

Abstract
We have synthesized new derivatives of the macrolide antibiotics erythromycin and azithromycin. Novel deoxysugar moieties were attached to these standard antibiotics by biotransformation using a heterologous host. The resulting compounds were tested against several standard laboratory and clinically isolated bacterial strains. In addition, they were also tested in vitro against standard and drug-resistant strains of human malaria parasites (Plasmodium falciparum) and the liver stages of the rodent malaria parasite (Plasmodium berghei). Antibacterial activity of modified erythromycin and azithromycin showed no improvement over the unmodified macrolides, but the modified compounds showed a 10-fold increase in effectiveness after a short-term exposure against blood stages of malaria. The new compounds also remained active against azithromycin-resistant strains of P. falciparum and inhibited growth of liver-stage parasites at concentrations similar to those used for primaquine. Our findings show that malaria parasites have two distinct responses to macrolide antibiotics, one reflecting the prokaryotic origin of the apicoplast and a second, as-yet uncharacterized response that we attribute to the eukaryotic nature of the parasite. This is the first report for macrolides that target two different functions in the Plasmodium parasites.
INTRODUCTION
The control of endemic pathogenic diseases remains a constant challenge mainly because of the emergence of resistance to existing drugs. Malaria, which is caused by five species of the parasite genus Plasmodium, is a striking example of this problem. Each new antimalarial has eventually lost effectiveness due to the appearance and spread of resistant parasites, sometimes with startling rapidity (1). Emerging parasite resistance to front-line antimalarial drugs highlights the urgent need to identify new drugs with suitable pharmacokinetic properties to facilitate their use in combination therapies that can slow the development of resistance.
Macrolide antibiotics have antimalarial properties (2) and act by targeting the bacterium-derived translational machinery in the relict plastid (apicoplast) present in Plasmodium spp. (3). Targeting the apicoplast translation machinery with macrolides and other antibiotics characteristically results in a delayed death effect, with the parasite surviving for one full life cycle before growth inhibition begins (4). Clinically, this delayed death response results in delayed parasite clearance and the need for prolonged exposures, thus making macrolides inappropriate as monotherapies (2). New approaches to improve the potency of azithromycin (Azi) by classical medicinal chemistry have yielded novel compounds with high potency and no delay in activity (5–9). The parasite response to these new compounds suggests that their target differs from that of existing macrolides, but this new target has not been identified and effects on apicoplast translation have not been assessed.
Deeper understanding of the biosynthetic pathways of glycosylated natural products, such as the macrolide antibiotics, has led to the emergence of new technologies able to diversify their glycosylation patterns. Modified glycosylation patterns frequently are crucial determinants of biological properties. Thus, altering the nature of the appended sugar residues can generate compounds with novel or improved biological activities (10). The strategies generally employed to prepare glycosylated natural-product variants include total synthesis, semisynthesis, glycorandomization, and in vivo pathway engineering (11). Although in vitro methods allow an almost limitless diversification of sugar precursors, the precursor engineering and the cost effectiveness of fermentation processes still renders in vivo methods a more realistic option for drug development. The in vivo approach involves the manipulation of sugar biosynthetic pathways to design hybrid glycoconjugates through the coupling of novel deoxysugar skeletons to various aglycones. This technology ultimately depends on the flexibility toward the TDP-sugar and aglycone substrates of the glycosyltransferases responsible for the final decoration step of the polyketide backbone (10). The use of a heterologous clean host lacking the genetic background for irrelevant sugar pathways and polyketide synthases is an attractive option for developing microbial cell factories to produce new glycosylated polyketides (11).
We recently elucidated the biosynthesis pathway of TDP-l-megosamine, a sugar component of the 14-membered macrolide megalomicin produced by the actinomycete Micromonospora megalomicea (12). Megalomicin is similar to erythromycin in terms of antibiotic activity, spectrum of action, and pharmacokinetic properties. However, the presence of l-megosamine in megalomicins is believed to provide these compounds with additional biological activities, such as antiviral and antiparasitic activities, not seen in erythromycin (13, 14) (Fig. 1). The heterologous expression of TDP-l-megosamine biosynthesis in Escherichia coli permitted the production of megalomicin A (MegA) and 12-deoxynucleoside triphosphate (dNTP)-megalomicin A (12dMegA) through bioconversion experiments using erythromycin C (EryC) and erythromycin D (EryD) as macrolide substrates (12). These experiments demonstrated the ability of the megosaminyltransferase pair MegDI/MegDVI to accept different macrolide substrates. Based on this approach, we generated new macrolide derivatives containing either megosamine or megosamine analogues and evaluated their biological activities.
Fig 1.

Structure of megalomicins, erythromycins, azithromycin, and roxithromycin.
Here, we describe the production of novel megosaminylated compounds using commercially available erythromycin-derivative macrolides by means of an E. coli-based fermentation process. The successful recognition of different combinations of macrolides and TDP-sugars indicated a relaxed specificity of the glycosyltransferase pair MegDI-MegDVI toward these substrates. The new megosaminylated macrolides showed an improved antimalarial activity against both standard and drug-resistant strains of Plasmodium falciparum and improved efficacy against the liver stages of Plasmodium berghei.
MATERIALS AND METHODS
Strains and culturing conditions.
E. coli strain LB19b, harboring pGro7 (TaKaRa) and a sugar expression plasmid (pM9, pM100, or pM103) (12), were grown at 37°C in LB to an optical density at 600 nm (OD600) of 0.6. Chaperones and sugar pathway gene expression were induced by addition of 2 mg/ml l-arabinose and 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG), respectively. Induced cultures were supplemented with 40 mg/ml of the macrolide and incubated at 23°C for a further 72 h.
Plasmids pM100 and pM103 were constructed using the same cloning strategies as those previously described for pM9 (12). For pM100, the dnmV gene from the daunosamine pathway (15) was used instead of megDV. For pM103, the dnmU and dnmV genes were used instead of megDIV and megDV genes. None of these plasmids carried the megDIII gene, encoding dimethyltransferase. The dnmV and dnmU genes from the l-daunosamine biosynthetic pathway (15) were amplified by PCR using Streptomyces peucetius genomic DNA as the template. The 5′ primers used were designed to have an NdeI site overlapping the translational initiation codon. The 3′ primers contained EcoRI and SpeI sites downstream of the stop codon. The following oligonucleotides were used: 5′-CATATGAAGGCGCGGGAACTG-3′ (forward) and 5′-ACTAGTCACCCGTCTCCGCGTGAC-3′ (reverse) for dnmU and 5′-CATATGCGGGTCGTGGTTCTG-3′ (forward) and 5′-ACTAGTCTAGGCCGGGGCGCCGTG-3′ (reverse) for dnmV. PCRs were performed using a DNA thermal cycler 480 (Perkin-Elmer) with the following cycling parameters: 30 cycles of 30 s at 94°C, 30 s at 56°C, and 80 s at 72°C. The PCR products were digested with NdeI and EcoRI and cloned into pET24b vector (Novagen) digested with the same enzymes, and the sequences were confirmed.
P. falciparum 3D7 and W2Mef are commonly used reference strains available through MR4 (Malaria Research and Reference Reagent Resource Center, ATCC, Manassas, VA). 3D7 is sensitive to most antimalarials, while W2Mef exhibits resistance to mefloquine (16). Laboratory-generated lines of 3D7 resistant to the macrolide azithromycin (3D7AziR) were obtained using the method described by Sidhu et al. (3). Parasites were cultured using standard techniques (4). To obtain tightly synchronized cultures, two sorbitol synchronizations were carried out 16 h apart. Forty-eight h after the initial synchronization, the cultures were synchronized again to eliminate any residual schizonts (4).
P. berghei ANKA sporozoites dissected from the salivary glands of Anopheles stephensi were seeded onto and grown in HepG2 cells on coverslips at 37°C and 8% CO2 in advanced minimum essential medium (MEM; Gibco) supplemented with 10% fetal calf serum (FCS; Bovogen), 1% l-glutamine, 1% penicillin-streptomycin, and 0.1% amphotericin B (Thermo Scientific).
Product identification and characterization.
Cultures were clarified by centrifugation at 5,000 × g for 10 min and their pH values adjusted to 9.5, and they were extracted with an equal volume of ethyl acetate (EtOAc). The organic layer was separated and concentrated under vacuum, and the presence of bioconversion products was further analyzed by thin-layer chromatography (TLC) in a solvent system consisting of ethyl acetate, ethanol, and ammonia in a ratio of 80:15:5.
Compounds were purified by flash column chromatography using silica gel 60 (230 to 400 mesh; Merck). The column was initially washed with ethyl acetate and then with a 3:2 dilution of chloroform-methanol. Megalomicins then were eluted with 3:2 chloroform-methanol with 2% triethylamine. Fractions assayed positively for megalomycin were combined and evaporated. The resulting crude product was further purified by reverse-phase high-performance liquid chromatography (HPLC) with a C18 semipreparative column (Prodigy ODS-3V; 5 μm particle size; 250 by 10 mm; Phenomonex) running a gradient of 5 mM NH4OAc in H2O and 5 mM NH4OAc in MeCN-MeOH (4:1) from an 85:15 dilution to 0:100 dilution for 15 min at a flow rate of 4 ml/min. LC-electrospray ionization (ESI)-high resolution mass spectrometry were recorded on an Agilent 1100 HPLC coupled with a Bruker micrOTOF-Q II mass spectrometer (MS). Nuclear magnetic resonance (NMR) spectra were acquired at 300 MHz for 1H and 75 MHz for 13C on samples dissolved in CDCl3 on a Bruker Avance 300 using standard software. The purity of all products was over 95% based on 1H NMR.
Biological assays.
Antibacterial activity data given in Table 1 were obtained by the agar 2-fold dilution method in plates with Mueller-Hinton agar (Difco) to determine the MIC. Test substance and standard were dissolved in ethanol at 5 mg/ml. Solutions of the substance were prepared in Mueller-Hinton agar with final concentrations ranging from 64 to 0.125 μg/ml. Strains were grown until they achieved a final concentration of 5 × 105 CFU/ml. Two μl of the microorganisms was incubated on agar plates containing antibiotics at 37°C for 16 to 18 h. The MIC is defined as the minimal concentration of the compound that shows 99% growth inhibition by visual determination (17).
Table 1.
Antimicrobial activities of the new compounds
| Microorganism | MIC (μg/ml) |
|||||
|---|---|---|---|---|---|---|
| EryA | 1 | 3 | 4 | Azi | 2 | |
| Acinetobacter baumannii ATCC 17978 | 16 | 32 | 4 | 16 | 16 | 32 |
| Enterococcus faecalis ATCC 29212 | 0.5 | 1 | 0.25 | 16 | 0.5 | 1 |
| E. coli ATCC 51447 | 4 | >32 | 8 | 16 | 4 | >32 |
| S. aureus ATCC 25923 | 0.125 | 2 | 0.06 | 16 | 0.125 | 2 |
| S. aureus (ermC) MLSi | 512 | >1,024 | >1,024 | >1,024 | 512 | >1,024 |
| S. aureus (ermA) MLSc | >1,024 | >1,024 | >1,024 | >1,024 | >1,024 | >1,024 |
| S. aureus (mrsA) Eflux | 512 | >1,024 | 64 | >1,024 | 512 | >1,024 |
P. falciparum blood-stage drug trials were carried out in triplicate in 96-well plates, with each well containing 2% washed red blood cells and the desired inhibitor concentration (diluted in ethanol) in 0.2 ml of media with 0.5 or 0.2% parasitemia using synchronized parasites (4). Growth medium was changed at 48 and 96 h for cultures grown to 120 h. Cells were washed with fresh medium prior to assay, and parasitemia was assessed using a previously described fluorescence assay (4).
P. berghei exoerythrocytic drug effects were assayed using HepG2 liver cells grown on coverslips in 24-well plates (18), and each well was infected with 15,000 sporozoites. One hour after sporozoite addition, the growth medium was replaced with fresh medium containing the desired inhibitor concentration. The media, including inhibitors, were replaced after 24 h. After 56 h, the cells were fixed using 4% paraformaldehyde in phosphate-buffered saline (PBS) followed by ice-cold methanol. Parasites were labeled with a polyclonal anti-P. berghei antibody (18) followed by Alexa Flour 488 secondary antibody (Invitrogen). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Infections were assessed by the number of infected cells observed in 100 fields at 100× magnification using a Leica epifluorescence microscope. Drug response curves were calculated using Sigmaplot (Systat Software, San Jose, CA).
RESULTS
Generation of new macrolides.
As we previously described, the megosaminyltransferase pair MegDI/MegDVI exhibited some flexibility toward their macrolide substrate, as it enabled the production of MegA or 12dMegA by bioconversion of EryC or EryD, respectively (12). This was achieved by using the genetically engineered E. coli strain LB19b (19), harboring plasmid pM9 (12). This plasmid expresses the entire TDP-l-megosamine biosynthetic pathway, the erythromycin resistance protein ErmE, and the pair of proteins, MegDI/MegDVI, involved in the transfer of megosamine to the C6-OH group of the macrolide substrate. To further explore its properties as a glycodiversification tool, we tested the substrate flexibility of the megosaminyltransferase MegDI/MegDVI with some of the most commonly used therapeutic macrolides containing a C6-OH group as an acceptor. We also screened alternative sugar donors by generating new TDP-sugar operons using enzymes from the TDP-daunosamine biosynthetic pathway. We focused on the well-known antibiotic erythromycin A (EryA) and on two extended-spectrum erythromycin derivatives, azithromycin (Azi) and roxythromycin. Thus, bioconversion experiments were performed in flask cultures of strain LB19b carrying the pM9 plasmid, adding each of these macrolides at a concentration of 20 mg/liter. After 3 days of fermentation, the polyketides were extracted with organic solvent and analyzed by TLC and LC-MS. Analysis of culture broth fed with EryA or Azi showed the presence of two new compounds with m/z 891.5 (M + H+) (Fig. 2B) and m/z 906.6 (M + H+) (Fig. 2C), consistent with the calculated molecular formula of the protonated form of 6-O-megosaminyl-erythromycin A (compound 1) and 6-O-megosaminyl-azithromycin (compound 2), respectively.
Fig 2.

(A) Structure of 6-O-megosaminyl-erythromycin A (compound 1), 6-O-megosaminyl-azithromycin (2), 6-O-epidigitoxosyl-erythromycin A (3), and 6-O-daunosaminyl-erythromycin A (4). Also shown is the LC-MS of the peak corresponding to compounds 1 (B), 2 (C), 3 (D), and 4 (E) present in the organic extract of E. coli fermentation broth. Intens., intensity (ion counts).
To characterize these new compounds, 1-liter bioconversion experiments were performed using 40 mg of macrolides, which yielded ∼10 mg of each new compound after purification. High-resolution MS analysis showed that compound 1 has a molecular formula of C45H82N2O15, while compound 2 has a molecular formula of C46H87N3O14. The structure of the new compounds was initially identified by comparison of the NMR spectra of the starting macrolide to those of the products (see the supplemental material). 1H NMR of compound 1 clearly shows that the product has an additional acetalic proton, in agreement with glycosylation having occurred. In fact, the spectrum shown by compound 1 has a double doublet at 4.87 ppm that is absent from erythromycin. Another unequivocal indication that a megosamine has been incorporated is the singlet integrated for 6H at 2.37 ppm. This is assignable to a new dimethylamino group incorporated on the C-3 of megosamine. Experiments using 13C NMR show seven additional signals, including a characteristic ketalic carbon at 90.1 ppm that is not present on the starting material. Heteronuclear single-quantum coherence (HSQC) experiments resulted in a correlation of the 4.87-ppm acetalic proton with the corresponding 90.1-ppm carbon.
To get additional evidence of megosamine incorporation, a series of total correlated spectroscopy (TOCSY) experiments were performed that showed clear evidence of the nature of the incorporated sugar. To that end, all of the ketalic signals of compound 1 were irradiated and the spectra acquired. The identity of the newly incorporated sugar was confirmed by the agreement of signals from irradiation of the new acetalic proton that are in concordance with those reported for the megosamine portion of megalomicin. The irradiation of the other two ketalic protons produced the expected signals for desosamine and mycarose (4.56 and 4.98 doublets, respectively).
Compound 2 was characterized using the same approach and was identified as the C6-O-megosaminyl analog of azithromycin. The spectrum of compound 2 had a double doublet at 5.07 ppm on the 1H NMR that correlates with the signal at 90.7 ppm. TOCSY experiments corroborate the nature of the new incorporated sugar.
Analysis of the culture broth supplemented with roxithromycin did not show the presence of the expected megosaminylated derivative of this macrolide. The fact that Azi, with an N-Me group over the aglycon, and EryA, with a C-9 ketone, are substrates of the transferase clearly shows that a hydrogen bond acceptor is required for enzyme recognition, and this site must be properly positioned to be metabolized. Roxithromycin, with an oxime in that position, is less polar and holds a larger substituent with higher steric requirements. Those differences may prevent proper access to, and orientation on, the MegDI/MegDVI active site, thereby preventing glycosylation. Despite this limitation, the LB19b/pM9 bioconversion platform proved to be a reliable system to generate megosaminylated polyketides that otherwise are impossible to obtain in a simple, fast, cheap, and scalable fashion.
Our next objective was to obtain new macrolides by attaching sugars other than megosamine at the C6-OH group of the macrolide substrate. Thus, we constructed new sugar operons by replacing the genes encoding the epimerase MegDIV and the 4-ketoreductase MegDV with daunosamine pathway homologues. Replacement of megDV with dnmV in the megosamine operon yield plasmid pM100, while replacement of both megDIV-megDV with dnmU-dnmV genes resulted in plasmid pM103. None of these plasmids contain the gene encoding the dimethyltransferase MegDIII. Bioconversion experiments were performed in flask cultures of strain LB19b carrying either pM100 or pM103 plasmids by feeding either erythromycin or azithromycin at a final concentration of 20 mg/liter. After 3 days of fermentation, the modified macrolides were extracted and analyzed as previously described. Analysis of organic extracts obtained from cultures of LB19b/pM100 or LB19b/pM103 fed with EryA showed the presence of two new compounds with m/z 863.5 (M + H+) (Fig. 2D) and m/z 864.5 (M + H+) (Fig. 2E). This is consistent with the calculated molecular formula of 6-O-epidigitoxosyl-erythromycin A (compound 3) and 6-O-daunosaminyl-erythromycin A (compound 4). No bioconversion products were obtained from cultures fed with azithromycin. To characterize these new compounds, 1-liter bioconversion experiments were performed by feeding 40 mg of EryA, which yielded ∼10 mg of each new compound after purification. High-resolution MS analysis showed that compound 3 has a molecular formula of C43H77NO16, while compound 4 has a molecular formula of C43H78N2O15.
The structures of the new compounds were confirmed by NMR studies by following the approach described before (see the supplemental material). Evidence of the newly incorporated sugar on compound 3 comes from the 1H NMR spectrum where the singlet of the N-dimethylamino at 2.28 ppm is missing. Additionally, a TOCSY experiment irradiating the ketalic proton at 5.10 ppm reveals the shift of the C3 proton signal from 2.94 ppm on compound 1 to 3.83 ppm, which is a clear indication of the hydroxyl for amino exchange. The stereochemistry of the new sugar was assigned by nuclear Overhauser effect (nOe) experiments in which the ketalic proton was irradiated. On those spectra it was observed to have correlation with only one of the C2 protons with no other interaction, giving some evidence of C5 epimerization. Compound 4 also does not show N-dimethyl signal on the 1H NMR spectrum. TOCSY experiments that involved irradiating the ketalic proton at 5.15 ppm showed a pattern similar to that of compound 1, indicating that both share a similar substitution over the ring. In this case, unfortunately, nOe experiments were inconclusive, requiring a further exhaustive study to determine the stereochemistry of the newly incorporated sugar.
Biological activities of new megosamine-containing polyketides.
Megalomicins and EryA exhibit different antimicrobial potencies against several bacterial strains, which could be related to either the different acylations at the l-mycarose residue or the presence of the l-megosamine residue (20). Thus, the attachment of l-megosamine to EryA and Azi allowed us to specifically determine the contribution of this sugar residue to the antibacterial activity of these molecules. In vitro antimicrobial assays were performed with EryA, Azi, and their corresponding megosaminylated derivatives, meg-EryA and meg-Azi, against a panel of Gram-negative and Gram-positive bacteria, including three macrolide-resistant Staphylococcus aureus strains (Table 1). The results showed that the addition of megosamine reduced the potency of both EryA and Azi against all strains tested. The lack of changes of the MICs of the S. aureus erythromycin-resistant strains suggests that the megosamine residue does not interfere with the binding of the macrolide to methylated ribosomes, but megosaminylated products seem to be pumped out more efficiently.
It has been hypothesized that, unlike the case for EryA, the presence of megosamine is the key determinant for the antiviral and antiparasitic activities exhibited by megalomicins (13, 14). To test this hypothesis, we evaluated the antiparasitic activity of compounds 1 and 2, as well as the new nonmegosamilylated macrolides (compounds 3 and 4), against in vitro cultures of P. falciparum. Like other drugs that inhibit apicoplast translation, both EryA and Azi antibiotics show much greater potency after prolonged exposure (3), so we compared the antiparasitic activity of the precursor macrolides to those of the new compounds at 48 and 120 h postinfection. Measurements of the antiparasitic activity after 48 h of infection showed a significant improvement of the 50% inhibitory concentration (IC50) against P. falciparum, with a 6- to 10-fold increase for both macrolide scaffolds containing the megosamine residue (Table 2). The antiparasitic activity after prolonged exposure was modestly changed by the addition of the megosamine sugar to these compounds, although they remained effective in the submicromolar range (Table 2).
Table 2.
Antiparasitic activities of the new macrolide compounds at 48 and 120 h
| Strain | IC50 (μM) at: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 48 h |
120 h |
|||||||||||
| EryA | 1 | 3 | 4 | Azi | 2 | EryA | 1 | 3 | 4 | Azi | 2 | |
| 3D7 | 82.2 ± 12.7 | 9.7 ± 1.3 | 60.9 ± 8.4 | 38.2 ± 8.5 | 18.2 ± 1.4 | 1.6 ± 0.1 | 1.8 ± 0.2 | 1.2 ± 0.4 | 10.1 ± 1.4 | 15.2 ± 1.1 | 0.06 ± 0.006 | 0.16 ± 0.009 |
| W2Mef | 86.7 ± 13.3 | 12.8 ± 1.7 | 58.6 ± 9.8 | 27.6 ± 10.0 | 18.9 ± 0.7 | 1.8 ± 0.2 | 2.7 ± 0.4 | 0.72 ± 0.06 | 5.1 ± 0.7 | 11.3 ± 0.5 | 0.05 ± 0.004 | 0.21 ± 0.01 |
| 3D7AziR | 9.7 ± 0.4 | 1.8 ± 0.14 | NDa | ND | 1.2 ± 0.07 | 0.21 ± 0.009 | ||||||
| W2 Mef AziR | 10.0 ± 0.3 | 0.93 ± 0.08 | ND | ND | 0.90 ± 0.08 | 0.22 ± 0.009 | ||||||
ND, not determined.
To further investigate the relevance of the megosamine residue for the improved antiparasitic properties of compounds 1 and 2, we tested different sugar moieties attached to the C6-OH group of erythromycin macrolides to assay their biological properties (compounds 3 and 4). These new sugars differ from megosamine at the dimethylamino and in the stereochemistry of the 4′′′-OH groups (Fig. 2A). As shown in Table 2, the best of these compounds showed only modest improvement of antiparasitic activity at 48 h compared to the activity of EryA, reinforcing the relevance of the megosamine residue for this activity.
To gain more information about the target of these novel compounds, we generated an azithromycin-resistant line of P. falciparum with a C-to-T mutation at base 2,409 of the apicoplast 23S rRNA (strain 3D7AziR). This is equivalent to base 2,611 in E. coli 23S rRNA and is a common mutation conferring macrolide resistance in bacteria, algal chloroplasts, and yeast mitochondria (21–23). As expected, this mutant line showed significant resistance (>10-fold) to prolonged exposure to azithromycin (Table 2). The short-term efficacy of both the precursor macrolides and megosaminylated derivatives remained unaffected by the resistance mutation, suggesting a distinct target for the short-term effects of these compounds. Somewhat surprisingly, after prolonged exposure, compounds 1 and 2 retained their efficacy against P. falciparum azithromycin-resistant strains (Table 2).
We also tested the effect of compound 2 on liver-stage growth of the parasite life cycle, using P. berghei as a model. Development of P. berghei parasites in cultured hepatoma cells was strongly inhibited by compound 2, with an IC50 of 6.9 μM (Fig. 3). This is in contrast to azithromycin, which had no effect at concentrations of up to 40 μM under our assay conditions (24).
Fig 3.
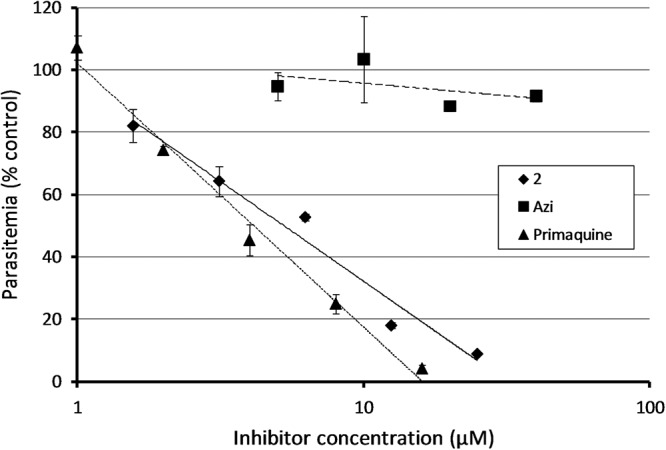
Response of P. berghei exoerythrocytic (liver stage) parasites to compound 2, Azi, and primaquine.
DISCUSSION
Macrolide antibiotics remain of interest as antimalarials, because they exhibit a good safety profile in children and pregnant women. The currently licensed macrolides are of limited use because they require prolonged exposure. This is directly related to the delayed effect that drugs specifically targeting apicoplast translation have on parasite growth (14, 16, 17). Recently, several new compounds generated by traditional medicinal chemistry have been described that improve on the relatively poor efficacy of currently licensed molecules in short-term treatment (5–9). Here, we used a simple plasmid/host microbial system as a reliable platform to generate megosaminylated macrolide derivatives that would be impossible to obtain by the traditional chemistry used in other studies. Our bioconversion system demonstrates the flexibility of the megosaminyltransferase pair MegDI-MegDVI toward the macrolide substrate with modifications of the neutral sugar mycarose or of the size of the macrolactone ring. This glycosyltransferase is also flexible toward the sugar substrate, suggesting that a much wider variety of macrolide derivatives could be created through combinatorial biosynthesis with this method. Moreover, it is feasible to use this bioconversion process to create new antimalarial macrolides through the addition of megosamine to the traditionally synthesized macrolide derivatives having a free 6-OH group.
Compared to their macrolide precursors, the two new compounds (1 and 2) tested here show a marked increase in effectiveness against P. falciparum after a short-term exposure while still remaining effective at nanomolar concentrations over longer exposures (Table 2). Improved efficacy after short-term exposures is unlikely to result from better inhibition of the prokaryotic translation system in the apicoplast, because targeting the apicoplast translation machinery with macrolides and other antibiotics characteristically results in a delayed death effect, with the parasite surviving for one full life cycle before growth inhibition begins (4). The improvement in short-term efficacy of our compounds, combined with the slight loss of activity after long-term exposure, suggest that these compounds are hitting a new target. The correlation between higher antiparasitic activity and lower antibacterial activity in these compounds (Table 1 and 2) indicates that the new target is unlikely to be the apicoplast.
A non-apicoplast target for the modified macrolides is also congruent with the response of exoerythrocytic parasites to these modified compounds. Treatment of liver cell infections with low micromolar concentrations of azithromycin has little effect on parasite development, and parasites treated with higher concentrations are inhibited only very late (64 to 72 h) in infection (24). In contrast, compound 2 showed significant effects after 56 h at low micromolar concentrations, a response more similar to that of primaquine treatment (Fig. 3). Again, this suggests that addition of a megosamine group causes these compounds to target something in addition to the apicoplast translation machinery.
Both our biotransformation process and traditional synthetic chemistry show that the antibacterial and antiparasitic activities of macrolides can be modulated individually. However, it remains unclear whether inhibition of apicoplast translation, with its canonical delayed effect, and the as-yet unidentified (non-apicoplast) target are mutually exclusive. Our data showing continued efficacy of the megosamine containing macrolides after 120 h (Table 2) suggest that apicoplast translation is still targeted by the new derivatives. If this is the case, it opens the possibility of developing a single compound that yields both fast- and slow-acting profiles by targeting two separate parasite metabolic processes, which is an attractive approach to mitigate parasite resistance.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the UNICEF/UNDP/WORLD BANK/WHO Special Programme for Research and Training in Tropical Diseases (TDR) to G.R.L., ANPCyT grants PICT 2006-01978 to E.R. and PICT2008-644 to H.G., a Program Grant from the National Health and Medical Research Council of Australia, an Australian Research Council Federation Fellowship, and a Howard Hughes Medical Institute International Research Scholar award to G.I.M.
We thank the Australian Red Cross for blood.
S.P., G.R.L., E.R., and H.G. are members of the Research Career and M.U. is a doctoral fellow of CONICET.
Footnotes
Published ahead of print 3 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01825-12.
REFERENCES
- 1. Parija SC, Praharaj I. 2011. Drug resistance in malaria. Indian J. Med. Microbiol. 29:243–248 [DOI] [PubMed] [Google Scholar]
- 2. Dahl EL, Rosenthal PJ. 2007. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 51:3485–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sidhu AB, Sun Q, Nkrumah LJ, Dunne MW, Sacchettini JC, Fidock DA. 2007. In vitro efficacy, resistance selection, and structural modeling studies implicate the malarial parasite apicoplast as the target of azithromycin. J. Biol. Chem. 282:2494–2504 [DOI] [PubMed] [Google Scholar]
- 4. Goodman CD, Su V, McFadden GI. 2007. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 152:181–191 [DOI] [PubMed] [Google Scholar]
- 5. Hutinec A, Rupcic R, Ziher D, Smith KS, Milhous W, Ellis W, Ohrt C, Schonfeld ZI. 2011. An automated, polymer-assisted strategy for the preparation of urea and thiourea derivatives of 15-membered azalides as potential antimalarial chemotherapeutics. Bioorg. Med. Chem. 19:1692–1701 [DOI] [PubMed] [Google Scholar]
- 6. Lee Y, Choi JY, Fu H, Harvey C, Ravindran S, Roush WR, Boothroyd JC, Khosla C. 2011. Chemistry and biology of macrolide antiparasitic agents. J. Med. Chem. 54:2792–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peric M, Fajdetic A, Rupcic R, Alihodzic S, Ziher D, Bukvic Krajacic M, Smith KS, Ivezic-Schonfeld Z, Padovan J, Landek G, Jelic D, Hutinec A, Mesic M, Ager A, Ellis WY, Milhous WK, Ohrt C, Spaventi R. 2012. Antimalarial activity of 9a-N substituted 15-membered azalides with improved in vitro and in vivo activity over azithromycin. J. Med. Chem. 55:1389–1401 [DOI] [PubMed] [Google Scholar]
- 8. Pesic D, Starcevic K, Toplak A, Herreros E, Vidal J, Almela MJ, Jelic D, Alihodzic S, Spaventi R, Peric M. 2012. Design, synthesis, and in vitro activity of novel 2′-o-substituted 15-membered azalides. J. Med. Chem. 55:3216–3227 [DOI] [PubMed] [Google Scholar]
- 9. Starcevic K, Pesic D, Toplak A, Landek G, Alihodzic S, Herreros E, Ferrer S, Spaventi R, Peric M. 2012. Novel hybrid molecules based on 15-membered azalide as potential antimalarial agents. Eur. J. Med. Chem. 49:365–378 [DOI] [PubMed] [Google Scholar]
- 10. Madduri K, Kennedy J, Rivola G, Inventi-Solari A, Filippini S, Zanuso G, Colombo AL, Gewain KM, Occi JL, MacNeil DJ, Hutchinson CR. 1998. Production of the antitumor drug epirubicin (4′-epidoxorubicin) and its precursor by a genetically engineered strain of Streptomyces peucetius. Nat. Biotechnol. 16:69–74 [DOI] [PubMed] [Google Scholar]
- 11. Blanchard S, Thorson JS. 2006. Enzymatic tools for engineering natural product glycosylation. Curr. Opin. Chem. Biol. 10:263–271 [DOI] [PubMed] [Google Scholar]
- 12. Useglio M, Peiru S, Rodriguez E, Labadie GR, Carney JR, Gramajo H. 2010. TDP-l-megosamine biosynthesis pathway elucidation and megalomicin a production in Escherichia coli. Appl. Environ. Microbiol. 76:3869–3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonay P, Duran-Chica I, Fresno M, Alarcon B, Alcina A. 1998. Antiparasitic effects of the intra-Golgi transport inhibitor megalomicin. Antimicrob. Agents Chemother. 42:2668–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. San Jose E, Munoz-Fernandez MA, Alarcon B. 1997. Megalomicin inhibits HIV-1 replication and interferes with gp160 processing. Virology 239:303–314 [DOI] [PubMed] [Google Scholar]
- 15. Otten SL, Gallo MA, Madduri K, Liu X, Hutchinson CR. 1997. Cloning and characterization of the Streptomyces peucetius dnmZUV genes encoding three enzymes required for biosynthesis of the daunorubicin precursor thymidine diphospho-l-daunosamine. J. Bacteriol. 179:4446–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cowman AF, Galatis D, Thompson JK. 1994. Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc. Natl. Acad. Sci. U. S. A. 91:1143–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clinical and Laboratory Standards Institute 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard–7th ed. CLSI document M7-A7, vol 26(2). CLSI, Wayne, PA [Google Scholar]
- 18. Nair SC, Brooks CF, Goodman CD, Strurm A, McFadden GI, Sundriyal S, Anglin JL, Song Y, Moreno SN, Striepen B. 2011. Apicoplast isoprenoid precursor synthesis and the molecular basis of fosmidomycin resistance in Toxoplasma gondii. J. Exp. Med. 208:1547–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peiru S, Rodriguez E, Menzella H, Carney J, Gramajo H. 2008. Metabolically engineered Escherichia coli for efficient production of glycosylated natural products. Microb. Biotechnol. 1:476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waitz JA, Moss EL, Jr, Oden EM, Weinstein MJ. 1971. Biological activity of megalomicin A phosphate, a water-soluble salt of megalomicin A. J. Antibiot. (Tokyo) 24:310–316 [DOI] [PubMed] [Google Scholar]
- 21. Harris EH, Burkhart BD, Gillham NW, Boynton JE. 1989. Antibiotic resistance mutations in the chloroplast 16S and 23S rRNA genes of Chlamydomonas reinhardtii: correlation of genetic and physical maps of the chloroplast genome. Genetics 123:281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matsuoka M, Narita M, Okazaki N, Ohya H, Yamazaki T, Ouchi K, Suzuki I, Andoh T, Kenri T, Sasaki Y, Horino A, Shintani M, Arakawa Y, Sasaki T. 2004. Characterization and molecular analysis of macrolide-resistant Mycoplasma pneumoniae clinical isolates obtained in Japan. Antimicrob. Agents Chemother. 48:4624–4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vannuffel P, Di Giambattista M, Morgan EA, Cocito C. 1992. Identification of a single base change in ribosomal RNA leading to erythromycin resistance. J. Biol. Chem. 267:8377–8382 [PubMed] [Google Scholar]
- 24. Shimizu S, Osada Y, Kanazawa T, Tanaka Y, Arai M. 2010. Suppressive effect of azithromycin on Plasmodium berghei mosquito stage development and apicoplast replication. Malar. J. 9:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.