenic Ther. Implications, Stresa, Italy [Google Scholar]
- 70. Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434:1093–1097 [DOI] [PubMed] [Google Scholar]
- 71. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC. 2004. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 200:749–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA. 1998. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280:427–431 [DOI] [PubMed] [Google Scholar]
- 73. Kewenig S, Schneider T, Hohloch K, Lampe-Dreyer K, Ullrich R, Stolte N, Stahl-Hennig C, Kaup FJ, Stallmach A, Zeitz M. 1999. Rapid mucosal CD4(+) T-cell depletion and enteropathy in simian immunodeficiency virus-infected rhesus macaques. Gastroenterology 116:1115–1123 [DOI] [PubMed] [Google Scholar]
- 74. Smit-McBride Z, Mattapallil JJ, McChesney M, Ferrick D, Dandekar S. 1998. Gastrointestinal T lymphocytes retain high potential for cytokine responses but have severe CD4(+) T-cell depletion at all stages of simian immunodeficiency virus infection compared to peripheral lymphocytes. J. Virol. 72:6646–6656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Verhoeven D, Sankaran S, Silvey M, Dandekar S. 2008. Antiviral therapy during primary simian immunodeficiency virus infection fails to prevent acute loss of CD4+ T cells in gut mucosa but enhances their rapid restoration through central memory T cells. J. Virol. 82:4016–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sankaran S, George MD, Reay E, Guadalupe M, Flamm J, Prindiville T, Dandekar S. 2008. Rapid onset of intestinal epithelial barrier dysfunction in primary human immunodeficiency virus infection is driven by an imbalance between immune response and mucosal repair and regeneration. J. Virol. 82:538–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li Q, Estes JD, Duan L, Jessurun J, Pambuccian S, Forster C, Wietgrefe S, Zupancic M, Schacker T, Reilly C, Carlis JV, Haase AT. 2008. Simian immunodeficiency virus-induced intestinal cell apoptosis is the underlying mechanism of the regenerative enteropathy of early infection. J. Infect. Dis. 197:420–429 [DOI] [PubMed] [Google Scholar]
- 78. Gordon SN, Klatt NR, Bosinger SE, Brenchley JM, Milush JM, Engram JC, Dunham RM, Paiardini M, Klucking S, Danesh A, Strobert EA, Apetrei C, Pandrea IV, Kelvin D, Douek DC, Staprans SI, Sodora DL, Silvestri G. 2007. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J. Immunol. 179:3026–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pandrea IV, Gautam R, Ribeiro RM, Brenchley JM, Butler IF, Pattison M, Rasmussen T, Marx PA, Silvestri G, Lackner AA, Perelson AS, Douek DC, Veazey RS, Apetrei C. 2007. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J. Immunol. 179:3035–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Paiardini M, Frank I, Pandrea I, Apetrei C, Silvestri G. 2008. Mucosal immune dysfunction in AIDS pathogenesis. AIDS Rev. 10:36–46 [PubMed] [Google Scholar]
- 81. Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, Scheinberg P, Price DA, Hage CA, Kholi LM, Khoruts A, Frank I, Else J, Schacker T, Silvestri G, Douek DC. 2008. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 112:2826–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brenchley JM, Knox KS, Asher AI, Price DA, Kohli LM, Gostick E, Hill BJ, Hage CA, Brahmi Z, Khoruts A, Twigg HL, Schacker TW, Douek DC. 2008. High frequencies of polyfunctional HIV-specific T cells are associated with preservation of mucosal CD4 T cells in bronchoalveolar lavage. Mucosal Immunol. 1:49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cecchinato V, Trindade CJ, Laurence A, Heraud JM, Brenchley JM, Ferrari MG, Zaffiri L, Tryniszewska E, Tsai WP, Vaccari M, Parks RW, Venzon D, Douek DC, O'Shea JJ, Franchini G. 2008. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol. 1:279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nigam P, Kwa S, Velu V, Amara RR. 2011. Loss of IL-17-producing CD8 T cells during late chronic stage of pathogenic simian immunodeficiency virus infection. J. Immunol. 186:745–753 [DOI] [PubMed] [Google Scholar]
- 85. Reeves RK, Rajakumar PA, Evans TI, Connole M, Gillis J, Wong FE, Kuzmichev YV, Carville A, Johnson RP. 2011. Gut inflammation and indoleamine deoxygenase inhibit IL-17 production and promote cytotoxic potential in NKp44+ mucosal NK cells during SIV infection. Blood 118:3321–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Klatt NR, Estes JD, Sun X, Ortiz AM, Barber JS, Harris LD, Cervasi B, Yokomizo LK, Pan L, Vinton CL, Tabb B, Canary LA, Dang Q, Hirsch VM, Alter G, Belkaid Y, Lifson JD, Silvestri G, Milner JD, Paiardini M, Haddad EK, Brenchley JM. 2012. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. 5:646–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, Mold J, Swainson L, Barbour JD, Baskin CR, Palermo R, Pandrea I, Miller CJ, Katze MG, McCune JM. 2009. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 5:e1000295 doi:10.1371/journal.ppat.1000295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kao CY, Chen Y, Thai P, Wachi S, Huang F, Kim C, Harper RW, Wu R. 2004. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. J. Immunol. 173:3482–3491 [DOI] [PubMed] [Google Scholar]
- 89. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203:2271–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kolls JK, Lindén A. 2004. Interleukin-17 family members and inflammation. Immunity 21:467–476 [DOI] [PubMed] [Google Scholar]
- 91. Pandrea I, Gaufin T, Brenchley JM, Gautam R, Monjure C, Gautam A, Coleman C, Lackner AA, Ribeiro RM, Douek DC, Apetrei C. 2008. Cutting edge: experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J. Immunol. 181:6687–6691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pandrea I, Cornell E, Wilson C, Ribeiro RM, Ma D, Kristoff J, Xu C, Haret-Richter GS, Trichel A, Apetrei C, Landay A, Tracy R. 2012. Coagulation biomarkers predict disease progression in SIV-infected nonhuman primates. Blood 120:1357–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sharpstone D, Neild P, Crane R, Taylor C, Hodgson C, Sherwood R, Gazzard B, Bjarnason I. 1999. Small intestinal transit, absorption, and permeability in patients with AIDS with and without diarrhoea. Gut 45:70–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Clayton F, Kapetanovic S, Kotler DP. 2001. Enteric microtubule depolymerization in HIV infection: a possible cause of HIV-associated enteropathy. AIDS 15:123–124 [DOI] [PubMed] [Google Scholar]
- 95. Kapembwa MS, Fleming SC, Sewankambo N, Serwadda D, Lucas S, Moody A, Griffin GE. 1991. Altered small-intestinal permeability associated with diarrhoea in human-immunodeficiency-virus-infected Caucasian and African subjects. Clin. Sci. (Lond.) 81:327–334 [DOI] [PubMed] [Google Scholar]
- 96. Kotler DP, Gaetz HP, Lange M, Klein EB, Holt PR. 1984. Enteropathy associated with the acquired immunodeficiency syndrome. Ann. Intern. Med. 101:421–428 [DOI] [PubMed] [Google Scholar]
- 97. Estes JD, Gordon SN, Zeng M, Chahroudi AM, Dunham RM, Staprans SI, Reilly CS, Silvestri G, Haase AT. 2008. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J. Immunol. 180:6798–6807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tlaskalová-Hogenová H, Stepánková R, Hudcovic T, Tucková L, Cukrowska B, Lodinová-Zádníková R, Kozáková H, Rossmann P, Bártová J, Sokol D, Funda DP, Borovská D, Reháková Z, Sinkora J, Hofman J, Drastich P, Kokesová A. 2004. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol. Lett. 93:97–108 [DOI] [PubMed] [Google Scholar]
- 99. Umesaki Y, Okada Y, Matsumoto S, Imaoka A, Setoyama H. 1995. Segmented filamentous bacteria are indigenous intestinal bacteria that activate intraepithelial lymphocytes and induce MHC class II molecules and fucosyl asialo GM1 glycolipids on the small intestinal epithelial cells in the ex-germ-free mouse. Microbiol. Immunol. 39:555–562 [DOI] [PubMed] [Google Scholar]
- 100. Brenchley JM, Douek DC. 2012. Microbial translocation across the GI tract. Annu. Rev. Immunol. 30:149–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, Liu Z, Lozupone CA, Hamady M, Knight R, Bushman FD. 2008. The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog. 4:e20 doi:10.1371/journal.ppat.0040020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gori A, Tincati C, Rizzardini G, Torti C, Quirino T, Haarman M, Ben Amor K, van Schaik J, Vriesema A, Knol J, Marchetti G, Welling G, Clerici M. 2008. Early impairment of gut function and gut flora supporting a role for alteration of gastrointestinal mucosa in human immunodeficiency virus pathogenesis. J. Clin. Microbiol. 46:757–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tøn H, Brandsnes, Dale S, Holtlund J, Skuibina E, Schjønsby H, Johne B. 2000. Improved assay for fecal calprotectin. Clin. Chim. Acta 292:41–54 [DOI] [PubMed] [Google Scholar]
- 104. Vaarala O, Atkinson MA, Neu J. 2008. The “perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes 57:2555–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. 2010. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328:228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lin YP, Thibodeaux CH, Peña JA, Ferry GD, Versalovic J. 2008. Probiotic Lactobacillus reuteri suppress proinflammatory cytokines via c-Jun. Inflamm. Bowel Dis. 14:1068–1083 [DOI] [PubMed] [Google Scholar]
- 107. Neaton JD, Neuhaus J, Emery S. 2010. Soluble biomarkers and morbidity and mortality among people infected with HIV: summary of published reports from 1997 to 2010. Curr. Opin. HIV AIDS 5:480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Redd AD, Dabitao D, Bream JH, Charvat B, Laeyendecker O, Kiwanuka N, Lutalo T, Kigozi G, Tobian AA, Gamiel J, Neal JD, Oliver AE, Margolick JB, Sewankambo N, Reynolds SJ, Wawer MJ, Serwadda D, Gray RH, Quinn TC. 2009. Microbial translocation, the innate cytokine response, and HIV-1 disease progression in Africa. Proc. Natl. Acad. Sci. U. S. A. 106:6718–6723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Redd AD, Eaton KP, Kong X, Laeyendecker O, Lutalo T, Wawer MJ, Gray RH, Serwadda D, Quinn TC, Rakai Health Sciences Program 2010. C-reactive protein levels increase during HIV-1 disease progression in Rakai, Uganda, despite the absence of microbial translocation. J. Acquir. Immune Defic. Syndr. 54:556–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. George MD, Sankaran S, Reay E, Gelli AC, Dandekar S. 2003. High-throughput gene expression profiling indicates dysregulation of intestinal cell cycle mediators and growth factors during primary simian immunodeficiency virus infection. Virology 312:84–94 [DOI] [PubMed] [Google Scholar]
- 111. Stone JD, Heise CC, Miller CJ, Halsted CH, Dandekar S. 1994. Development of malabsorption and nutritional complications in simian immunodeficiency virus-infected rhesus macaques. AIDS 8:1245–1256 [DOI] [PubMed] [Google Scholar]
- 112. Dandekar S. 2007. Pathogenesis of HIV in the gastrointestinal tract. Curr. HIV/AIDS Rep. 4:10–15 [DOI] [PubMed] [Google Scholar]
- 113. Zoufaly A, an der Heiden M, Kollan C, Bogner JR, Fätkenheuer G, Wasmuth JC, Stoll M, Hamouda O, van Lunzen J, ClinSurv Study Group 2011. Clinical outcome of HIV-infected patients with discordant virological and immunological response to antiretroviral therapy. J. Infect. Dis. 203:364–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Loutfy MR, Genebat M, Moore D, Raboud J, Chan K, Antoniou T, Milan D, Shen A, Klein MB, Cooper C, Machouf N, Rourke SB, Rachlis A, Tsoukas C, Montaner JS, Walmsley SL, Smieja M, Bayoumi A, Mills E, Hogg RS, CANOC Collaboration 2010. A CD4+ cell count <200 cells per cubic millimeter at 2 years after initiation of combination antiretroviral therapy is associated with increased mortality in HIV-infected individuals with viral suppression. J. Acquir. Immune Defic. Syndr. 55:451–459 [DOI] [PubMed] [Google Scholar]
- 115. Deeks SG. 2011. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 62:141–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D. 2008. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One 3:e2516 doi:10.1371/journal.pone.0002516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kamat A, Lyons JL, Misra V, Uno H, Morgello S, Singer EJ, Gabuzda D. 2012. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. J. Acquir. Immune Defic. Syndr. 60:234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ellis R, Gelman B, Letendre S, Clifford D, Simpson D, Marra C, McArthur J, McCutchan A, Grant I, the CHARTER Group 2012. Soluble CD14 in cerebrospinal fluid in HIV sensory neuropathy, abstr 496. Abstr. 19th Conf. Retroviruses Opportun. Infect., Seattle, WA [Google Scholar]
- 119. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, Estes JD, Dodiya HB, Keshavarzian A. 2011. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson's disease. PLoS One 6:e28032 doi:10.1371/journal.pone.0028032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jirillo E, Caccavo D, Magrone T, Piccigallo E, Amati L, Lembo A, Kalis C, Gumenscheimer M. 2002. The role of the liver in the response to LPS: experimental and clinical findings. J. Endotoxin Res. 8:319–327 [DOI] [PubMed] [Google Scholar]
- 121. Jerala R. 2007. Structural biology of the LPS recognition. Int. J. Med. Microbiol. 297:353–363 [DOI] [PubMed] [Google Scholar]
- 122. Caradonna L, Mastronardi ML, Magrone T, Cozzolongo R, Cuppone R, Manghisi OG, Caccavo D, Pellegrino NM, Amoroso A, Jirillo E, Amati L. 2002. Biological and clinical significance of endotoxemia in the course of hepatitis C virus infection. Curr. Pharm. Des. 8:995–1005 [DOI] [PubMed] [Google Scholar]
- 123. Balagopal A, Philp FH, Astemborski J, Block TM, Mehta A, Long R, Kirk GD, Mehta SH, Cox AL, Thomas DL, Ray SC. 2008. Human immunodeficiency virus-related microbial translocation and progression of hepatitis C. Gastroenterology 135:226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Thurman RG. 1998. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am. J. Physiol. 275:G605–G611 [DOI] [PubMed] [Google Scholar]
- 125. Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. 2003. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 37:1043–1055 [DOI] [PubMed] [Google Scholar]
- 126. Rubio-Tapia A, Murray JA. 2007. The liver in celiac disease. Hepatology 46:1650–1658 [DOI] [PubMed] [Google Scholar]
- 127. Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. 1997. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood 90:3204–3213 [PubMed] [Google Scholar]
- 128. Feld JJ, Meddings J, Heathcote EJ. 2006. Abnormal intestinal permeability in primary biliary cirrhosis. Dig. Dis. Sci. 51:1607–1613 [DOI] [PubMed] [Google Scholar]
- 129. Marchetti G, Nasta P, Bai F, Gatti F, Bellistrì GM, Tincati C, Borghi F, Carosi G, Puoti M, Monforte A. 2012. Circulating sCD14 is associated with virological response to pegylated-interferon-alpha/ribavirin treatment in HIV/HCV co-infected patients. PLoS One 7:e32028 doi:10.1371/journal.pone.0032028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Sandler NG, Koh C, Roque A, Eccleston JL, Siegel RB, Demino M, Kleiner DE, Deeks SG, Liang TJ, Heller T, Douek DC. 2011. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology 141:1220–1230.e3 doi:10.1053/j.gastro.2011.06.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. McGovern BH. 2007. Hepatitis C in the HIV-infected patient. J. Acquir. Immune Defic. Syndr. 45(Suppl 2):S47–S56; discussion S66–S67. doi:10.1097/QAI.0b013e318068d190 [DOI] [PubMed] [Google Scholar]
- 132. Gonzalez VD, Falconer K, Blom KG, Reichard O, Mørn B, Laursen AL, Weis N, Alaeus A, Sandberg JK. 2009. High levels of chronic immune activation in the T-cell compartments of patients coinfected with hepatitis C virus and human immunodeficiency virus type 1 and on highly active antiretroviral therapy are reverted by alpha interferon and ribavirin treatment. J. Virol. 83:11407–11411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, Sulkowski M, McHutchison JG, Goldstein DB. 2009. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401 [DOI] [PubMed] [Google Scholar]
- 134. Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, Bochud M, Battegay M, Bernasconi E, Borovicka J, Colombo S, Cerny A, Dufour JF, Furrer H, Günthard HF, Heim M, Hirschel B, Malinverni R, Moradpour D, Müllhaupt B, Witteck A, Beckmann JS, Berg T, Bergmann S, Negro F, Telenti A, Bochud PY, Swiss Hepatitis C Cohort Study, Swiss HIV Cohort Study 2010. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology 138:1338–1345.e7 doi:10.1053/j.gastro.2009.12.056 [DOI] [PubMed] [Google Scholar]
- 135. Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, Bassendine M, Spengler U, Dore GJ, Powell E, Riordan S, Sheridan D, Smedile A, Fragomeli V, Müller T, Bahlo M, Stewart GJ, Booth DR, George J. 2009. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 41:1100–1104 [DOI] [PubMed] [Google Scholar]
- 136. Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, Nakagawa M, Korenaga M, Hino K, Hige S, Ito Y, Mita E, Tanaka E, Mochida S, Murawaki Y, Honda M, Sakai A, Hiasa Y, Nishiguchi S, Koike A, Sakaida I, Imamura M, Ito K, Yano K, Masaki N, Sugauchi F, Izumi N, Tokunaga K, Mizokami M. 2009. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 41:1105–1109 [DOI] [PubMed] [Google Scholar]
- 137. Dill MT, Duong FH, Vogt JE, Bibert S, Bochud PY, Terracciano L, Papassotiropoulos A, Roth V, Heim MH. 2011. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology 140:1021–1031 [DOI] [PubMed] [Google Scholar]
- 138. Wurfel MM, Hailman E, Wright SD. 1995. Soluble CD14 acts as a shuttle in the neutralization of lipopolysaccharide (LPS) by LPS-binding protein and reconstituted high density lipoprotein. J. Exp. Med. 181:1743–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Marchetti G, Cozzi-Lepri A, Bellistri GM, Merlini E, Ceccherini-Silberstein F, De Luca A, Antinori A, Castagna A, Puoti M, d'Arminio Monforte A, the Icona Fndn Study Group 2011. Role of immune activation and microbial translocation in liver disease progression in HIV/HCV co-infected antiretroviral-naïve patients with high CD4+ count, abstr 936. Abstr. 18th Conf. Retroviruses Opportun. Infect., Boston, MA [Google Scholar]
- 140. Ross AC, Rizk N, O'Riordan MA, Dogra V, El-Bejjani D, Storer N, Harrill D, Tungsiripat M, Adell J, McComsey GA. 2009. Relationship between inflammatory markers, endothelial activation markers, and carotid intima-media thickness in HIV-infected patients receiving antiretroviral therapy. Clin. Infect. Dis. 49:1119–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Calmy A, Gayet-Ageron A, Montecucco F, Nguyen A, Mach F, Burger F, Ubolyam S, Carr A, Ruxungtham K, Hirschel B, Ananworanich J, STACCATO Study Group 2009. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 23:929–939 [DOI] [PubMed] [Google Scholar]
- 142. Mangili A, Polak JF, Quach LA, Gerrior J, Wanke CA. 2011. Markers of atherosclerosis and inflammation and mortality in patients with HIV infection. Atherosclerosis 214:468–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Stoll LL, Denning GM, Weintraub NL. 2004. Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24:2227–2236 [DOI] [PubMed] [Google Scholar]
- 144. Libby P. 2002. Inflammation in atherosclerosis. Nature 420:868–874 [DOI] [PubMed] [Google Scholar]
- 145. Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F, Muggeo M, Xu Q, Wick G, Poewe W, Willeit J. 2001. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation 103:1064–1070 [DOI] [PubMed] [Google Scholar]
- 146. Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, Willeit J. 1999. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck study. J. Am. Coll. Cardiol. 34:1975–1981 [DOI] [PubMed] [Google Scholar]
- 147. Merlini E, Bai F, Suardi E, Bellistrì GM, Negri S, Cristina M, Bini T, d'Arminio Monforte A, Marchetti G. 2011. Microbial translocation-induced immune activation associates to atherosclerosis in CART-treated HIV+ patients, abstr 309. Abstr. 18th Conf. Retroviruses Opportun. Infect., Boston, MA [Google Scholar]
- 148. Kelesidis T, Yang O, Kendall M, Hodis H, Currier J. 2012. Biomarkers of microbial translocation and macrophage activation are associated with progression of atherosclerosis in HIV infection: ACTG NWCS 332/A5078 Study, abstr 122. Abstr. 19th Conf. Retroviruses Opportun. Infect., Seattle, WA [Google Scholar]
- 149. Irvine SL, Hummelen R, Hekmat S, Looman CW, Habbema JD, Reid G. 2010. Probiotic yogurt consumption is associated with an increase of CD4 count among people living with HIV/AIDS. J. Clin. Gastroenterol. 44:e201–e205 doi:10.1097/MCG.0b013e3181d8fba8 [DOI] [PubMed] [Google Scholar]
- 150. Klatt N, Canary L, Sun X, Vinton C, Perkins M, Hazuda D, Lifson J, Haddad E, Estes J, Brenchley J. 2012. Probiotic supplementation of ARV treatment during SIV infection of pigtail macaques results in enhanced GI tract CD4+ T cell frequency and immunological function, abstr 95. Abstr. 19th Conf. Retroviruses Opportun. Infect., Seattle, WA [Google Scholar]
- 151. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. 2008. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118:534–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Sereti I, Estes J, Thompson W, Fischl M, Croughs T, Beq S, Yao M, Boulassel R, Lederman M, Routy JP. 2012. Gut mucosa T lymphocyte restoration in chronically HIV+ patients treated with recombinant interleukin-7, abstr 94. Abstr. 19th Conf. Retroviruses Opportun. Infect., Seattle, WA [Google Scholar]
- 153. Hauser AB, Azevedo IR, Gonçalves S, Stinghen A, Aita C, Pecoits-Filho R. 2010. Sevelamer carbonate reduces inflammation and endotoxemia in an animal model of uremia. Blood Purif. 30:153–158 [DOI] [PubMed] [Google Scholar]