

Abstract
SM-295291 and SM-369926 are new parenteral 2-aryl carbapenems with strong activity against major causative pathogens of community-acquired infections such as methicillin-susceptible Staphylococcus aureus, Streptococcus pneumoniae (including penicillin-resistant strains), Streptococcus pyogenes, Enterococcus faecalis, Klebsiella pneumoniae, Moraxella catarrhalis, Haemophilus influenzae (including β-lactamase-negative ampicillin-resistant strains), and Neisseria gonorrhoeae (including ciprofloxacin-resistant strains), with MIC90s of ≤1 μg/ml. Unlike tebipenem (MIC50, 8 μg/ml), SM-295291 and SM-369926 had no activity against hospital pathogens such as Pseudomonas aeruginosa (MIC50, ≥128 μg/ml). The bactericidal activities of SM-295291 and SM-369926 against penicillin-resistant S. pneumoniae and β-lactamase-negative ampicillin-resistant H. influenzae were equal or superior to that of tebipenem and greater than that of cefditoren. The therapeutic efficacies of intravenous administrations of SM-295291 and SM-369926 against experimentally induced infections in mice caused by penicillin-resistant S. pneumoniae and β-lactamase-negative ampicillin-resistant H. influenzae were equal or superior to that of tebipenem and greater than that of cefditoren, respectively, reflecting their in vitro activities. SM-295291 and SM-369926 showed intravenous pharmacokinetics similar to those of meropenem in terms of half-life in monkeys (0.4 h) and were stable against human dehydropeptidase I. SM-368589 and SM-375769, which are medoxomil esters of SM-295291 and SM-369926, respectively, showed good oral bioavailability in rats, dogs, and monkeys (4.2 to 62.3%). Thus, 2-aryl carbapenems are promising candidates that show an ideal broad spectrum for the treatment of community-acquired infections, including infections caused by penicillin-resistant S. pneumoniae and β-lactamase-negative ampicillin-resistant H. influenzae, have low selective pressure on antipseudomonal carbapenem-resistant nosocomial pathogens, and allow parenteral, oral, and switch therapies.
INTRODUCTION
Community-acquired infections caused by extended-spectrum β-lactamase (ESBL) producers, quinolone-resistant pathogens, penicillin-resistant Streptococcus pneumoniae (PRSP), and β-lactamase-negative ampicillin-resistant (BLNAR) Haemophilus influenzae are of great concern (1). In moderate or severe cases, inpatient parenteral antibiotic therapy is needed, and carbapenems are often used to treat infections refractory to parenteral penicillin or cephalosporin therapy (e.g., ESBL producer, PRSP, and BLNAR organism infections); however, current practices in antipseudomonal carbapenem use are a risk factor for the emergence of carbapenem-resistant nosocomial pathogens, especially Pseudomonas aeruginosa (2). The non-antipseudomonal carbapenem ertapenem (ERM) is used for the treatment of community-acquired infections, but it has little or moderate activity against P. aeruginosa (3), implying that there is a risk of selection for resistance to antipseudomonal carbapenem in P. aeruginosa (4, 5). In the case of tebipenem (TBM)-pivoxil, an oral carbapenem, because there may be concern about the development of cross-resistance to existing parenteral carbapenems in nosocomial pathogens, TBM-pivoxil has been used only as salvage therapy for pediatric patients who are expected to be refractory to another oral antimicrobial agent.
Therefore, there is a need to develop a new class of carbapenems that have adequate antibacterial properties for the treatment of community-acquired infections and low selective pressure on conventional carbapenem-resistant bacteria based on structural differences from the existing carbapenems. Combinational development of parenteral and oral formulations of the same new class carbapenem, allowing a switch from parenteral to oral treatment, could contribute to early hospital discharge, decrease the cost of treatment (6–8), and reduce the risk of selection for cross-resistance to existing parenteral carbapenems in nosocomial pathogens.
We previously reported that 2-aryl carbapenems, which are desmethyl-carbapenems with a structurally unique C-2 side chain, exhibited well-balanced antibacterial activities against important pathogens of community-acquired infections (9).
Based on structure-activity relationship studies, we identified attractive 2-aryl carbapenems, SM-295291, (5R,6S)-6-[(R)-1-hydroxyethyl]-3-{4-[(methoxycarbonylamino)methyl]phenyl}-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid, and SM-369926, (5R,6S)-6-[(R)-1-hydroxyethyl]-3-{4-[(methylcarbamoyloxy)methyl]phenyl}-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid (Fig. 1), and in this study, we investigated their in vitro antibacterial activities compared with those of TBM, cefditoren (CDN), faropenem (FRM), clarithromycin (CLR), and levofloxacin (LVX). We evaluated the therapeutic efficacies of intravenous administration of SM-295291 and SM-369926 in several PRSP and BLNAR organism infection models and their pharmacokinetics in various animals. Esterification of β-lactams is one of the ways to improve their oral absorption (10); therefore, we synthesized medoxomil esters of SM-295291 and SM-369926 (SM-368589 and SM-375769, respectively) (Fig. 1) and assessed their oral bioavailability in various animals.
Fig 1.
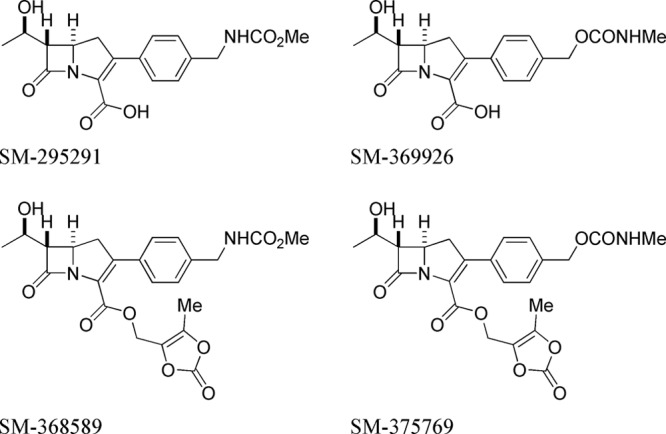
Chemical structures of 2-aryl carbapenems.
(This study was presented in part at the 51st Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, September 2011 [11, 12].)
MATERIALS AND METHODS
Organisms.
Clinical isolates used in this study were collected from different patients in various hospitals in Japan from 1996 to 2009. The β-lactamase-producing organisms were from our bacterial collections (13).
Antibacterial agents.
SM-295291, SM-369926, SM-368589, SM-375769, TBM, FRM, and meropenem (MEM) were synthesized in our laboratories. Imipenem (IPM) and cilastatin were prepared from TIENAM (MSD K.K., Tokyo, Japan), and CDN, CLR, and LVX were prepared from MEIACT MS (Meiji Seika Pharma Co., Ltd., Tokyo, Japan), Klaricid (Abbott Japan Co., Ltd., Tokyo, Japan), and CRAVIT (Daiichi Sankyo Company, Limited, Tokyo, Japan), respectively, in our laboratories. ERM (Invanz; Merck & Co., Inc., Whitehouse Station, NJ) was purchased.
Animals.
Male ICR mice (Japan SLC, Inc., Shizuoka, Japan), male Sprague-Dawley (SD) rats (Charles River Laboratories Japan, Kanagawa, Japan), male New Zealand White rabbits (Kitayama Labes, Co., Ltd., Nagano, Japan), and male beagles and cynomolgus monkeys (Japan Laboratory Animals, Inc., Tokyo, Japan) were used. All animal procedures were performed in accordance with the institutions' guidelines for the humane handling, care, and treatment of research animals in Dainippon Sumitomo Pharma Co., Ltd., and Astellas Pharma Inc.
Susceptibility testing.
MICs were determined by the 2-fold serial agar dilution method recommended by the Japanese Society of Chemotherapy and the Clinical and Laboratory Standards Institute guidelines (14) with Mueller-Hinton agar (MHA) (Becton, Dickinson and Company, Tokyo, Japan) unless otherwise specified. Susceptibility testing was performed with MHA supplemented with 5% defibrinated horse blood for streptococci and Mueller-Hinton chocolate agar (5% defibrinated horse blood) for H. influenzae and Neisseria gonorrhoeae. Modified GAM agar (Nissui Pharmaceutical Co., Ltd., Tokyo, Japan) was used for the culture of anaerobic bacteria. The final inocula comprised approximately 106 CFU/ml. Agar plates were incubated at 35°C for 18 and 48 h for aerobic and anaerobic bacteria, respectively. Incubation was carried out anaerobically for anaerobes and in an atmosphere of 5% CO2 for streptococci, H. influenzae, and N. gonorrhoeae. The MIC was defined as the lowest drug concentration that completely prevented visible growth.
Determination of MBC.
MIC tests were performed by the broth microdilution method, with the organism grown in Mueller-Hinton broth. Minimal bactericidal concentration (MBC) was determined by aspirating the antibiotic-containing medium in an MIC assay well and adding antibiotic-free MHA. The MBC was defined as the lowest antibiotic concentration that reduced the number of viable cells by >99.9%.
Time-kill curves.
An aliquot of 4.5 ml bacterial suspension in medium (about 106 CFU/ml) was mixed with 0.5 ml drug solution in medium and incubated at 35°C in an atmosphere of 5% CO2. Viable cell counts were determined by a general plating method 2, 4, and 6 h after drug addition.
In vitro evaluation of the emergence of carbapenem-resistant P. aeruginosa.
P. aeruginosa PAO1 was incubated in Mueller-Hinton broth (MHB) (Becton, Dickinson and Company) containing various concentrations of drugs at 37°C for 1 day. The MIC was defined as the lowest concentration of drug that resulted in no visible growth in the broth. Serial passages of P. aeruginosa PAO1 were done daily for 14 days in MHB in the presence of SM-295291 or SM-369926 at 8 μg/ml. In the case of TBM and ERM, P. aeruginosa PAO1 in MHB containing the highest concentration of drug at which the optical density was almost the same as that of the drug-free control was transferred to fresh medium containing various drug concentrations. This passage was performed daily for 14 days.
Stability against hydrolysis by DHP-I.
The stability of carbapenems to hydrolysis by dehydropeptidase I (DHP-I) was determined with purified rat and dog renal DHP-I and recombinant human DHP-I, as reported previously (13, 15). The activity of DHP-I was spectrophotometrically determined by measuring the hydrolysis of glycyldehydrophenylalanine as a substrate (13). The relative rate of hydrolysis was also calculated as a ratio against the hydrolysis rate for IPM or MEM, which was assigned a value of 1.
Stability in human plasma.
An aliquot of 1 ml human plasma (Rockland Immunochemicals Inc., Gilbertsville, PA; catalog no. D519-06) was mixed with 10 μl drug solution in 1/15 M phosphate buffer (pH 7.4) at a concentration of 30 μg/ml. The plasma sample was kept at 37°C for 4 h. The sample was mixed with 3 volumes of methanol, vortex mixed, and centrifuged at 10,000 × g for 10 min at 4°C. The supernatant was collected. The levels of drugs in human plasma were determined by a high-pressure liquid chromatography (HPLC)-UV detection assay method using an LC-2010C system and CLASS-VP workstation (Shimadzu Co., Kyoto, Japan). Chromatography was performed using an Xterra MS C18 reverse-phase column (3.5 μm, 4.6 by 20 mm; Nihon Waters K.K., Tokyo, Japan). The mobile phase consisted of 5 mM phosphate buffer (pH 7.0) and methanol. The flow rate was 1.5 ml/min. The wavelength for drug detection was 302 nm.
Protein binding.
Percent binding to rat, dog, and human plasma protein was determined by the ultrafiltration method (16). The degree of binding was measured using a drug concentration of 30 μg/ml. The concentration of each drug in the flowthrough fraction was measured by HPLC.
Murine PRSP and BLNAR organism infection models.
Male ICR mice were used at 4 weeks of age. At each administration, 100 mg/kg (of body weight) cilastatin, a DHP-I inhibitor, was administered subcutaneously 5 min before administration of SM-295291, SM-369926, and TBM in the murine infection models, because the hydrolyzing activities of DHP-I for a carbapenem differ greatly among animal species.
(i) Systemic infection.
Mice were inoculated intraperitoneally with 0.5 ml of a 5% mucin (Nacalai Tesque Inc., Kyoto, Japan) suspension of PRSP strain 18280 (1.4 × 104 CFU/mouse). Drugs in saline were administered intravenously 1 and 3 h after infection.
(ii) Pulmonary infection.
To induce neutropenia, cyclophosphamide was administered intraperitoneally 4 days before (200 mg/kg/day, PRSP and BLNAR organism infection models) and 4 h before (100 mg/kg/day, BLNAR organism infection model only) infection. For airway impairment, 50 μl influenza virus A/PR8 suspension was instilled intranasally into mice 5 days before BLNAR organism infection. Fifty microliters of PRSP strain 18280 (1.7 × 106 CFU/mouse) or BLNAR strain 17051 (4.4 × 107 CFU/mouse) suspension was inoculated intranasally. Drugs in saline were administered intravenously thrice daily at 1 and 2 days after infection.
(iii) Meningitis.
Mice were challenged intracerebrally with 2 × 104 CFU of PRSP strain 18280. Drugs in saline were administered intravenously at 5 h after infection and twice daily at 1 and 2 days after infection.
Pharmacokinetic study.
Male SD rats, beagles, and cynomolgus monkeys were used at 8 weeks, 20 months, and 2 years of age, respectively. Three animals were used for each group. SM-295291 or SM-369926 in saline at a dose of 1 mg/kg was administered intravenously to fasted rats given cilastatin at 100 mg/kg, fasted dogs, and fasted monkeys. SM-368589 or SM-375769 in 50% propylene glycol at a dose of 1 mg/kg was administered intraduodenally to fasted rats with cilastatin at 100 mg/kg and orally to fasted dogs and monkeys with omeprazole at 1 mg/kg. The plasma drug concentrations were determined by the liquid chromatography-mass spectrometric (LC-MS-MS) method consisting of API2000 (AB Sciex, Tokyo, Japan) with the Agilent 1100 series (Agilent Technologies, Santa Clara, CA). Chromatography was performed using Atlantis dC18 columns (5.0-μm particle size, 4.6 by 50 mm; Waters K.K.). The mobile phase consisted of 10 mM ammonium acetate and acetonitrile. The flow rate was 0.2 ml/min. Plasma samples were deproteinized using acetonitrile prior to LC-MS-MS analysis. Compound was detected by selective reaction monitoring under the positive ionization electrospray mode. The pharmacokinetic parameters were calculated according to moment analysis.
Testing of convulsant activity.
The convulsant stability of carbapenems was determined as reported previously (17). Seven-week-old male ICR mice were intracerebroventricularly injected with each dose (50 to 400 μg/mouse) of drugs dissolved in 5 μl phosphate-buffered saline. Immediately after injection, incidences of clonic and tonic convulsions were recorded for 30 min.
Assessment of renal nephrotoxicity.
SM-295291 at a dose of 100 mg/kg was administered intravenously to two rabbits. Blood and urine were collected at 1 (urine only), 2, and 4 days after administration. The kidneys were removed 4 days after the dose. The following parameters were investigated: blood urea nitrogen, blood creatinine, urinary glucose, urinary protein, urinary pH, renal weight, macroscopic examination of kidneys, and histopathological examination of renal sections. Because the synthetic quantity of SM-369926 was insufficient, we were not able to evaluate the nephrotoxicity of SM-369926.
Statistical analysis.
The 50% effective dose (ED50) and the convulsant activity were calculated by probit analysis. Dunnett's test for multiple comparisons was used to assess significant differences in the bacterial burden. All analyses were performed using the Statistical Analysis System for Windows (SAS Institute Inc., Cary, NC).
RESULTS
In vitro antimicrobial activity.
Strong antibacterial activity is required for oral antibiotics because of a relatively low achievable concentration in blood compared to that achieved with a parenteral drug. Therefore, to determine whether our 2-aryl carbapenems could be attractive candidates for alternative oral antibiotics, the antimicrobial activities of SM-295291 and SM-369926 were compared with those of conventional oral antibiotics (TBN, CDN, FRM, CLR, and LVX) (Tables 1 and 2).
Table 1.
In vitro activities of SM-295291, SM-369926, and selected antimicrobial agents against clinical isolates
| Organism | n | Antimicrobial agent | MIC (μg/ml) |
||
|---|---|---|---|---|---|
| Range | 50% | 90% | |||
| Staphylococcus aureus (methicillin susceptible) | 50 | SM-295291 | 0.125 to 0.25 | 0.125 | 0.125 |
| 26 | SM-369926 | ≤0.0313 to 0.125 | 0.0625 | 0.0625 | |
| 50 | Tebipenem | ≤0.0313 to 0.0625 | ≤0.0313 | ≤0.0313 | |
| 50 | Cefditoren | 0.5 to 2 | 1 | 1 | |
| 50 | Clarithromycin | 0.25 to >128 | 0.25 | >128 | |
| 50 | Levofloxacin | 0.125 to 16 | 0.25 | 0.5 | |
| Staphylococcus epidermidis (methicillin susceptible) | 50 | SM-295291 | 0.0625 to 0.125 | 0.0625 | 0.125 |
| 27 | SM-369926 | ≤0.0313 to 0.0625 | 0.0625 | 0.0625 | |
| 50 | Tebipenem | ≤0.0313 | ≤0.0313 | ≤0.0313 | |
| 50 | Cefditoren | 0.125 to 0.5 | 0.25 | 0.25 | |
| 50 | Clarithromycin | 0.125 to >128 | 0.25 | 0.25 | |
| 50 | Levofloxacin | 0.125 to 4 | 0.25 | 0.5 | |
| Streptococcus pneumoniae (penicillin susceptible; penicillin MIC, ≤0.06 μg/ml) | 48 | SM-295291 | ≤0.0039 to 0.0156 | 0.0078 | 0.0078 |
| 27 | SM-369926 | ≤0.0039 to 0.0156 | ≤0.0039 | 0.0078 | |
| 48 | Tebipenem | ≤0.0039 to 0.0156 | ≤0.0039 | ≤0.0039 | |
| 48 | Faropenem | 0.0078 to 0.125 | 0.0156 | 0.0313 | |
| 48 | Cefditoren | 0.0078 to 0.25 | 0.0625 | 0.125 | |
| 48 | Clarithromycin | 0.0625 to >128 | 2 | 128 | |
| 48 | Levofloxacin | 0.5 to 16 | 1 | 2 | |
| Streptococcus pyogenes | 48 | SM-295291 | ≤0.0039 to 0.0078 | 0.0078 | 0.0078 |
| 24 | SM-369926 | ≤0.0039 to 0.0078 | ≤0.0039 | 0.0078 | |
| 48 | Tebipenem | ≤0.0039 | ≤0.0039 | ≤0.0039 | |
| 48 | Cefditoren | ≤0.0039 to 0.0156 | 0.0078 | 0.0156 | |
| 48 | Clarithromycin | 0.0313 to >128 | 0.0625 | 4 | |
| 48 | Levofloxacin | 0.25 to 2 | 0.5 | 2 | |
| Streptococcus agalactiae | 49 | SM-295291 | ≤0.0039 to 0.0313 | 0.0156 | 0.0156 |
| 24 | SM-369926 | ≤0.0039 to 0.0313 | 0.0156 | 0.0156 | |
| 49 | Tebipenem | ≤0.0039 to 0.0156 | 0.0078 | 0.0156 | |
| 49 | Cefditoren | 0.0156 to 0.125 | 0.0313 | 0.0313 | |
| 49 | Clarithromycin | 0.0625 to >128 | 0.125 | >128 | |
| 49 | Levofloxacin | 0.5 to >16 | 1 | >16 | |
| Enterococcus faecalis | 51 | SM-295291 | 0.125 to 2 | 0.5 | 1 |
| 27 | SM-369926 | 0.0625 to 1 | 0.25 | 0.5 | |
| 51 | Tebipenem | 0.25 to 8 | 1 | 4 | |
| 51 | Cefditoren | 8 to >128 | >128 | >128 | |
| 51 | Clarithromycin | 0.25 to >128 | >128 | >128 | |
| 51 | Levofloxacin | 1 to 64 | 2 | 64 | |
| Enterococcus faecium | 41 | SM-295291 | 2 to >128 | 32 | 64 |
| 27 | SM-369926 | 1 to 128 | 32 | 64 | |
| 41 | Tebipenem | 4 to >128 | 128 | >128 | |
| 41 | Cefditoren | 128 to >128 | >128 | >128 | |
| 41 | Clarithromycin | 0.125 to >128 | >128 | >128 | |
| 41 | Levofloxacin | 1 to >128 | 32 | 128 | |
| Moraxella catarrhalis | 44 | SM-295291 | ≤0.0313 to 0.25 | 0.125 | 0.25 |
| 24 | SM-369926 | ≤0.0313 to 0.125 | ≤0.0313 | 0.0625 | |
| 44 | Tebipenem | ≤0.0313 to 0.0625 | ≤0.0313 | 0.0625 | |
| 44 | Cefditoren | ≤0.0313 to 2 | 0.25 | 1 | |
| 44 | Clarithromycin | 0.0625 to 1 | 0.125 | 0.5 | |
| 44 | Levofloxacin | ≤0.0313 to 4 | 0.0625 | 0.125 | |
| Haemophilus influenzae | 41 | SM-295291 | 0.0625 to 0.5 | 0.0625 | 0.25 |
| 27 | SM-369926 | 0.0313 to 1 | 0.0625 | 0.5 | |
| 41 | Tebipenem | 0.0313 to 1 | 0.125 | 0.5 | |
| 41 | Faropenem | 0.25 to 16 | 1 | 8 | |
| 41 | Cefditoren | 0.0078 to 0.5 | 0.0313 | 0.125 | |
| 41 | Clarithromycin | 4 to 32 | 8 | 16 | |
| 41 | Levofloxacin | 0.0078 to 16 | 0.0156 | 0.0313 | |
| Klebsiella pneumoniae | 47 | SM-295291 | 0.125 to 2 | 0.25 | 0.5 |
| 26 | SM-369926 | 0.125 to 2 | 0.25 | 0.5 | |
| 47 | Tebipenem | ≤0.0313 to 0.0625 | ≤0.0313 | ≤0.0313 | |
| 47 | Cefditoren | 0.125 to 1 | 0.25 | 0.5 | |
| 47 | Clarithromycin | 32 to >128 | 128 | 128 | |
| 47 | Levofloxacin | 0.0625 to 2 | 0.125 | 0.125 | |
| Escherichia coli | 50 | SM-295291 | 0.25 to 8 | 0.5 | 1 |
| 26 | SM-369926 | 0.125 to 8 | 0.5 | 1 | |
| 50 | Tebipenem | ≤0.0313 to 1 | ≤0.0313 | ≤0.0313 | |
| 50 | Cefditoren | 0.125 to >128 | 0.25 | 0.5 | |
| 50 | Clarithromycin | 16 to >128 | 64 | >128 | |
| 50 | Levofloxacin | ≤0.0313 to 64 | 0.0625 | 16 | |
| Enterobacter cloacae | 48 | SM-295291 | 1 to 16 | 4 | 8 |
| 27 | SM-369926 | 1 to 16 | 4 | 8 | |
| 48 | Tebipenem | ≤0.0313 to 0.125 | ≤0.0313 | 0.125 | |
| 48 | Cefditoren | 0.25 to >128 | 1 | 64 | |
| 48 | Clarithromycin | 64 to >128 | 128 | 128 | |
| 48 | Levofloxacin | ≤0.0313 to 8 | 0.0625 | 1 | |
| Enterobacter aerogenes | 50 | SM-295291 | 0.125 to 16 | 4 | 8 |
| 26 | SM-369926 | 0.0625 to 8 | 2 | 8 | |
| 50 | Tebipenem | ≤0.0313 to 0.125 | ≤0.0313 | 0.0625 | |
| 50 | Cefditoren | 0.125 to 128 | 1 | 32 | |
| 50 | Clarithromycin | 32 to >128 | 128 | >128 | |
| 50 | Levofloxacin | ≤0.0313 to 1 | 0.125 | 0.125 | |
| Pseudomonas aeruginosa | 50 | SM-295291 | 64 to >128 | >128 | >128 |
| 27 | SM-369926 | 32 to >128 | 128 | >128 | |
| 50 | Tebipenem | 1 to 128 | 8 | 64 | |
| 50 | Cefditoren | 16 to >128 | 64 | >128 | |
| 50 | Clarithromycin | 32 to >128 | >128 | >128 | |
| 50 | Levofloxacin | 0.125 to >128 | 2 | 64 | |
| Acinetobacter spp. | 27 | SM-295291 | 2 to 64 | 16 | 32 |
| 27 | SM-369926 | 1 to 64 | 16 | 32 | |
| 27 | Tebipenem | 0.25 to 16 | 4 | 4 | |
| 27 | Faropenem | 1 to 64 | 16 | 32 | |
| 27 | Cefditoren | 4 to 64 | 32 | 32 | |
| 27 | Clarithromycin | 2 to >128 | 16 | 32 | |
| 27 | Levofloxacin | 0.0625 to 16 | 0.125 | 8 | |
| Neisseria gonorrhoeae | 35 | SM-295291 | 0.0313 to 1 | 0.5 | 1 |
| 14 | SM-369926 | 0.0156 to 0.25 | 0.25 | 0.25 | |
| 35 | Tebipenem | 0.0156 to 0.5 | 0.25 | 0.25 | |
| 35 | Faropenem | 0.0156 to 2 | 2 | 2 | |
| 35 | Cefditoren | ≤0.0039 to 0.25 | 0.0313 | 0.125 | |
| 35 | Clarithromycin | ≤0.0313 to 64 | 0.5 | 4 | |
| 35 | Levofloxacin | 0.0156 to 8 | 4 | 8 | |
| Peptostreptococcus sp. | 38 | SM-295291 | ≤0.0313 to 1 | 0.0625 | 0.125 |
| 26 | SM-369926 | ≤0.0313 to 1 | ≤0.0313 | 0.0625 | |
| 38 | Tebipenem | ≤0.0313 to 0.25 | ≤0.0313 | 0.125 | |
| 38 | Cefditoren | ≤0.0313 to 32 | 0.25 | 8 | |
| 38 | Clarithromycin | ≤0.0313 to >128 | 0.5 | >128 | |
| 38 | Levofloxacin | 0.5 to 128 | 4 | 64 | |
| Bacteroides fragilis | 45 | SM-295291 | ≤0.0313 to 32 | 0.25 | 2 |
| 27 | SM-369926 | 0.0625 to 16 | 0.5 | 2 | |
| 45 | Tebipenem | 0.0625 to 32 | 0.25 | 2 | |
| 45 | Cefditoren | 1 to >128 | 2 | 64 | |
| 45 | Clarithromycin | 0.5 to >128 | 2 | >128 | |
| 45 | Levofloxacin | 0.5 to 32 | 2 | 8 | |
Table 2.
In vitro activities of SM-295291, SM-369926, and selected antimicrobial agents against drug-resistant clinical pathogens
| Organism | n | Antimicrobial agent | MIC (μg/ml) |
||
|---|---|---|---|---|---|
| Range | 50% | 90% | |||
| S. aureus (methicillin resistant; oxacillin MIC, ≥4 µg/ml) | 49 | SM-295291 | 0.5 to >128 | 64 | 128 |
| 27 | SM-369926 | 0.5 to 128 | 64 | 128 | |
| 49 | Tebipenem | 0.5 to 16 | 4 | 16 | |
| 49 | Cefditoren | 8 to >128 | 128 | >128 | |
| 49 | Clarithromycin | 0.25 to >128 | >128 | >128 | |
| 49 | Levofloxacin | 0.25 to >128 | 16 | >128 | |
| S. epidermidis (methicillin resistant; oxacillin MIC, ≥0.5 µg/ml) | 36 | SM-295291 | 0.5 to >128 | 64 | >128 |
| 27 | SM-369926 | 1 to 128 | 16 | 128 | |
| 36 | Tebipenem | 0.25 to 16 | 8 | 16 | |
| 36 | Cefditoren | 1 to 128 | 64 | 128 | |
| 36 | Clarithromycin | 0.25 to >128 | 128 | >128 | |
| 36 | Levofloxacin | 0.25 to 32 | 4 | 16 | |
| S. pneumoniae (penicillin resistant; penicillin MIC, ≥2 µg/ml) | 54 | SM-295291 | 0.0625 to 0.25 | 0.0625 | 0.125 |
| 54 | SM-369926 | 0.0313 to 0.25 | 0.0625 | 0.125 | |
| 54 | Tebipenem | 0.0313 to 0.25 | 0.0625 | 0.125 | |
| 54 | Faropenem | 0.25 to 2 | 0.5 | 1 | |
| 54 | Cefditoren | 0.5 to 8 | 1 | 2 | |
| 54 | Clarithromycin | 0.0625 to >128 | 2 | >128 | |
| 54 | Levofloxacin | 0.5 to >16 | 1 | 2 | |
| H. influenzae (BLNAR; ampicillin MIC, ≥2 μg/ml) | 22 | SM-295291 | 0.125 to 0.5 | 0.25 | 0.25 |
| 22 | SM-369926 | 0.125 to 1 | 0.25 | 1 | |
| 22 | Tebipenem | 0.0625 to 2 | 0.5 | 1 | |
| 22 | Faropenem | 2 to 16 | 8 | 16 | |
| 22 | Cefditoren | 0.0313 to 1 | 0.25 | 0.5 | |
| 22 | Clarithromycin | 4 to 16 | 8 | 16 | |
| 22 | Levofloxacin | 0.0156 to 0.0313 | 0.0156 | 0.0313 | |
| E. coli (ciprofloxacin and ceftazidime resistant; ciprofloxacin MIC, ≥4 μg/ml; ceftazidime MIC, ≥16 μg/ml) | 9 | SM-295291 | 1 to 16 | ||
| 9 | SM-369926 | 1 to 16 | |||
| 9 | Tebipenem | 0.0156 to 1 | |||
| 9 | Faropenem | 1 to 16 | |||
| 9 | Cefditoren | 4 to >128 | |||
| 9 | Clarithromycin | 32 to >128 | |||
| 9 | Levofloxacin | 8 to 64 | |||
| N. gonorrhoeae (ciprofloxacin resistant; ciprofloxacin MIC, ≥1 μg/ml) | 16 | SM-295291 | 0.0313 to 1 | ||
| 16 | SM-369926 | 0.0156 to 1 | |||
| 16 | Tebipenem | ≤0.0039 to 0.25 | |||
| 16 | Faropenem | 0.0078 to 2 | |||
| 16 | Cefditoren | ≤0.0039 to 0.125 | |||
| 16 | Clarithromycin | 0.25 to 2 | |||
| 16 | Levofloxacin | 2 to 32 | |||
The MICs of SM-295291 and SM-369926 against methicillin-susceptible staphylococci ranged from ≤0.0313 to 0.25 μg/ml and were lower than those of all other comparators except TBM. Against the streptococci (except PRSP), the maximum MIC observed for SM-295291 and SM-369926 was 0.0313 μg/ml, which was lower than for all comparators except TBM (0.0156 μg/ml). SM-295291 and SM-369926 had the lowest MICs against Enterococcus faecalis of any tested comparators. SM-295291 and SM-369926 exhibited low to moderate activity against Enterococcus faecium, and the MIC50 and MIC90 for this organism were 32 and 64 μg/ml, respectively.
SM-295291 and SM-369926 demonstrated strong activities, with MIC90s of ≤1 μg/ml against most Gram-negative species, Escherichia coli, Klebsiella pneumoniae, H. influenzae, Moraxella catarrhalis, and N. gonorrhoeae. Unlike TBM (MIC50, 8 μg/ml), SM-295291 and SM-369926 had no activity against P. aeruginosa (MIC50, ≥128 μg/ml). SM-295291 and SM-369926 showed very poor activity against Acinetobacter spp.
SM-295291 and SM-369926 showed potent activity against peptostreptococci and Bacteroides fragilis, with an MIC90 of ≤2 μg/ml, which was similar to that of TBM and lower than those of CDN, CLR, and LVX.
SM-295291 and SM-369926 were less active against methicillin-resistant staphylococci (MIC90, ≥128 μg/ml). The SM-295291 and SM-369926 MIC90 of 0.125 μg/ml for PRSP was comparable to that of TBM and ≥8-fold more potent than FRM, CDN, CLR, and LVX. The MIC90s of SM-295291 and SM-369926 against BLNAR organisms were 0.25 and 1 μg/ml, respectively, which were less than that of LVX but comparable to those of TBM and CDN and ≥16-fold more potent than those of FRM and CLR. Against the ciprofloxacin-resistant isolates of N. gonorrhoeae, the in vitro activities of SM-295291 and SM-369926 were higher than those of FRM and LVX and comparable to that of CLR, but they were lower than those of TBM and CDN. Against ciprofloxacin- and ceftazidime-resistant E. coli, the in vitro activities of SM-295291 and SM-369926 were similar to that of FRM and higher than those of the other comparators, with the exception of TBM. As shown in Table 3, SM-295291 and SM-369926 maintained activity against E. coli and K. pneumoniae producing ESBL, although the MICs of SM-295291 and SM-369926 were higher than those of IPM. For these ESBL-producing isolates, no inoculum effect was observed for SM-295291 and SM-369926 or for IPM.
Table 3.
In vitro antibacterial activities of SM-295291, SM-369926, and IPM against extended-spectrum-β-lactamase-producing bacteria
| Organism | β-Lactamase | MIC (μg/ml) at indicated inoculum size (CFU/ml) |
|||||
|---|---|---|---|---|---|---|---|
| SM-295291 |
SM-369926 |
IPM |
|||||
| 106 | 108 | 106 | 108 | 106 | 108 | ||
| E. coli TL-3138 | CTX-M-44 | 1 | 1 | 1 | 1 | 0.062 | 0.125 |
| E. coli TL-3135 | CTX-M-14 | 1 | 2 | 1 | 2 | 0.125 | 0.125 |
| E. coli TL-3141 | CTX-M-1 | 2 | 2 | 2 | 2 | 0.125 | 0.25 |
| E. coli TL-3180 | SHV-12 | 0.5 | 0.5 | 0.25 | 0.5 | 0.062 | 0.125 |
| K. pneumoniae TL-3139 | CTX-M-1 | 1 | 1 | 1 | 1 | 0.062 | 0.125 |
| K. pneumoniae TL-3149 | SHV | 2 | 4 | 2 | 4 | 0.5 | 1 |
Bactericidal activity.
SM-295291 and SM-369926 were bactericidal against S. aureus, E. coli, and K. pneumoniae in terms of an MBC/MIC ratio of 1 (Table 4).
Table 4.
MICs, MBCs, and MBC/MIC ratios of SM-295291, SM-369926, and TBM
| Strain | SM-295291 |
SM-369926 |
TBM |
||||||
|---|---|---|---|---|---|---|---|---|---|
| MIC (μg/ml) | MBC (μg/ml) | MBC/MIC ratio | MIC (μg/ml) | MBC (μg/ml) | MBC/MIC ratio | MIC (μg/ml) | MBC (μg/ml) | MBC/MIC ratio | |
| S. aureus ATCC 6538P | 0.0313 | 0.0313 | 1 | 0.0156 | 0.0156 | 1 | 0.0039 | 0.0156 | 4 |
| E. coli ATCC 25404 | 0.5 | 0.5 | 1 | 0.5 | 0.5 | 1 | 0.0156 | 0.0313 | 2 |
| K. pneumoniae ATCC 10031 | 0.0625 | 0.0625 | 1 | 0.0313 | 0.0313 | 1 | 0.0156 | 0.0313 | 2 |
In time-kill assays, SM-295291 caused a 2-log reduction in the CFU of S. pneumoniae 18280 (PRSP) at more than the MIC until 2 h, whereas the killing rate of CDN was relatively low (Fig. 2A). After 6 h of incubation, SM-295291 and TBM resulted in a 4-log reduction at more than the MIC and 2× the MIC, respectively. For H. influenzae 17051 (BLNAR), four times the MIC of SM-295291 caused a 2-log reduction after 6 h; its killing kinetics were similar to those of IPM (Fig. 2B). The killing rate of SM-295291 was higher than that of TBM. The killing rate of CDN was lower than that of SM-295291 until 4 h, although CDN only achieved a 3-log reduction at 4× the MIC 6 h after incubation.
Fig 2.

Bactericidal activities of SM-295291 and reference antimicrobial agents against PRSP strain 18280 (A) and BLNAR strain 17051(B). Symbols: ○, control; ▲, 1/4× the MIC; ■, 1/2× the MIC; ●, 1× the MIC; ◆, 2× the MIC; □, 4× the MIC; ♢, IPM concentration at 32 μg/ml.
In vivo efficacy against PRSP and BLNAR organisms.
Prior to all murine experiments, we determined the pharmacokinetics of SM-295291, SM-369926, and MEM administered intravenously at a dose of 10 mg/kg with 2 mg cilastatin in ICR male mice. The concentrations 5 min after administration (C5 mins) of SM-295291, SM-369926, and MEM were 49.8, 53.6, and 21.0 μg/ml, respectively. The areas under the concentration (in serum)-time curve (AUC) for SM-295291, SM-369926, and MEM were 1,383, 1,942, and 481 μg · min/ml, respectively. The half-lives (t1/2s) of SM-295291, SM-369926, and MEM were 21.5, 30.5, and 10.9 min, respectively. SM-295291 and SM-369926 exhibited better pharmacokinetics than MEM in mice.
The MICs of SM-295291, SM-369926, TBM, FRM, CDN, CLR, and LVX against PRSP strain 18280 were 0.125, 0.0625, 0.0625, 0.5, 0.5, 2, and 1 μg/ml, respectively.
In mouse systemic and meningitis infection models with PRSP strain 18280, the ED50s of SM-295291 and SM-369926 were comparable to that of TBM and much lower than that of CDN (Table 5). In a murine pneumonia model, the bacterial count in the lungs of untreated mice on day 3 after infection was 7.19 log CFU (Fig. 3A). SM-295291 and SM-369926 dose dependently reduced bacterial numbers in the lungs following six intravenous injections of 0.32, 1, and 3.2 mg/kg/dose and caused >5-log reduction of bacterial numbers at the 3.2-mg/kg/dose.
Table 5.
In vivo efficacies of SM-295291, SM-369926, TBM, and CDN against PRSP 18280 systemic infection and meningitis in mice
| Antimicrobial agent | ED50, mg/kg (95% confidence interval)a |
|
|---|---|---|
| Systemic infectionb | Meningitisc | |
| SM-295291 | 0.20 (0.083 to 0.48) | 0.72 (0.14 to 1.72) |
| SM-369926 | Not tested | 1.01 (0.45 to 2.25) |
| TBM | 0.34 (0.15 to 0.68) | 1.01 (0.45 to 2.25) |
| CDN | 5.42 (not determined) | 3.23 (0.95 to 10.2) |
ED50 was calculated from survival rates 7 days after infection. Cilastatin at a dose of 100 mg/kg was administered subcutaneously 5 min before carbapenem treatment.
Mice were inoculated intraperitoneally with 1.4 × 104 CFU of PRSP strain 18280. Antimicrobial agents were administered intravenously 1 and 3 h after infection (n = 8).
Mice were challenged intracerebrally with 2 × 104 CFU of PRSP strain 18280. Antimicrobial agents were administered intravenously 5 h after infection and twice daily 1 and 2 days after infection (n = 8).
Fig 3.
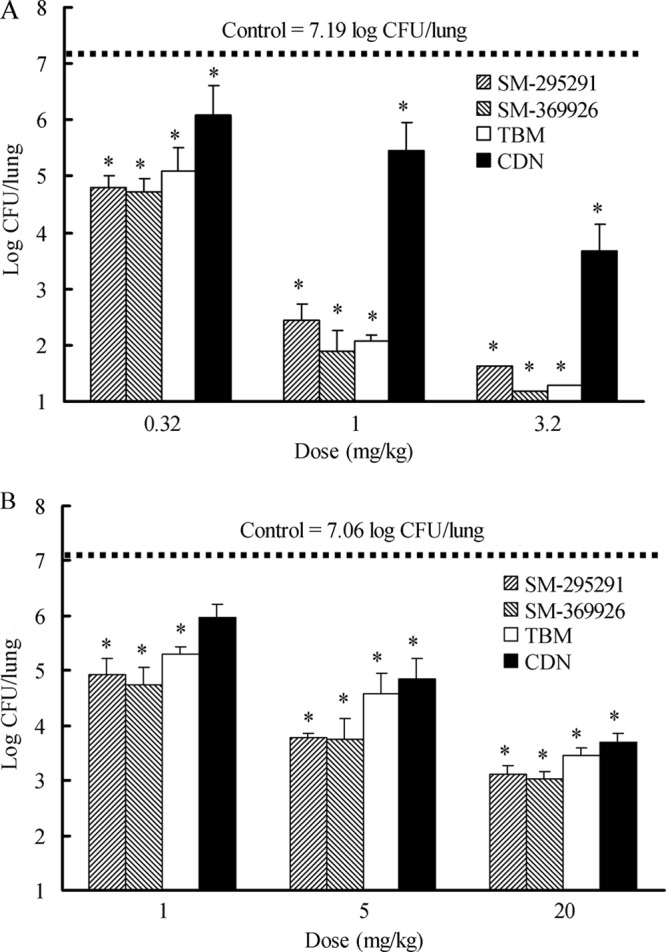
Effects of antimicrobial agents on the bacterial burden in the lungs of mice infected intranasally with PRSP strain 18280 at 1.7 × 106 CFU/mouse (A) and BLNAR strain 17051 at 4.4 × 107 CFU/mouse (B). Antibacterial agents were administered intravenously thrice daily 1 day and 2 days after infection (n = 6). Cilastatin at a dose of 100 mg/kg was administered subcutaneously 5 min before carbapenem treatment. The lungs were removed 3 days after infection. The values represent the means and standard deviations. The dotted line represents the mean bacterial burden in the lungs for the control group. *, significantly different from control (P < 0.01 by Dunnett's test for multiple comparisons).
The MICs of SM-295291, SM-369926, TBM, and CDN against BLNAR strain 17051 were 0.125, 0.125, 0.5, and 0.25 μg/ml, respectively. The bacterial count in the lungs of untreated mice on day 3 after infection was 7.06 log CFU (Fig. 3B). Dose-dependent effects of SM-295291 and SM-369926 were observed: treatment with 1, 5, and 20 mg/kg resulted in 2-log, 3-log, and 4-log reductions of bacterial numbers in the lungs, respectively.
In vitro serial passage of P. aeruginosa.
We evaluated the risk of the emergence of carbapenem-resistant P. aeruginosa after the clinical use of our 2-aryl carbapenems with oral application. The MICs of SM-295291, SM-369926, TBM, ERM, IPM, and MEM against P. aeruginosa PAO1 were 128, 64, 2, 4, 1, and 0.25 μg/ml, respectively. Since the blood concentration of oral antibiotics generally achieves less than 8 μg/ml, serial passages of P. aeruginosa PAO1 were done in the presence of SM-295291 (8 μg/ml), SM-369926 (8 μg/ml), TBM (initial concentration, 1 μg/ml), or ERM (initial concentration, 2 μg/ml). Against P. aeruginosa PAO1, the MICs of SM-295291 and SM-369926 against were always within 2-fold of the initial values during 14 daily passages, whereas the MICs of TBM and ERM increased 32-fold from 2 to 64 μg/ml and 16-fold from 4 to 64 μg/ml, respectively (Fig. 4). Exposure to SM-295291 and SM-369926 had little to no impact on the MICs of IPM and MEM (1 and 0.5 μg/ml, respectively); in contrast, passages in subinhibitory levels of TBM resulted in development of cross-resistance to IPM and MEM (both MICs were 16 μg/ml). The exposure to ERM subinhibitory concentrations yielded a 2-fold increase in IPM MIC (2 μg/ml) and an 8-fold increase in MEM MIC (2 μg/ml).
Fig 4.
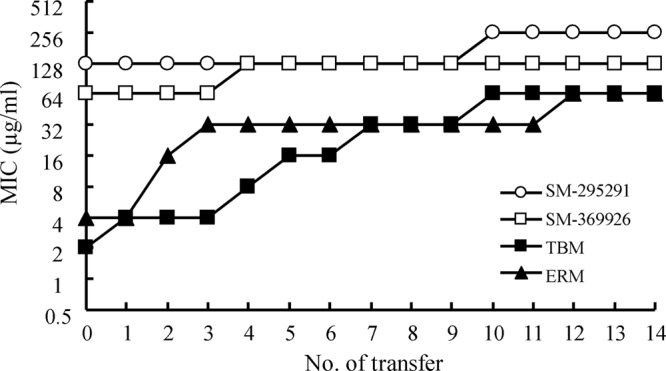
In vitro serial passage study of P. aeruginosa PAO1.
Pharmacokinetics in laboratory animals.
SM-295291 and SM-369926 were more stable to hydrolysis by human DHP-I than IPM: the relative hydrolysis rates of SM-295291 and SM-369926 were 0.55 and 0.46, respectively, compared to which the rate of IPM was assigned a value of 1. The stability of SM-295291 and SM-369926 to hydrolysis by human DHP-I was equal to that of MEM: the relative hydrolysis rates of SM-295291 and SM-369926 were 0.85 and 0.99, respectively, compared to which the rate of MEM was assigned a value of 1. In contrast to human and dog DHP-I (0.29, ratio of susceptibility compared with IPM), rat DHP-I rapidly hydrolyzed SM-295291, and the ratio of susceptibility was 9.24.
To avoid this species-specific effect by rat DHP-I on the metabolism of carbapenems in rats, SM-295291 and SM-369926 were administered with the DHP-I inhibitor cilastatin to rats in subsequent pharmacokinetic analysis. The AUCs from 0 to 3 h (AUC0–3s) and t1/2s of SM-295291 and SM-369926 at a dose of 1 mg/kg were 1.13 to 1.69 μg · h/ml and 0.39 to 0.56 h, respectively, in dogs and monkeys (Table 6). The AUC0–3s (6.41 and 5.35 μg · h/ml) and t1/2s (0.85 and 0.45 h) of SM-295291 and SM-369926 in rats were higher than or equal to those in dogs and monkeys, probably due to the coadministratiion of cilastatin and/or higher rat plasma protein binding (87.3% in rats versus 32.2% in dogs for SM-295291). Sumita et al. reported that the t1/2s of MEM at a dose of 20 mg/kg in dogs and monkeys were 0.68 and 0.52 h, respectively (18), i.e., almost equal to those of SM-295291 and SM-369926.
Table 6.
Pharmacokinetic parameters of intravenous administration of SM-295291 and SM-369926 and intraduodenal or oral administration of their ester prodrugs in animals
| Carbapenema | Parameter | Rat | Dog | Monkey |
|---|---|---|---|---|
| SM-295291 | C5 min (μg/ml) | 9.64 | 2.73 | 3.90 |
| AUC0–3 (μg · h/ml) | 6.41 | 1.49 | 1.59 | |
| t1/2 (h) | 0.85 | 0.56 | 0.40 | |
| Vssb (liter/kg) | 0.12 | 0.33 | 0.21 | |
| SM-368589 (ester prodrug) | Cmaxc (μg/ml) | 0.42 | 0.96 | 0.18 |
| AUC0–3 (μg · h/ml) | 0.51 | 0.93 | 0.21 | |
| Fd (%) | 8.0 | 62.3 | 12.9 | |
| SM-369926 | C5 min (μg/ml) | 7.91 | 2.82 | 3.23 |
| AUC0–3 (μg · h/ml) | 5.35 | 1.69 | 1.13 | |
| t1/2 (h) | 0.45 | 0.56 | 0.39 | |
| Vss (liter/kg) | 0.11 | 0.32 | 0.20 | |
| SM-375769 (ester prodrug) | Cmax (μg/ml) | 0.78 | 0.63 | 0.05 |
| AUC0–3 (μg · h/ml) | 0.93 | 0.57 | 0.05 | |
| Fd (%) | 17.1 | 34.2 | 4.2 |
Three animals in each group were administered carbapenem at 1 mg/kg.
Vss, volume of distribution at steady state.
Cmax, maximum concentration of drug in serum.
F, bioavailability.
SM-295291 and SM-369926 were relatively stable after a 4-h incubation at 37°C in human plasma, although their residual percentages (44% and 22%, respectively) were lower than those of IPM and MEM (60% and 70%, respectively). Considering the half-life of IPM and MEM in humans (1 h) (19), the pharmacokinetics of SM-295291 and SM-369926 in humans may not be greatly influenced by their stability in human plasma over 4 h. SM-295291 and SM-369926 were not highly bound to human plasma protein (43% and 64%, respectively), although the protein binding rates of IPM and MEM were low (2% and 16%, respectively).
These results suggested that SM-295291 and SM-369926 may show pharmacokinetics similar to those of MEM in humans.
Pharmacokinetic parameters following intraduodenal or oral administration of ester prodrugs.
Since carbapenems are very slightly lipophilic and are hardly absorbed from the gastrointestinal tract when administered orally (20), no non-prodrug carbapenems are being developed for use as oral therapy. Based on our previous study of a suitable series of ester prodrug, we selected medoxomil ester, because of good oral absorption and the risk of formaldehyde generation from a pivoxil ester (21), although pivoxil esters of 2-aryl carbapenems also showed good oral absorption.
Because SM-295291 and SM-369926 were unstable at normal gastric pH, SM-368589 and SM-375769, which are medoxomil esters of SM-295291 and SM-369926, were administered intraduodenally to rats with cilastatin and orally to dogs and monkeys with omeprazole, which inhibits gastric acid secretion. The oral bioavailabilities of SM-368589 were 8.0, 62.3, and 12.9% and of SM-375769 were 17.1, 34.2, and 4.2% in rats, dogs, and monkeys, respectively (Table 6).
Toxicity study.
Since carbapenems have been suggested to induce convulsive side effects and have nephrotoxicity in experimental animals and humans (19, 22), preliminary toxicity studies of SM-295291 and SM-369926 were carried out. Intracerebroventricular administration of 50 μg IPM resulted in convulsions in all mice. The administration of 200, 280, and 400 μg SM-295291 or SM-369926 resulted in incidence rates of 0, 20, and 70% or 0, 0, and 20%, respectively. The administration of 50, 100, and 200 μg TBM resulted in incidence rates of 10, 80, and 90%, respectively. The ED50s of SM-295291 and SM-369926, which induced convulsions in 50% of mice, were 348.8 and >400 μg/mouse, respectively, and were higher than that of TBM (82.2 μg/mouse), suggesting that SM-295291 and SM-369926 had low central nervous system (CNS) toxicity. SM-295291 at 100 mg/kg did not change blood urea nitrogen, blood creatinine, urinary pH, or ratio of kidney weight to body weight (SM-295291 versus pretreatment, 15 versus 20.5 mg/dl, 0.65 versus 0.70 mg/dl, 8.5 versus 8.5, and 0.5%, respectively). Urinary glucose, urinary protein, macroscopic abnormalities, and histopathological abnormalities were not detected in rabbits administered SM-295291 at 100 mg/kg. Therefore, no renal toxicity was seen with SM-295291 at a dose of at least 100 mg/kg in rabbits. In our general safety assessments, all studies indicated that SM-295291 and SM-369926 had no major adverse effects.
DISCUSSION
SM-295291 and SM-369926 have ideal drug properties for the treatment of community-acquired infections, because these compounds show strong (bactericidal), broad-spectrum antibacterial activity against important pathogens of community-acquired infections such as staphylococci, streptococci (including PRSP), E. faecalis, M. catarrhalis, H. influenzae (including BLNAR H. influenzae), Enterobacteriaceae (including ESBL producers), N. gonorrhoeae (including ciprofloxacin-resistant strains), and anaerobes but no activity against nontarget nosocomial pathogens such as P. aeruginosa and Acinetobacter spp. These profiles were due to construction from a unique carbapenem skeleton (desmethyl-carbapenems) and a unique C-2 side chain (having 2-aryl moiety).
The excellent antimicrobial activities of SM-295291 and SM-369926 could be confirmed in PRSP and BLNAR organism infection models. The therapeutic efficacies of SM-295291 and SM-369926 were equal or superior to that of TBM, which is the only oral carbapenem agent on the market, and were greater than that of CDN, which is a representative oral cephalosporin, in PRSP and BLNAR organism infection models, suggesting that SM-295291 and SM-369926 could be effective in clinical infections due to these resistant bacteria. The in vivo activities of SM-295291, SM-369926, TBM, and CDN against PRSP and BLNAR organisms showed a correlation with their MIC and in vitro early bactericidal activity.
The unbound fraction of the drug (non-protein bound) is only available for inhibiting bacterial cell growth, and thus the protein-binding properties of antibiotics need to be considered in order to predict their clinical efficacies (23). Since SM-295291 and SM-369926 had moderate degrees of protein binding (43 to 64%), the effect of 4% human serum albumin on their MICs was assessed against the type strains of Gram-positive and -negative bacteria. The presence of 4% human serum albumin had a small effect; the MICs of SM-295291 and SM-369926 against S. aureus ATCC 6538P, E. coli ATCC 25404, K. pneumoniae ATCC 10031, S. pneumoniae ATCC 6305, and H. influenzae ATCC 9334 were within 1 dilution, except for MIC of SM-369926 against S. aureus ATCC 6538P (2 dilutions). Although CDN is a highly protein-bound antibiotic (about 90%), it shows clinical efficacy in respiratory tract infection (24). These observations suggest that the protein binding rates of SM-295291 and SM-369926 may not significantly affect their clinical antimicrobial activities.
Our study suggests that SM-295291 and SM-369926 with parenteral application could have pharmacokinetics similar to those of the existing carbapenems in humans. In the ester prodrug approach, SM-368589 and SM-375769 showed good oral bioavailability in all animals, although the oral bioavailabilities of SM-368589 and SM-375769 differed among animal species. Other groups of investigators reported that the bioavailabilities of TBM-pivoxil were 59, 35, and 45% and of cefcapene-pivoxil were 14, 6, and 21% in rats, dogs, and monkeys, respectively (25, 26). For CDN-pivoxil, these were 20 and 10% in rats and dogs, respectively (27). Based on these literature data for the bioavailability of existing ester prodrugs of β-lactam agents in animals, it could be expected that SM-368589 and SM-375769 will show oral absorption in humans.
We found that SM-295291 and SM-369926 had good safety profiles. IPM and panipenem (PAM) at a higher dose cause acute renal injuries in animals (22). These renal injuries are closely related to the high intracellular concentration of these agents in renal tubules (22). To inhibit IPM and PAM uptake into the tubular epithelium and prevent their nephrotoxicity, IPM and PAM are coadministered with the anion transport inhibitors cilastatin and betamipron, respectively (22). Besides the higher stability against human DHP-I, SM-295291 had low renal toxicity in rabbits; therefore, coadministration of cilastatin or betamipron may not be necessary with SM-295291 in humans.
Biologically active β-lactam antibiotics in the gut lumen can affect the intestinal microbial flora, causing postantibiotic diarrhea (28, 29). SM-368589 and SM-375769 may have no antibacterial activity before their ester bond is hydrolyzed; this may occur during its passage through the small-intestine wall (10, 29, 30). In addition to improved oral bioavailability, esterification of SM-295291 and SM-369926 would make them likely to have little impact on the intestinal microbial flora compared with non-prodrug agents.
Owing to a simple synthetic route, 2-aryl carbapenems are expected to have markedly low manufacturing costs compared to carbapenems with a thiol side chain and 1β-methyl, for example, MEM and TBM. Besides a good safety and pharmacokinetic profile because of a unique carbapenem skeleton and a unique side chain, this economic advantage is also a key point in the development of oral antibiotics.
Since the adequate antibacterial properties (with typically an MIC90 of ≤1 μg/ml against clinically important pathogens), safety properties, and pharmacokinetic properties of our 2-aryl carbapenems seem favorable for not only parenteral formulation but also oral formulation, we believe that combinational development of these formulations is the best way to make effective use of their properties. Hospitalized patients with severe community-acquired infections should be treated initially with parenteral agents and could be switched to oral therapy when the clinical status improves. This switch therapy is gaining acceptance as a means of facilitating early discharge of patients from the hospital and reducing the overall costs of antimicrobial therapy (6–8). In the case of our 2-aryl carbapenems, this treatment strategy for community-acquired infections could also contribute to preserving the therapeutic efficacy of existing antipseudomonal carbapenems, which are key antibiotics for hospital-acquired infections. Our serial-passage study suggests that there is a low risk of selection of antipseudomonal carbapenem-resistant P. aeruginosa after the clinical use of our 2-aryl carbapenems with oral application and supports the above expectation. However, because our 2-aryl carbapenems may be hydrolyzed by carbapenemase of Enterobacteriaceae such as KPC and OXA-48-like, there is a possibility of selection of carbapenem-resistant P. aeruginosa via carbapenemase-producing Enterobacteriaceae due to use of our 2-aryl carbapenems.
We are continuing preclinical investigations of SM-295291, SM-369926, and their ester prodrugs for development into potential therapeutic agents for community-acquired infections.
In conclusion, a new category of antibiotic, 2-aryl carbapenems, showed an ideal broad spectrum for the treatment of community-acquired infections, including infections caused by conventional antibiotic-resistant pathogens, but no activity against hospital pathogens such as P. aeruginosa, and had good safety and pharmacokinetic profiles. These results suggest that these new 2-aryl carbapenems are promising candidates as novel therapeutic agents for parenteral, oral, and switching from parenteral to oral treatment of community-acquired infections.
ACKNOWLEDGMENTS
We thank K. Kubota for suggestions and acknowledge the excellent technical assistance of K. Urasaki and Y. Hirai.
Footnotes
Published ahead of print 12 November 2012
REFERENCES
- 1. Saga T, Yamaguchi K. 2009. History of antimicrobial agents and resistant bacteria. Jpn. Med. Assoc. J. 52:103–108 [Google Scholar]
- 2. Pakyz AL, Oinonen M, Polk RE. 2009. Relationship of carbapenem restriction in 22 university teaching hospitals to carbapenem use and carbapenem-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53:1983–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Livermore DM, Carter MW, Bagel S, Wiedemann B, Baquero F, Loza E, Endtz HP, van Den Braak N, Fernandes CJ, Fernandes L, Frimodt-Moller N, Rasmussen LS, Giamarellou H, Giamarellos-Bourboulis E, Jarlier V, Nguyen J, Nord CE, Struelens MJ, Nonhoff C, Turnidge J, Bell J, Zbinden R, Pfister S, Mixson L, Shungu DL. 2001. In vitro activities of ertapenem (MK-0826) against recent clinical bacteria collected in Europe and Australia. Antimicrob. Agents Chemother. 45:1860–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Daigle DM, Xerri L. 2008. Susceptibility of Pseudomonas aeruginosa to imipenem is unaffected by exposure to PZ-601 (SMP-601), poster C1-1064. Abstr. 48th Intersci. Conf. Antimicrob. Agents Chemother., Washington, DC, 25 to 28 October 2008 American Society for Microbiology, Washington, DC [Google Scholar]
- 5. Livermore DM, Mushtaq S, Warner M. 2005. Selectivity of ertapenem for Pseudomonas aeruginosa mutants cross-resistant to other carbapenems. J. Antimicrob. Chemother. 55:306–311 [DOI] [PubMed] [Google Scholar]
- 6. Lee RWW, Lindstrom ST. 2007. Early switch to oral antibiotics and early discharge guidelines in the management of community-acquired pneumonia. Respirology 12:111–116 [DOI] [PubMed] [Google Scholar]
- 7. Ramirez JA. 1995. Switch therapy in community-acquired pneumonia. Diagn. Microbiol. Infect. Dis. 22:219–223 [DOI] [PubMed] [Google Scholar]
- 8. Ramirez JA, Srinath L, Ahkee S, Huang A, Raff MJ. 1995. Early switch from intravenous to oral cephalosporins in the treatment of hospitalized patients with community-acquired pneumonia. Arch. Intern. Med. 155:1273–1276 [PubMed] [Google Scholar]
- 9. Sunagawa M, Ueda Y, Okada S, Watanabe S, Hashizuka T, Hori S, Sasaki A, Eriguchi Y, Kanazawa K. 2005. Orally active carbapenem antibiotics. I. Antibacterial and pharmacokinetic potential of 2-phenyl and 2-thienylcarbapenems. J. Antibiot. 58:787–803 [DOI] [PubMed] [Google Scholar]
- 10. Mizen L, Burton G. 1998. The use of esters as prodrugs for oral delivery of beta-lactam antibiotics. Pharm. Biotechnol. 11:345–365 [DOI] [PubMed] [Google Scholar]
- 11. Takemoto K, Hatano K, Eguchi K, Hirai Y, Kanazawa K, Sunagawa M, Ueda Y. 2011. A new parenteral and oral 2-aryl carbapenem (CB): I. In vitro activity against antibiotic-susceptible and -resistant causative pathogens of community-acquired infections (CAIs), poster F1-139. Abstr. 51st Intersci. Conf. Antimicrob. Agents Chemother., Chicago, IL, 17 to 20 September 2011 American Society for Microbiology, Washington, DC [Google Scholar]
- 12. Fujimoto K, Hatano K, Nakai T, Terashita S, Eriguchi Y, Urasaki K, Shimizudani T, Sato K, Kanazawa K, Sunagawa M, Ueda Y. 2011. A new parenteral and oral 2-aryl carbapenem (CB): II. Therapeutic efficacy against PRSP and BLNAR infection models and pharmacokinetics (PKs) in laboratory animals, poster F1-140. Abstr. 51st Intersci. Conf. Antimicrob. Agents Chemother., Chicago, IL, 17 to 20 September 2011 American Society for Microbiology, Washington, DC [Google Scholar]
- 13. Ueda Y, Kanazawa K, Eguchi K, Takemoto K, Eriguchi Y, Sunagawa M. 2005. In vitro and in vivo antibacterial activities of SM-216601, a new broad-spectrum parenteral carbapenem. Antimicrob. Agents Chemother. 49:4185–4196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. National Committee for Clinical Laboratory Standards 2001. Methods for antimicrobial susceptibility testing of anaerobic bacteria, 5th ed; approved standard M11-A5. National Committee for Clinical Laboratory Standards, Wayne, PA [Google Scholar]
- 15. Fukasawa M, Sumita Y, Harabe ET, Tanio T, Nouda H, Kohzuki T, Okuda T, Matsumura H, Sunagawa M. 1992. Stability of meropenem and effect of 1β-methyl substitution on its stability in the presence of renal dehydropeptidase I. Antimicrob. Agents Chemother. 36:1577–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sunagawa M, Itoh M, Kubota K, Sasaki A, Ueda Y, Angehrn P, Bourson A, Goetschi E, Hebeisen P, Then RL. 2002. New anti-MRSA and anti-VRE carbapenems; synthesis and structure-activity relationships of 1beta-methyl-2-(thiazol-2-ylthio) carbapenems. J. Antibiot. 55:722–757 [DOI] [PubMed] [Google Scholar]
- 17. Sunagawa M, Matsumura H, Sumita Y, Nouda H. 1995. Structural features resulting in convulsive activity of carbapenem compounds: effect of C-2 side chain. J. Antibiot. 48:408–416 [DOI] [PubMed] [Google Scholar]
- 18. Sumita Y, Nouda H, Tada E, Kohzuuki T, Kato M, Okuda T, Fukasawa M. 1992. Pharmacokinetics of meropenem, a new carbapenem antibiotic, parenterally administrated to laboratory animals. Chemotherapy 40(Suppl 1):123–131 [Google Scholar]
- 19. Nicolau DP. 2008. Carbapenems: a potent class of antibiotics. Expert Opin. Pharmacother. 9:23–37 [DOI] [PubMed] [Google Scholar]
- 20. Torii M, Takiguchi Y, Izumi M, Fukushima T, Yokota M. 2002. Carbapenem antibiotics inhibit valproic acid transport in Caco-2 cell monolayers. Int. J. Pharm. 233:253–256 [DOI] [PubMed] [Google Scholar]
- 21. Nakashima M, Uematsu T, Oguma T, Yoshida T, Mizojiri K, Matsuno S, Yamamoto S. 1992. Phase I clinical studies of S-1108: safety and pharmacokinetics in a multiple-administration study with special emphasis on the influence on carnitine body stores. Antimicrob. Agents Chemother. 36:762–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hirouchi Y, Naganuma H, Kawahara Y, Okada R, Kamiya A, Inui K, Hori R. 1994. Preventive effect of betamipron on nephrotoxicity and uptake of carbapenems in rabbit renal cortex. Jpn. J. Pharmacol. 66:1–6 [DOI] [PubMed] [Google Scholar]
- 23. Andes D, Craig WA. 2002. Animal model pharmacokinetics and pharmacodynamics: a critical review. Int. J. Antimicrob. Agents 19:261–268 [DOI] [PubMed] [Google Scholar]
- 24. Sevillano D, Aguilar L, Alou L, Gimenez Gonzalez M-JN, Torrico M, Cafini F, Fenoll A, Coronel P, Prieto J. 2008. High protein binding and cidal activity against penicillin-resistant S. pneumoniae: a cefditoren in vitro pharmacodynamic simulation. PLoS One 3:e2717 doi:10.1371/journal.pone.0002717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kijima K, Morita J, Suzuki K, Aoki M, Kato K, Hayashi H, Shibasaki S, Kurosawa T. 2009. Pharmacokinetics of tebipenem pivoxil, a novel oral carbapenem antibiotic, in experimental animals. Jpn. J. Antibiot. 62:214–240 [PubMed] [Google Scholar]
- 26. Kimura Y, Nakano M, Nakashimizu H, Nakamoto S, Watanabe Y, Otubo S, Matubara T, Sato S, Nara H, Yoshida T. 1993. Pharmacokinetics of S-1108, a new oral cephalosporin, in experimental animals. Chemotherapy 41(Suppl 1):163–176 [Google Scholar]
- 27. Matsumoto T, Okamoto J, Saito K, Aizawa K, Komiya I. 1992. Pharmacokinetics of ME1207, a novel oral cephem antibiotic, in experimental animals. Chemotherapy 40(Suppl 2):120–130 [Google Scholar]
- 28. Mitropoulos IF, Rotschafer JC, Rodvold KA. 2007. Adverse events associated with the use of oral cephalosporins/cephems. Diagn. Microbiol. Infect. Dis. 57:67S–76S [DOI] [PubMed] [Google Scholar]
- 29. Sjövall J, Huitfeldt B, Magni L, Nord CE. 1986. Effect of beta-lactam prodrugs on human intestinal microflora. Scand. J. Infect. Dis. Suppl. 49:73–84 [PubMed] [Google Scholar]
- 30. Stoeckel K, Hayton WL, Edwards DJ. 1995. Clinical pharmacokinetics of oral cephalosporins. Antibiot. Chemother. 47:34–71 [DOI] [PubMed] [Google Scholar]