

Abstract
New drugs for the treatment of human African trypanosomiasis are urgently needed. A number of 2-aminopyrazines/2-aminopyridines were identified as promising leads following a focused screen of 5,500 compounds for Trypanosoma brucei subsp. brucei viability. Described compounds are trypanotoxic in the submicromolar range and show comparably low cytotoxicity on representative mammalian cell lines. Specifically, 6-([6-fluoro-3,4-dihydro-2H-1-benzopyran-4-yl)]oxy)-N-(piperidin-4-yl)pyrazin-2-amine (CBK201352) is trypanotoxic for T. brucei subsp. brucei, T. brucei subsp. gambiense, and T. brucei subsp. rhodesiense and is nontoxic to mammalian cell lines, and in vitro preclinical assays predict promising pharmacokinetic parameters. Mice inoculated intraperitoneally (i.p.) with 25 mg/kg CBK201352 twice daily for 10 days, starting on the day of infection with T. brucei subsp. brucei, show complete clearance of parasites for more than 90 days. Thus, CBK201352 and related analogs are promising leads for the development of novel treatments for human African trypanosomiasis.
INTRODUCTION
Trypanosoma brucei is the causative agent of human African trypanosomiasis (HAT), also known as sleeping sickness, a vector-borne parasitic disease transmitted by the tsetse fly, Glossina sp. Two subspecies cause the human disease. T. brucei subsp. gambiense is prevalent in central and western Africa and causes the Gambian form of the disease with a course of months to years, while T. brucei subsp. rhodesiense causes the Rhodesian form of the disease with a faster course of weeks to months (1).
The pathophysiology of HAT as a disease is first characterized by an early hemolymphatic stage of infection. After an initial phase of local replication at the site of the tsetse fly bite, trypanosomes spread into the lymphatic system and bloodstream. Current first-line therapies of the early stage of HAT are pentamidine and/or suramin, both of which require parenteral dosing regimens.
If untreated, the disease progresses to a late meningoencephalitic stage, during which the parasites cross the blood-brain barrier (BBB) to infect the central nervous system (CNS). The latter phase causes CNS dysfunctions that, in addition to sleep disruption, are manifested as movement disturbances, neuropsychiatric symptoms, and pain. If left untreated, the infection is inevitably lethal (2, 3). Late-stage treatment of HAT relies primarily on melarsoprol and eflornithine (α-difluoromethylornithine [DFMO]) (4). Melarsoprol treatment is highly toxic; up to 5% of the second-stage patients treated with melarsoprol die of a reactive encephalopathy. Eflornithine, which displays a better safety profile, is unfortunately effective only against T. brucei subsp. gambiense. Nifurtimox-eflornithine combination therapy (NECT) has recently been approved and allows a simplified administration schedule compared to that of eflornithine monotherapy, improving the possibilities for late-stage treatment (5).
Despite continuous control efforts that have curbed the number of infected cases in the last decade, existing treatments need to be improved. Current therapy is highly dependent on the stage of infection, and drugs efficient to control the late-stage disease need to cross the blood-brain barrier to reach the parasite, presenting a challenge of exposure. Although there are therapies available for the treatment of HAT, most drugs are associated with severe side effects or are complicated to administer (4).
In addition to the currently available therapy, several new exciting lead compounds against both stages of T. brucei subspecies infection have recently been described (6–8). Many of these are, however, not effective in animal models of late-stage disease (9, 10). Until these exploratory compounds have been validated and demonstrate efficacy in human, there is a need to pursue additional alternatives.
This work describes a focused small-molecule screen for compounds affecting T. brucei subsp. brucei viability and proliferation. Identified hits in the screen were followed by testing of available analogs from our compound collection and new synthetic analogs with the aim of identifying novel chemotypes from which new therapies for HAT can be developed. The screen and follow-up studies resulted in the identification of a series of 2-aminopyrazines/2-aminopyridines as novel T. brucei subsp. brucei inhibitors, the properties of which are described here.
MATERIALS AND METHODS
Parasite cultures.
T. brucei subsp. brucei (AnTat1.1E) (an animal-infecting subspecies of T. brucei), T. brucei subsp. rhodesiense (STIB 851), T. brucei subsp. gambiense (MBA), and stable Renilla luciferase-expressing recombinant parasite T. brucei subsp. brucei (Rluc AnTat1.1E) (11, 12), kindly provided by P. Büscher (Institute of Tropical Medicine, Antwerp, Belgium), were cultured at 37°C and 5% CO2 in Iscove's modified Dulbecco's medium (IMDM; HyClone) containing 10% heat-inactivated calf serum (Invitrogen, Carlsbad, CA), 28 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 0.14% glucose, 1.5% sodium bicarbonate, 2 mM l-glutamine, 0.14 mg/ml gentamicin, 0.3 mM dithiothreitol (DTT), 1.4 mM sodium pyruvate, 0.7 mM l-cysteine, 28 μM adenosine, and 14 μM guanosine (13). T. brucei forms were harvested by centrifugation at 2,500 rpm for 6 min followed by resuspension in fresh medium. Parasites were utilized when in the log phase of growth (14).
Mammalian cell cultures.
Human MOLT4 (ATCC CRL-1582) and mouse fibroblast L-929 (ATCC CCL-1) cells were cultured at 37°C and 5% CO2 in RPMI medium (HyClone) supplemented with 10% fetal calf serum (FCS), 100 units/ml penicillin, and 0.1 mg/ml streptomycin. Cells were harvested by centrifugation at 1,500 rpm for 6 min followed by resuspension in fresh medium.
WST-1 in vitro proliferation assays and compound screen. (i) Trypanotoxicity assay.
Compounds evaluated were prepared as 10 mM stock solutions in dimethyl sulfoxide (DMSO) and diluted to the appropriate concentrations with IMDM and 10% FCS (HyClone). For dose-response evaluation, compound stocks were initially serial diluted with 100% DMSO in 11 steps from 10 mM to 0.17 μM final concentrations prior to dilution. Diluted compounds were then transferred to 96-well assay-ready plates. Maximum final DMSO concentration in each assay was 0.25%. Suspensions of trypanosomes (for T. brucei subsp. brucei, gambiense, and rhodesiense, 6,000 bloodstream T. brucei subspecies forms/well) in IMDM and 10% FCS (HyClone) were next added to each well using a multipipette, and plates were incubated at 37°C and 5% CO2 for 72 h. DMSO (0.25% final concentration) and 3′-deoxyadenosine (10 μM final concentration) were used as negative and positive controls, respectively.
(ii) Compound screen.
The following modifications to the above-described WST-1 assay were employed for the liquid handling during the screening campaign: 2 μl of 10 mM DMSO stock solutions of the 5,500 compounds was dispensed into columns 1 to 11 of round-bottomed 96-well plates (Nunc). Two microliters of a 10 mM stock solution of 3′-deoxyadenosine and 2 μl DMSO were used as positive and negative controls, respectively. The compound solutions were diluted in a two-step procedure to 10 μM with IMDM (HyClone) using a FlexDrop (PerkinElmer) for noncontact dispensing. A CybiWell liquid handling station with a 96-well head was employed to transfer 10 μl of the compound solution into a transparent 96-well assay plate (Sigma-Aldrich). Parasite suspensions (6,000 forms, 90 μl/well) were then added in accordance to the above-described in vitro trypanotoxicity assay procedure to yield a final compound concentration of 1.0 μM/well.
(iii) Mammalian cell cytotoxicity assay.
Using a multichannel pipette, 90 μl of a suspension of 104 MOLT4 cells or 5 × 103 L929 cells was seeded in 96-well transparent plates containing compound solutions prepared as indicated above. Plates were incubated at 37°C and 5% CO2 for 72 h.
For detection of cell viability in all of the above-described assays, 10 μl WST-1 reagent (Roche) was then added to each well, and the plates were incubated for 2 h at 37° and 5% CO2. Absorbance was recorded from the plates using a multiwell spectrophotometer Victor2 (PerkinElmer) at an excitation wavelength of 450 nm. For dose-response measurements, half-maximum inhibitory concentrations (IC50s) and standard errors were calculated by fitting resulting data as the log of inhibitor concentration versus normalized response with nonlinear regression analysis (variable slope) using GraphPad Prism software.
Chemistry. (i) General.
All starting materials were purchased from Sigma-Aldrich and used without further purification unless otherwise noted. Intermediates and end products were prepared as previously reported (15), besides CBK201352, which was prepared as described below. Flash chromatography was performed using silica gel 60 from Merck. Compound purity was determined by reverse-phase high-pressure liquid chromatography-coupled mass spectrometry (HPLC-MS) on an Agilent/HP 1200 system fitted with a Waters XBridge C18 3.5-μm column (3.0 by 50 mm) using acetonitrile-0.1% trifluoroacetic acid in water as mobile phases. 1H nuclear magnetic resonance (NMR) spectra were recorded using a Bruker DPX400 spectrometer (400 MHz) using dimethyl sulfoxide-d6 or methanol-d4 as the solvent.
CBK201352 was prepared as follows: tert-butyl 4-([{tert-butoxy}carbonyl][6-chloropyrazin-2-yl]amino)piperidine-1-carboxylate (1.85 g, 4.48 mmol) (15) and 6-fluoro-3,4-dihydro-2H-1-benzopyran-4-ol (1.13 g, 6.72 mmol) were dissolved in dimethyl sulfoxide (20 ml), and potassium tert-butoxide (1.01 g, 8.96 mmol) was added while being stirred at room temperature. After 1 h, the reaction mixture was poured onto 100 ml water and extracted with dichloromethane (3 × 50 ml). The organic fractions were combined, washed with brine, and then dried over magnesium sulfate and concentrated to yield 3.0 g of an oil that was dissolved in dichloromethane (100 ml). Lutidine (2.36 g, 22.0 mmol) was added to the solution, followed by the dropwise addition of trimethylsilyl trifluoromethanesulfonate (3.67 g, 17.0 mmol) while being stirred. After 1 h, saturated aqueous ammonium chloride (20 ml) was added in one portion. The pH was adjusted to 10 with 2 M aqueous sodium hydroxide solution. The organic phase was separated and the residual aqueous phase extracted with two portions of dichloromethane. Combined organic phases were washed with water, dried over anhydrous sodium sulfate, and concentrated to yield 1.9 g crude product. Flash chromatography on silica with a gradient from 100% dichloromethane to dichloromethane-5% NH3 in methanol (9:1) gave analytically pure CBK201352 (0.6 g, 33%). HPLC (UV) purity was 98%; electrospray ionization (ESI)-MS m/z [M+H]+ was 345; 1H NMR (CD3OD) δ was 1.50 to 1.66 (m, 2H), 2.07 to 2.20 (m, 2H), 2.20 to 2.32 (m, 2H), 2.77 to 2.90 (m, 2H), 3.15 to 3.25 (m, 2H), 3.93 to 4.05 (m, 1H), 4.24 to 4.35 (m, 2H), 6.15 (t, 1H), 6.81 to 6.87 (dd, 1H), 6.95 to 7.03 (m, 1H), 7.07 to 7.14 (dd, 1H), 7.26 (s, 1H), and 7.46 (s, 1H) (data are reported as follows: chemical shift, multiplicity [s, singlet; d, doublet; t, triplet; q, quartet; br, broad; m, multiplet], integration).
T. brucei subsp. brucei (Rluc) trypanocidal assay.
Serial dilutions of the studied compounds were incubated with 200 μl T. brucei subsp. brucei (Rluc), 1.25 × 106 forms/ml, in 96-well plates, and parasite luminescence was measured with respect to the untreated control between 0 and 10 h after initial drug exposure. Luminescence was measured 5 min after the addition of 10 μM substrate coelenterazine (Nanolight). Luminescence was quantified in a MicroBeta Jet luminometer (PerkinElmer, formerly Wallac) in triplicate samples at each time point and drug dilution. Half-maximum effective trypanocidal activity (EC50,killing) was calculated by fitting resulting data as the log of inhibitor concentration versus normalized response with nonlinear regression analysis (variable slope) using GraphPad Prism software.
In vitro and in silico preclinical assays. (i) General.
All samples referred to below were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a Waters XEVO TQ triple-quadrupole mass spectrometer (ESI) coupled to a Waters Acquity ultra-high-performance liquid chromatography (Waters Corp.). For chromatographic separation, a general gradient was used (5% mobile phase B to 90% over a 2-min total run) on a C18 BEH 1.7-μm column, 2 by 50 mm (Waters Corp.). Mobile phase A consisted of 5% acetonitrile and 0.1% formic acid in water, and mobile phase B consisted of 0.1% formic acid in acetonitrile. The flow rate was 0.5 ml/min. Five microliters of the sample was injected and run in MRM mode detection (CBK201352 parent/daughter ion m/z of 345.2/195.1). Plasma protein binding samples were precipitated (methanol, 4:1) and centrifuged prior to analysis. A standard curve was constructed using the spiked frozen plasma sample. Caco-2 samples were quantified using an external standard curve between 1 and 1,000 nM. Analysis of metabolic stability does not require a standard curve since the individual time points were analyzed on a relative basis. In silico analysis of CBK201352 was performed using ADMET Predictor version 5.5 (Simulations Plus, Inc.).
(ii) Plasma protein binding.
Plasma protein binding/stability of CBK201352 was performed using rapid equilibrium dialysis (RED device inserts; Thermo Scientific). Pooled human plasma, collected from two male and two female donors (nonsmoking), was provided by Uppsala Academic Hospital. In brief, 0.2 ml of the plasma test solution, spiked with 10 μM (final compound concentration) CBK201352, was dialyzed against 0.35 ml isotonic phosphate buffer (pH 7.4) for 4 h on a Kisker rotational incubator at 37°C (≈900 rpm) to achieve equilibrium (16). A separate stability test of the spiked plasma test solution was also prepared (to allow detection of drug degradation) and was incubated separately on a sealed plate at 37°C for 4 h. After incubation, the contents of each plasma (dialysis and stability test) and buffer compartment were removed and immediately frozen until analysis. The samples were analyzed by LC-MS/MS as described above.
(iii) Microsomal metabolic stability.
Pooled human or male CD-1 mouse liver microsomes (Xenotech LLC) were used with a supplemented cofactor (NADPH) to primarily facilitate CYP450 reactivity against the target compound. CBK201352 (1 μM incubation concentration) and microsomes (0.5 mg/ml incubation concentration) were diluted in 0.1 M phosphate buffer (pH 7.4), and the reaction was initiated with the addition of NADPH (1 mM incubation concentration). Control samples were precipitated prior to the addition of NADPH. The incubation times were 0, 5, 15, and 40 min, after which the reactions were quenched by the addition of ice-cold acetonitrile (40% [vol/vol] final concentration) containing warfarin as the analytical internal standard. The plate was then sealed, centrifuged, and frozen at −20°C until LC-MS/MS analysis. The intrinsic clearance was calculated using the well-stirred model (17).
(iv) Permeability.
Caco-2 membrane permeability was performed in accordance with published protocols (18). Caco-2 cell monolayers (passages 94 to 105) were grown on permeable filter support and used for the transport study on day 21 after seeding. Prior to the experiment, a drug solution of 10 μM was prepared and warmed to 37°C. The Caco-2 filters were washed with prewarmed Hanks balanced salt solution (HBSS), and thereafter the donor solution was applied on the apical side. The transport experiments were carried out at pH 6.5 in the apical chamber, reflecting the pH of the intestinal lumen, and pH 7.4 in the basolateral chamber, reflecting the pH of the blood. The experiments were performed at 37°C and with a stirring rate of 500 rpm. The receiver compartment was sampled at 15, 30, and 60 min, and at 60 min also a final sample from the donor chamber was taken in order to calculate the mass balance of the compound. Directly after the termination of the experiment, the filter inserts were washed with prewarmed HBSS, and the membrane integrity was checked. This was performed by transepithelial electrical resistance (TEER) measurement and by measurement of 14C-mannitol permeability, which is a paracellular marker used for integrity measurements. All samples were analyzed by LC-MS/MS as described above.
In vivo studies.
All experiments were conducted following protocols that received institutional authorization by the Stockholm Region's animal protection committee. Adult C57BL6J (B6) mice 8 to 10 weeks old were kept with food and water ad libitum under specific pathogen-free conditions prior to infection. Mice (four in each group) were infected intraperitoneally (i.p.) with 2,000 T. brucei subsp. brucei (Antat1.1E) bloodstream forms, and compounds were administered i.p. starting on the day of infection and consecutively once daily for 5 days (CBK3974) or twice daily for 10 days (CBK201352). A control group was infected but remained untreated. Weight and mortality were recorded every other day. Parasitemia was determined once or twice per week by microscopic examination of tail vein blood diluted in phosphate-buffered saline (PBS) in a hemocytometer. Mice with undetectable levels of parasitemia on day 90 after infection were considered cured.
RESULTS
Focused screen.
In search for novel trypanotoxic compounds, we screened a library of 5,500 compounds, selected based on diversity from the compound collection at the Laboratories for Chemical Biology at Karolinska Institutet (LCBKI) (www.cbcs.se/lcbki). The screen was performed at a 1 μM final compound concentration and resulted in a total of 33 preliminary hits showing a ≥40% reduction of T. brucei subsp. brucei proliferation based on the negative (DMSO) and positive (3′-deoxyadenosine) controls.
The half-maximum inhibitory concentrations (IC50) of preliminary hits were determined in dose-response mode in the described WST-1 assay, as well as in an alamarBlue viability assay to exclude compounds interfering with absorbance of the WST-1 reagent (results not presented). In total, 27 of the 33 preliminary positive hits were confirmed in the dose-response assay, showing submicromolar potency for inhibition of T. brucei subsp. brucei proliferation. Among these, the 2-aminopyridine CBK3987 ([3R]-1-[6-(benzyloxy)-5-(trifluoromethyl)pyridin-2-yl]-3-methylpiperazine), known previously to elicit pharmacological effects as a selective serotonin receptor 5-HT2c modulator (15), showed a trypanotoxic IC50 of 0.50 μM (compound 1a; Table 1).
Table 1.
2-Aminopyridines and 2-aminopyrazines with in vitro activity against T. brucei subsp. brucei and selectivity over mammalian cell lines
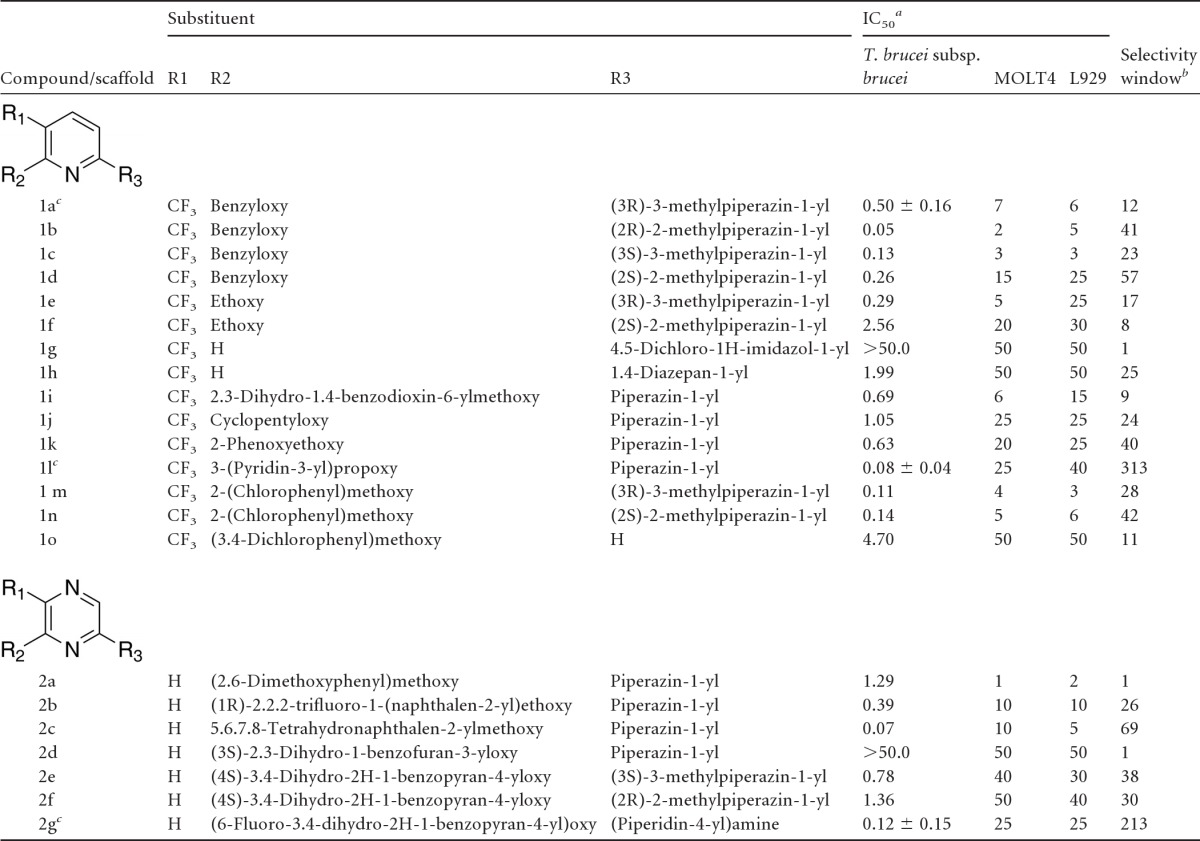
IC50s and standard deviations reported in micromolar units and based on averages from two independent dose-response evaluations, except for 1a, 1l, and 2g, which are based on averages from triplicate independent dose-response evaluations.
Selectivity window was calculated as the ratio of the IC50 against mammalian cells (using the lowest value obtained for MOLT4 or L929 cells) over the IC50 against T. brucei subsp. brucei in vitro.
1a, CBK3987; 1l, CBK3974; 2g, CBK201352.
In vitro activity of 2-aminopyrazines/2-aminopyridines on T. brucei subsp. brucei.
Based on the aminopyridine CBK3987, a number of related pyridine and pyrazine analogs from the LCBKI compound collection were evaluated in the WST-1 assay (Fig. 1). Chemical synthesis was also employed to expand compound diversity beyond what was available in our compound collection. The results from the analog evaluation are summarized in Table 1. A number of the compounds showed potent in vitro activity against T. brucei subsp. brucei proliferation (1b, 1l, 2c, and 2g), while others showed poor trypanotoxic effect (1g and 2d). Due to the complexity of parameters that may affect activity of the compounds in the whole-organism-based in vitro assay (i.e., direct pharmacological versus pharmacokinetic effects), direct structure-activity relationships were difficult to assess. Further ranking of compounds was based on the selectivity window of trypanotoxicity against T. brucei subsp. brucei versus cytotoxicity toward representative mammalian cell lines.
Fig 1.

Structures and in vitro trypanotoxic dose-response activities of CBK3974 (A), CBK3987 (B), and CBK201352 (C). Serial dilutions of compounds were incubated with T. brucei subsp. brucei for 72 h, and viability was determined with the WST-1 assay. IC50 calculations are based on averages from triplicate measurements with standard errors.
Mammalian cell cytotoxicity assay.
All compounds in Table 1 were tested for cytotoxicity using the WST-1 assay against two mammalian cell lines, L929 and MOLT4, in order to rank them for further evaluation. Many of the compounds showed a selectivity window of >20-fold over mammalian cells (see Table 1). Notably, 2-aminopyridine 1l and 2-aminopyrazine 2g (here named CBK3974 and CBK201352, respectively) showed a >200-fold selectivity window over both mammalian cell lines while retaining promising trypanotoxic effects in vitro. The original hit CBK3987 (1a), on the contrary, showed poor selectivity for trypanosomes (12-fold) compared to mammalian cell lines and was thus not evaluated further.
In vivo activity of CBK201352 in an acute infection model of HAT.
The ability of compounds CBK3974 and CBK201352, with a desirable selectivity window toward mammalian cells, to cure a murine infection with T. brucei subsp. brucei was next investigated in an acute infection model of HAT. Mice infected with T. brucei subsp. brucei were treated i.p. with 20 mg/kg CBK3974 or with 20 mg/kg CBK201352 once daily for 5 days. Mice inoculated with CBK3974 showed reduced parasitemia but did not clear the infection; higher doses than 20 mg/kg of CBK3974 were toxic (data not shown). Two out of four mice treated with 20 mg/kg CBK201352 cleared parasitemia effectively (Fig. 2B) and showed no signs of morbidity. As CBK201352 was well tolerated, treatment with this compound was escalated to 25 mg/kg twice daily for 10 days. All mice treated with this regimen were cured from infection (Fig. 2B), and no parasites were detected 90 days after the end of treatment (Fig. 2A). Neither weight loss nor other signs of morbidity were observed during treatment with CBK201352 (Fig. 2C). In contrast, all untreated animals died 30 to 40 days after infection (Fig. 2B).
Fig 2.

(A) Detectable parasitemia in mice treated with vehicle control (●) or treated with 25 mg/kg CBK201352 (■) twice daily for 10 days; (B) percent survival plots of mouse cohorts treated with 20 mg/kg (▲) once daily and 25 mg/kg (■) twice daily of CBK201352 and untreated (●); (C) weight gain of mice untreated (●) and treated (■) with 25 mg/kg CB201352 twice daily; (D) concentration- and time-dependent killing effect of CBK201352 in vitro.
In vitro activity of CBK201352 against T. brucei subsp. rhodesiense and T. brucei subsp. gambiense.
The cytotoxicity of CBK201352 for the human-pathogenic subspecies of T. brucei subsp. brucei was tested next. In vitro assessment in the described WST-1 assay showed that CBK201352 is roughly equipotent against T. brucei subsp. brucei, T. brucei subsp. gambiense, and T. brucei subsp. rhodesiense (results not shown).
In vitro/in silico preclinical evaluation of CBK201352.
Parameters predictive of the pharmacokinetics of CBK201352 were evaluated in silico (pKa, 9.36; logD, 0.87; cell permeability, 21; and solubility at pH 7.4, 83 mg/ml) and in vitro (apparent permeability [Papp] a-b, 39 ± 5 × 10−6 cm/s; Papp b-a, 66 ± 14 × 10−6 cm/s; efflux ratio, 1.7; Clint HLM, 4.4 μl/min ′ mg; Clint MLM, 7.5 μl/min ′ mg; plasma protein binding, 0.4 ± 0.03 FU; and plasma stability, 110%). ADMET Predictor (version 5.0; SimulationsPlus, Lancaster, CA) was used to predict general physicochemical parameters and permeability. The in vitro assays were performed as previously described (16, 18, 19) to give a preliminary prediction of in vivo oral absorption, clearance, and distribution of CBK201352. The Papp values obtained for CBK201352 indicated a very high passive permeability in both directions. Although a slight net efflux effect (ratio of 1.7) was acquired, it is not believed that transport across the intestinal barrier will limit systemic exposure. Human and mouse microsomal metabolism was very low with CBK201352, indicating a low first-pass hepatic extraction by oxidative enzymes. CBK201352 binding was low in human plasma (fraction unbound = 0.4), and the compound was stable in media. In summary, the in silico and in vitro pharmacokinetic parameters for CBK201352 predicted an acceptable in vivo exposure of the compound.
In vitro time-dependent trypanocidal effect of CBK201352.
As our primary WST-1 assay was unable to differentiate whether drugs are trypanotoxic or trypanocidal, we next decided to assay the direct trypanocidal activity of CBK201352 on parasites. T. brucei subsp. brucei tagged with Renilla sp. luciferase (Rluc AnTat1.1E) was used to determine the killing effect of the compound on concentrated parasite cultures (1.25 × 106/ml) at different time points after compound exposure. The recombinant Renilla enzyme is constitutively expressed, and the luminescence emitted efficiently monitors cell viability as opposed to proliferation (13).
A trypanocidal effect of CBK201352 for T. brucei subsp. brucei was initially detected 4 h after incubation with serial dilutions of the drug. Maximum trypanocidal activity was reached after 8 to 10 h of exposure to CBK201352. The maximum obtained activity of CBK201352 in this assay (EC50,killing = 2.6 ± 0.15 μM) was roughly 20-fold reduced in comparison to the antiproliferative effects of the compound.
DISCUSSION
Due to adverse side effects and toxicity of current first-line therapies, and the increasing development of resistance toward these agents (20, 21), there is a growing need for development of novel treatments of human African trypanosomiasis. Fortunately, there have been a number of interesting compounds developed and novel targets identified within the past few years which hold promise for the development of better and more efficient medicines with which to combat this devastating disease. Brand et al. have described the first T. brucei N-myrisoyl transferase (TbNMT) inhibitor DDD85646 and its related analogs as potent inhibitors of bloodstream-borne T. brucei proliferation (22). In addition, TbNMT has been extensively validated as a relevant target for the development of trypanosomiasis therapy in a number of models of HAT (9, 23).
Mercer and coworkers (10) have recently described the diaminopyrimidine SCYX-5070 as a promising candidate for HAT treatment with a number of mitogen-activated protein kinases and cdc2-related kinases as putative targets, as well as the benzoxaborole SCYX-7158 (24). Screening of nitroheterocyclics against T. brucei rediscovered fexinidazole as a drug candidate. Fexinidazole is effective for intraperitoneal treatment of the late-stage infection in mouse at 100 mg/kg body weight given over several days (7, 25).
Finally, we have previously shown that i.p. or oral treatment of the nucleoside analogue cordycepin (3′-deoxyadenosine) at doses of 2 mg/kg together with the adenosine deaminase inhibitor deoxycoformycin cures mice after parasites have penetrated into the brain in a late-stage infection model of human African trypanosomiasis (8). Although showing promise in the lab, until these and other candidates have proven effective in a clinical setting, there is a sustained need to continuously identify new chemotypes from which novel therapies can be developed.
This work describes the identification of CBK201352 and related analogs as promising lead compounds, providing a novel scaffold on which to further develop compounds with efficacious trypanocidal activity. Compounds of this class have previously been described by Nilsson as 5-HT2C serotonin receptor agonists for treatment of obesity (15). 5-HT2c agonists have been implicated as potential therapies for a number of indications, such as obesity, sexual dysfunction, epilepsy, hot flashes, and a number of central nervous system disorders (26). A number of safe, selective modulators (e.g., lorcaserin) have been evaluated in late-stage clinical trials with minimal side effects. The effect of compounds on 5-HT2C receptors in models of obesity requires CNS exposure, suggesting that the described 2-aminopyridines/2-aminopyrazines are suitable chemotypes for the development of BBB-permeable drugs.
The focused screen of 5,500 compounds from the Laboratories for Chemical Biology at Karolinska Institutet (LCBKI) at 1 μM single point concentration yielded a number of interesting clusters of compounds with potent in vitro effect on T. brucei subsp. brucei proliferation. Some of these are currently being pursued in additional studies outside the scope of this article. One of the clusters included the described initial hit CBK3987 (1a; Table 1) which was confirmed in dose-response as a potent inhibitor of parasite proliferation in vitro but did not exhibit significant selectivity versus mammalian cell cytotoxicity, determined in L929 (mouse) and MOLT4 (human) cell lines. Based on this hit, a number of related analogs available in the LCBKI compound collection were assayed for both in vitro activity against T. brucei subsp. brucei proliferation and concomitantly also mammalian cell line cytotoxicity.
The related pyridine analog CBK3974 (1l; Table 1) was identified in the described hit expansion. This molecule also exhibited potent in vitro activity and, in addition, an improved cytotoxicity window compared to those of mammalian cell lines. Based on these data, CBK3974 was advanced into in vivo studies in an acute infection model of human African trypanosomiasis. This compound, however, failed to effectively cure mice infected with T. brucei subsp. brucei even at the highest tolerated dose of 20 mg/kg, once daily. Although in vivo pharmacokinetic data for this compound is not available, it is reasonable to assume that the compound suffers from insufficient exposure in vivo. Escalating to higher doses of CBK3974 was not tolerated.
Related 2-aminopyrazine analog CBK201352, however, effectively cured all infected animals at 25 mg/kg twice daily for 10 days. Treated mice gained weight and showed no obvious adverse effects. Lowering the dose of administered CBK201352 to 20 mg/kg once daily lowered parasitemia in all animals but failed to effectively clear the infection in a fraction of treated mice. This compound showed potent in vitro activity against T. brucei subsp. brucei (IC50 = 0.12 ± 0.05 μM) and had >200-fold selectivity versus observed cytotoxicity in mammalian cell lines.
Investigations into the pharmacokinetics-related molecular properties of CBK201352 in silico and in vitro suggest that the compound has promising preclinical characteristics: high metabolic and plasma stability, high membrane permeability, and low protein binding, which predict good bioavailability of the compound. The in vivo pharmacokinetics of CBK201352 is currently being investigated in more detail.
Finally, evaluation of CBK201352 in a killing assay of T. brucei subsp. brucei indicates that its cytotoxic potency is roughly 20-fold lower than the corresponding cytostatic potency as determined by the antiproliferation assay. This may account for the relatively high dose required to cure mice in vivo. Further chemical optimization of this series of compounds may allow improvement of in vivo efficacy.
In summary, a focused compound screen and subsequent chemical optimization of hits resulted in the identification of CBK201352, which is a potent trypanocidal molecule, is active against human-pathogenic subspecies of the parasite, and efficiently cures mice in an acute in vivo model of HAT. Additionally, in silico and in vitro evaluations of preclinical properties of CBK201352 predict that the compound has high bioavailability and is a suitable lead for the development of novel therapies for HAT. CBK201352 and analogs are currently being evaluated in more advanced models of human African trypanosomiasis.
ACKNOWLEDGMENTS
T.L., B.S., R.S., and L.G.J.H. were supported by the Swedish National Research Council, Uppsala University, and Karolinska Institutet as a part of Chemical Biology Consortium Sweden (CBCS; www.cbcs.se). S.V. and M.E.R. were supported by grants from the Swedish International Development Agency, the Swedish National Research Council, and Karolinska Institutet.
JChem Cartridge was used for enabling chemical structure search and management within Oracle, JChem version 5.6.0, 2011, ChemAxon.
Footnotes
Published ahead of print 17 December 2012
REFERENCES
- 1. Bacchi CJ. 2009. Chemotherapy of human African trypanosomiasis. Interdiscip. Perspect. Infect. Dis. 2009:195040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bruzzone R, Dubois-Dalcq M, Grau GE, Griffin DE, Kristensson K. 2009. Infectious diseases of the nervous system and their impact in developing countries. PLoS Pathog. 5:e1000199 doi:10.1371/journal.ppat.1000199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Masocha W, Rottenberg ME, Kristensson K. 2007. Migration of African trypanosomes across the blood-brain barrier. Physiol. Behav. 92:110–114 [DOI] [PubMed] [Google Scholar]
- 4. Simarro PP, Franco J, Diarra A, Postigo JA, Jannin J. 2012. Update on field use of the available drugs for the chemotherapy of human African trypanosomiasis. Parasitology 139:842–846 [DOI] [PubMed] [Google Scholar]
- 5. Burri C. 2010. Chemotherapy against human African trypanosomiasis: is there a road to success? Parasitology 137:1987–1994 [DOI] [PubMed] [Google Scholar]
- 6. Jacobs RT, Plattner JJ, Keenan M. 2011. Boron-based drugs as antiprotozoals. Curr. Opin. Infect. Dis. 24:586–592 [DOI] [PubMed] [Google Scholar]
- 7. Kaiser M, Ma Bray Cal M, Bourdin Trunz B, Torreele E, Brun R. 2011. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 55:5602–5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vodnala SK, Ferella M, Lundén-Miguel H, Betha E, van Reet N, Amin DN, Oberg B, Andersson B, Kristensson K, Wigzell H, Rottenberg ME. 2009. Preclinical assessment of the treatment of second-stage African trypanosomiasis with cordycepin and deoxycoformycin. PLoS Negl. Trop. Dis. 3:e495 doi:10.1371/journal.pntd.0000495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frearson JA, Brand S, McElroy SP, Cleghorn LA, Smid O, Stojanovski L, Price HP, Guther ML, Torrie LS, Robinson DA, Hallyburton I, Mpamhanga CP, Brannigan JA, Wilkinson AJ, Hodgkinson M, Hui R, Qiu W, Raimi OG, van Aalten DM, Brenk R, Gilbert IH, Read KD, Fairlamb AH, Ferguson MA, Smith DF, Wyatt PG. 2010. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 464:728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mercer L, Bowling T, Perales J, Freeman J, Nguyen T, Bacchi C, Yarlett N, Don R, Jacobs R, Nare B. 2011. 2,4-Diaminopyrimidines as potent inhibitors of Trypanosoma brucei and identification of molecular targets by a chemical proteomics approach. PLoS Negl. Trop. Dis. 5:e956 doi:10.1371/journal.pntd.0000956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Njiru ZK, Traub R, Ouma JO, Enyaru JC, Matovu E. 2011. Detection of group 1 Trypanosoma brucei gambiense by loop-mediated isothermal amplification. J. Clin. Microbiol. 49:1530–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Radwanska M, Magez S. 2002. Direct detection and identification of African trypanosomes by fluorescence in situ hybridization with peptide nucleic acid probes. J. Clin. Microbiol. 40:4295–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Claes F, Vodnala SK, van Reet N, Boucher N, Lunden-Miguel H, Baltz T, Goddeeris BM, Büscher P, Rottenberg ME. 2009. Bioluminescent imaging of Trypanosoma brucei shows preferential testis dissemination which may hamper drug efficacy in sleeping sickness. PLoS Negl. Trop. Dis. 3:e486 doi:10.1371/journal.pntd.0000486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hesse F, Selzer PM, Mühlstädt K, Duszenko M. 1995. A novel cultivation technique for long-term maintenance of bloodstream form trypanosomes in vitro. Mol. Biochem. Parasitol. 70:157–166 [DOI] [PubMed] [Google Scholar]
- 15. Nilsson B. May 2002. Piperazinylpyrazine compounds as agonist or antagonist of serotonin 5HT-2 receptor. US patent WO0240456A1
- 16. Waters NJ, Jones R, Williams G, Sohal B. 2008. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J. Pharmaceut. Sci. 97:4586–4595 [DOI] [PubMed] [Google Scholar]
- 17. Houston J. 1994. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 47:1469–1479 [DOI] [PubMed] [Google Scholar]
- 18. Hubatsch I, Ragnarsson EGE, Artursson P. 2007. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2:2111–2119 [DOI] [PubMed] [Google Scholar]
- 19. Baranczewski P, Stańczak A, Sundberg K, Svensson R, Wallin A, Jansson J, Garberg P, Postlind H. 2006. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 58:453–472 [PubMed] [Google Scholar]
- 20. Balasegaram M, Young H, Chappuis F, Priotto G, Raguenaud M-E, Checchi F. 2009. Effectiveness of melarsoprol and eflornithine as first-line regimens for gambiense sleeping sickness in nine Médecins Sans Frontières programmes. Trans R Soc. Trop. Med. Hyg. 103:280–290 [DOI] [PubMed] [Google Scholar]
- 21. Bernhard SC, Nerima B, Mäser P, Brun R. 2007. Melarsoprol- and pentamidine-resistant Trypanosoma brucei rhodesiense populations and their cross-resistance. Int. J. Parasitol. 37:1443–1448 [DOI] [PubMed] [Google Scholar]
- 22. Brand S, Cleghorn LA, McElroy SP, Robinson DA, Smith VC, Hallyburton I, Harrison JR, Norcross NR, Spinks D, Bayliss T, Norval S, Stojanovski L, Torrie LS, Frearson JA, Brenk R, Fairlamb AH, Ferguson MA, Read KD, Wyatt PG, Gilbert IH. 2012. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 55:140–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bell AS, Mills JE, Williams GP, Brannigan JA, Wilkinson AJ, Parkinson T, Leatherbarrow RJ, Tate EW, Holder AA, Smith DF. 2012. Selective inhibitors of protozoan protein N-myristoyltransferases as starting points for tropical disease medicinal chemistry programs. PLoS Negl. Trop. Dis. 6:e1625 doi:10.1371/journal.pntd.0001625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, Jenks MX, Noe RA, Bowling TS, Mercer LT, Rewerts C, Gaukel E, Owens J, Parham R, Randolph R, Beaudet B, Bacchi CJ, Yarlett N, Plattner JJ, Freund Y, Ding C, Akama T, Zhang Y-K, Brun R, Kaiser M, Scandale I, Don R. 2011. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 5:e1151 doi:10.1371/journal.pntd.0001151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Torreele E, Bourdin Trunz B, Tweats D, Kaiser M, Brun R, MazuÉ G, Bray MA, Pécoul B. 2010. Fexinidazole—a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 4:e923 doi:10.1371/journal.pntd.0000923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bishop M, Nilsson B. 2003. New 5-HT2C receptor agonists. Exp. Opin. Therapeutic Patents 13:1691–1705 [Google Scholar]