

Abstract
Although artesunate is clearly superior, parenteral quinine is still used widely for the treatment of severe malaria. A loading-dose regimen has been recommended for 30 years but is still often not used. A population pharmacokinetic study was conducted with 75 Tanzanian children aged 4 months to 8 years with severe malaria who received quinine intramuscularly; 69 patients received a loading dose of 20 mg quinine dihydrochloride (salt)/kg of body weight. Twenty-one patients had plasma quinine concentrations detectable at baseline. A zero-order absorption model with one-compartment disposition pharmacokinetics described the data adequately. Body weight was the only significant covariate and was implemented as an allometric function on clearance and volume parameters. Population pharmacokinetic parameter estimates (and percent relative standard errors [%RSE]) of elimination clearance, central volume of distribution, and duration of zero-order absorption were 0.977 liters/h (6.50%), 16.7 liters (6.39%), and 1.42 h (21.5%), respectively, for a typical patient weighing 11 kg. Quinine exposure was reduced at lower body weights after standard weight-based dosing; there was 18% less exposure over 24 h in patients weighing 5 kg than in those weighing 25 kg. Maximum plasma concentrations after the loading dose were unaffected by body weight. There was no evidence of dose-related drug toxicity with the loading dosing regimen. Intramuscular quinine is rapidly and reliably absorbed in children with severe falciparum malaria. Based on these pharmacokinetic data, a loading dose of 20 mg salt/kg is recommended, provided that no loading dose was administered within 24 h and no routine dose was administered within 12 h of admission. (This study has been registered with Current Controlled Trials under registration number ISRCTN 50258054.)
INTRODUCTION
Malaria kills over 2,000 people each day. Young children in Africa account for over 85% of the malaria-associated mortality worldwide (1). The case-fatality rate of pediatric severe malaria usually exceeds 10%, with the highest mortality rates within the first 24 h (2, 3). Parenteral quinine has been the mainstay of severe malaria treatment for much of the past century (4). Although parenteral artesunate is now firmly established as the treatment of choice, availability is still limited, and so quinine remains very widely used (2).
Modern quinine dosing regimens were initially based on studies with Asian adults (5, 6) and have been extrapolated to African children, although there is relatively little detailed pharmacokinetic information for this important group (7–11). The currently recommended dosing regimen is a loading dose of 20 mg quinine dihydrochloride (salt)/kg of body weight followed by 10 mg/kg every 8 h, given by rate-controlled intravenous (i.v.) infusion or intramuscular injection (4). Peak quinine concentrations are achieved within 4 h after intramuscular injection, which is more practicable and may be safer in resource-limited settings where intravenous infusions cannot be established or reliably monitored (8, 10, 12).
Quinine is metabolized primarily via the cytochrome P450 enzyme CYP3A4, and the more polar metabolites are eliminated mainly by renal excretion (13–15). The major metabolite, 3-hydroxyquinine, contributes approximately 10% of the antimalarial activity of the parent compound (16). The pharmacokinetic properties of quinine are affected by the severity of infection as well as age (5, 10). The apparent volume of distribution (V/F) and elimination clearance (CL/F) values are significantly reduced in proportion to increased disease severity (5, 17), partly because of increased quinine binding to plasma proteins, mainly alpha 1-acid glycoprotein (AGP) (18, 19). Quinine clearance is also altered by some drug interactions (20). The unbound plasma fraction of quinine determines the therapeutic and toxic activity of the drug (21). In children under 2 years of age with severe malaria, unbound quinine concentrations were found to be slightly higher than those in older children and adults (10). Although quinine is generally well tolerated, the therapeutic window of unbound drug is relatively narrow, and toxic side effects include hypoglycemia (even in the therapeutic range), cardiotoxicity, ototoxicity, and ocular toxicity (22–25).
The rapid achievement of therapeutic quinine concentrations while avoiding toxicity is of vital importance in the treatment of severe malaria. Despite its extensive use in millions of critically ill children at risk of death, there are very few detailed assessments of the pharmacokinetic properties in the target population (7). The primary aim of this study was to characterize the population pharmacokinetic profile of intramuscular quinine after a loading dose in children with severe malaria in Tanzania. A limited pharmacodynamic assessment was included as a secondary aim.
MATERIALS AND METHODS
Study design, patients, and procedures.
This pharmacokinetic assessment of quinine was part of the AQUAMAT trial (Current Controlled Trials registration number ISRCTN 50258054), a large multinational trial comparing quinine and artesunate for the treatment of severe malaria, the results of which were reported elsewhere previously (2). This substudy was conducted in the Teule Hospital in Muheza, Tanzania, from May 2009 to July 2010. Apart from the additional blood sampling, procedures of the current study were part of the AQUAMAT study protocol. The clinical assessment was reported in full previously (2). Approval for the study, including the pharmacokinetic substudy, was obtained from the Tanzania Medical Research Coordinating Committee and the Oxford Tropical Research Ethics Committee. A total of 21 patients were coenrolled in the FEAST trial, evaluating fluid bolus therapy in children with compensated shock (26).
Children ≤14 years of age with a clinical diagnosis of severe malaria confirmed by a parasite lactate dehydrogenase (pLDH)-based rapid diagnostic test (Optimal; Diamed, Cressier, Switzerland) were recruited, provided that written informed consent was given by their parent or guardian. Severe malaria was defined by at least one of the following criteria: coma (Glasgow coma scale [GCS] score of ≤10 or Blantyre coma scale [BCS] score of ≤2 in preverbal children), convulsions (with a duration of >30 min or ≥2 convulsions in the 24 h before admission), respiratory distress (nasal alar flaring, costal indrawing/recession or use of accessory muscles, and severe tachypnea) or acidotic breathing (“deep” labored breathing), shock (capillary refill time of ≥3 s, temperature gradient, and/or systolic blood pressure of <70 mm Hg), severe symptomatic anemia (hemoglobin level of <5 g/dl with respiratory distress), hypoglycemia (blood glucose level of <3 mmol/liter), hemoglobinuria, severe jaundice, or a convincing history of anuria or oliguria in older children. Patients who had received full treatment with parenteral quinine or a parenteral artemisinin derivative for more than 24 h before admission were excluded.
A physical examination was performed at admission, and a venous blood sample was taken for an assessment of peripheral blood parasitemia, quantitative assessment of plasma Plasmodium falciparum histidine-rich protein 2 (PfHRP2) (a marker of the total-body parasite burden) (27), HIV serology (SD Bio-Line HIV 1/2 3.0 [Standard Diagnostics Inc., Kyonggi-do, South Korea] and Determine HIV-1/2 [Abbott Laboratories, IL]), blood culture, liver function tests (aspartate transaminase [AST], alanine aminotransferase [ALT], gamma-glutamyl transpeptidase, total bilirubin, creatinine, and urea) (Reflotron; Roche Diagnostics), hematocrit (Hct), biochemistry, and acid-base parameters (EC8+ cartridge for an i-STAT handheld blood analyzer). Hematocrit was reported by the i-STAT instrument or, when not available, derived from hemoglobin (Hb) measured by a Haemocue instrument (n = 5). At discharge, a neurological examination was performed.
Antimalarial treatment.
Quinine dihydrochloride (Indus Pharma, Karachi, Pakistan) was given as an intramuscular injection. A loading dose (20 mg salt/kg) was given shortly after admission, followed by 10 mg/kg every 8 h. In cases where the patient had received pretreatment with either a quinine loading dose (20 mg/kg) within 24 h or a maintenance dose (10 mg/kg) within 12 h before enrollment, quinine treatment after study enrollment was continued with 10 mg/kg (i.e., no loading dose). Quinine was diluted with normal saline to a concentration of 60 mg salt/ml and injected into the anterior thigh. The dosing was based on body weight, and injection volumes of over 3 ml were split and divided over both thighs. When the patient was well enough to take oral medication, but after a minimum of 24 h (3 doses) of intramuscular quinine, a full course of oral artemether-lumefantrine (Co-artem; Novartis, Basel, Switzerland) was given to complete the treatment.
Patient management.
The majority of patients receiving quinine received an intravenous infusion with 5% glucose. Vital signs and glucose levels were monitored at least every 6 h and with any deterioration in clinical condition. Low blood glucose levels (defined here as blood glucose levels of <3 mmol/liter) were treated with an i.v. 5-ml/kg 10% glucose bolus. A blood transfusion (20 ml/kg) was given to children with a hematocrit value of ≤15% or an Hb level of <5 g/dl. All children were treated empirically with i.v. antibiotics (ampicillin and gentamicin or ceftriaxone if clinically suspected of having sepsis or meningitis) until blood culture results were known or changed according to antibiotic sensitivity analysis. Convulsions were treated with diazepam or phenobarbitone if they persisted. A peripheral blood smear was repeated after 24 h.
Blood sampling.
Blood samples (1.5 ml) were drawn from an indwelling catheter into lithium-heparin tubes before the first dose (at baseline), and 4 subsequent samples were taken from each patient at preset random times in the time windows of 1 to 4 h, 4 to 8 h, 12 to 16 h, and 20 to 24 h after the injection of the first dose. The sampling windows were selected in order to cover the whole concentration-time profile during the first 24 h. More intensive sampling was practically difficult and ethically challenging considering the clinical setting and the age and disease severity of the patients. The randomization of sample times was done by computer-generated randomization with STATA, version 10 (Stata Corp., TX).
Blood samples were centrifuged at 2,000 × g for 10 min to obtain plasma. Duplicate plasma samples (0.5 ml) were stored at −80°C and sent to the AMBRELA/NIMR laboratory in Tanga, Tanzania, for plasma quinine quantification. Quinine drug content and quality were checked in ampoules taken randomly from the purchase lots (see the supplemental material in reference 2).
Drug analysis.
Total quinine in plasma samples was quantified by using high-performance liquid chromatography with UV detection (28). Quinine was extracted from plasma samples by liquid-liquid extraction using ethyl acetate-hexane (1:1, vol/vol). Separation was performed by isocratic elution from a reverse-phase Synergi Max-RP (250- by 4.6-mm, 4-μm) column (Phenomenex) with an acidic (adjusted to pH 2.8 with o-phosphoric acid) mobile phase (25 mM KH2PO4-methanol [80:20, vol/vol] plus 1% [vol/vol] triethylamine) at a flow rate of 1.2 ml/min. A quinidine internal standard (25-μl aliquot of 100 μg/ml quinidine as a working standard) and quinine were detected at 254 nm and resolved to baseline with retention times of 9.3 min and 11.8 min, respectively. The lower limit of quantification was set to 100 ng/ml. Quality control samples at 1, 10, and 20 mg/liter were prepared by spiking drug-free plasma. Intra-assay and interassay coefficients of variation (CV) ranged from 5.4% to 12.7%.
Population pharmacokinetic-pharmacodynamic analysis.
Quinine concentrations were transformed into their natural logarithms and modeled by using NONMEM, version 7 (Icon Development Solutions, Hanover, MD) (29). Automation and model diagnostics were performed by using Pearl-speaks-NONMEM (PsN) and Xpose (30–32). The Laplacian method (pharmacodynamic model) and the first-order conditional estimation method with interaction (pharmacokinetic model) were used throughout modeling. The difference in the objective function value (ΔOFV) computed by NONMEM as −2× the log likelihood was used as a statistical criterion for hierarchical models (an ΔOFV of >3.84 was considered statistically significant at a P value of <0.05, with a difference of 1 degree of freedom). Goodness-of-fit plots and simulation-based diagnostics were used for model evaluations. Only two postdose concentrations were quantified to be below the lower limit of quantification and were therefore omitted.
Population pharmacokinetic models were parameterized initially by using a first-order rate constant (ka) or a duration of a zero-order absorption (DUR), elimination clearance (CL/F), intercompartment clearance (Q/F), and apparent volume of distribution(s) (V/F). Limited data were available in the absorption phase, and the injection sites were therefore considered to be a single-depot compartment and bioavailability (F) was assumed to be 100%. Several absorption models were tried, i.e., first- and zero-order absorption with or without a lag time as well as a more flexible transit compartment absorption (33). Predose concentrations were handled by flagging patients with predose concentrations to allow a baseline value to be estimated for these individuals. Between-subject variability (BSV) and between-dose occasion variability (BOV) were modeled exponentially. One- and two-compartment disposition models were evaluated. First-order and Michaelis-Menten eliminations were evaluated. A Box-Cox transformation was tried on individual population parameters to assess formally the assumption that pharmacokinetic parameters are log-normally distributed (34). The residual random variability was assumed to be additive, since data were transformed into their natural logarithms (i.e., essentially equivalent to an exponential error model on an arithmetic scale).
The implementation of body weight as a covariate on the clearance and volume of distribution in the final structural model was assessed by using an allometric function [individual clearance value = typical clearance value × (individual body weight/median body weight in the population)EXP and individual volume value = typical volume value × (individual body weight/median body weight in the population)EXP]. An estimated exponent (EXP) for the allometric implementation of body weight on clearance and volume parameters was tried, as were fixed exponents of 1, 3/4, and 2/3 (35). An age-related enzyme maturation effect on clearance was also investigated (36).
Demographic, clinical, and laboratory data upon admission were considered potential covariates and investigated by using a stepwise forward-addition and backward-elimination approach. A P value of 0.05 was used in the forward step and a P value of 0.001 was used in the backward step to compensate for the relatively small population studied. The following admission covariates were investigated by using the stepwise approach: age (years), weight-for-age Z score (37, 38), temperature (°C), heart rate (beats/min), coma (continuous variable based on the GCS/BCS coma score), cerebral malaria (coma and/or convulsions), blood urea nitrogen (mg/dl), hemoglobin (g/dl), base excess (mmol/liter), parasitemia (parasites/μl), plasma PfHRP2 (ng/ml) as a marker of the total parasite burden (27), total bilirubin (μmol/liter), creatinine (high, ≥44.2 μmol/liter at <1 year of age and ≥62 μmol/liter at ≥1 year of age; low, <44.2 μmol/liter at <1 year of age and <62 μmol/liter at ≥1 year of age), HIV coinfection, shock (compensated or decompensated), fluid bolus treatment, and/or blood transfusion.
Numerical and visual predictive checks were used to assess the predictive performance of the final model. The final model with included variability was used to simulate 2,000 concentrations at each sampling time point, and the 95% confidence intervals (CIs) around the simulated 5th, 50th, and 95th percentiles were overlaid with the same percentiles of observed data to evaluate the predictive power of the model (visual predictive check). The percentages of observations below and above the simulated 5th and 95th percentiles were also calculated for a numerical predictive check. Nonparametric bootstrap diagnostics (n = 2,000), stratified by median body weight (above or below 11 kg), were performed for accurate relative standard errors and nonparametric confidence intervals of population parameter estimates. Stratification by body weight was performed to maintain an equal distribution of high and low body weights in the resampled data. The final model was also used for Monte Carlo simulations evaluating quinine exposure in children at different body weights with or without a loading dose.
Survival data were modeled in NONMEM using a time-to-event analysis. Patients were censored at 12 h after the last intramuscular quinine administration. The survival data were modeled by using a constant hazard function, a Weibull distribution hazard function, or an exponential hazard function. OFV and simulation-based diagnostics were used to compare models. There were 13 deaths in the 75 patients in this study, which were too few for a formal covariate analysis of the time to event. Drug concentration-response relationships were evaluated by a direct effect driven by plasma concentrations or a delayed effect by cumulative quinine exposure.
RESULTS
Clinical details.
Seventy-five children were included, 28 (37%) of whom were under 2 years of age. Demographic, clinical, and laboratory characteristics are described in Table 1. Severe prostration, convulsions, severe acidosis, severe anemia, and coma were the most common severity criteria. None of the 18 patients who presented with hypoglycemia at admission and only 4 out of 7 patients with hemoglobinuria had a history of quinine pretreatment. Seven (9.3%) patients had blood culture-confirmed bacteremia (12.5% in shocked patients versus 8.5% in nonshocked patients; P = 0.623). The identified organisms were non-serovar Typhi Salmonella spp., Enterobacter cloacae, Klebsiella spp., Escherichia coli, Burkholderia cepacia, and group A streptococci. HIV coinfection was found in 5/75 (6.7%) patients. None of these patients was receiving antiretroviral treatment. Out of 75 patients, 13 (17%) died, 10 (77%) of whom died within 24 h. Children who survived had no neurological sequelae at discharge.
Table 1.
Demographic, clinical, and laboratory characteristics of children admitted with severe malaria
| Variable | Value for children with severe malaria (n = 75) |
|---|---|
| Median age (yr) (range) | 2.4 (0.33–8.1) |
| Median wt (kg) (range) | 11 (5.5–27) |
| Median wt-for-age Z score (range) | −1.0 (−4.1–1.0) |
| No. (%) of patients with: | |
| Coma | 27 (36) |
| Prostration | 46 (61) |
| Convulsions | 34 (45) |
| Shocka | 16 (21) |
| Respiratory distress | 7 (9) |
| Acidosis (base excess of <8 mmol/liter) | 32 (46) |
| Hypoglycemia (glucose level of <3 mmol/liter) | 18 (24) |
| Anemia | 27 (36) |
| Black water fever | 7 (9) |
| Median axillary temp (°C) (range) | 38.2 (35.4–41.8) |
| Median heart rate (beats/min) (range) | 154 (98–202) |
| Median respiratory rate (breaths/min) (range) | 50 (24–98) |
| Median glucose level (mg/dl) (range) | 95 (15–240) |
| Median blood urea nitrogen level (mg/dl) (range) | 13 (4–97) |
| Median hemoglobin level (g/dl) (range) | 7.1 (2.6–13.2) |
| Median pH (range) | 7.36 (7.28–7.42) |
| Median HCO3 level (mmol/liter) (range) | 17.8 (3.5–25.6) |
| Median base excess (mmol/liter) (range) | −8 (−28–2) |
| Median AST level (U/liter) (range) | 85 (7–3,180) |
| Median ALT level (U/liter) (range) | 22 (3–1,490) |
| Median total bilirubin level (μmol/liter) (range) | 38 (5–250) |
| No. (%) of HIV-positive patients | 5 (6.7) |
| Geometric mean parasitemia level (parasites/μl) (95% CI) | 31,900 (17,300–58,900) |
| Geometric mean plasma PfHRP2 level (ng/ml) (95% CI) | 2,070 (1,470–2,900) |
Compensated and decompensated shock combined.
Sixty-nine (92%) patients received a quinine loading dose at the start of the study, and the remainder started with 10 mg/kg. During the first 24 h of admission, 37 patients received a blood transfusion, and 19 patients received a fluid bolus. Eleven patients (15%) developed hypoglycemia after admission, including 5/18 (27.7%) of those who presented with hypoglycemia at admission. Hypoglycemia occurred in 6/11 children despite an intravenous 5% dextrose infusion, 5 of whom subsequently died.
Coma after admission occurred in 7 (9%) patients. Geometric mean peripheral parasite densities upon admission were 31,900 parasites/μl (95% CI, 17,300 to 58,900 parasites/μl), and after 24 h of treatment, these values were 3,681 parasites/μl (95% CI, 1,422 to 10,790 parasites/μl), with a median fractional reduction of 78% (interquartile range [IQR], 38% to 99%) in 60 patients (1 and 14 patients with missing baseline and 24-h parasitemia, respectively, including 10 fatal cases).
A history of oral antimalarial treatment before admission was given for 41 (55%) patients (8 with quinine, 5 with amodiaquine, 17 with artemether-lumefantrine, 10 with sulfadoxine-pyrimethamine, and 1 with sulfadoxine-pyrimethamine followed by artemether-lumefantrine). Parenteral quinine pretreatment within 24 h prior to admission was reported for 15 patients, with a maximum of 3 doses and a mean dose of 10 mg/kg (standard deviation [SD], 1.8 mg/kg). Of these patients, 12 had detectable baseline plasma quinine concentrations ranging from 1.56 to 17.38 mg/liter. Three patients treated earlier with oral quinine had baseline drug concentrations of 3.34, 5.10, and 8.26 mg/liter. Another 6 patients without a history of quinine treatment before admission had detectable plasma quinine concentrations ranging from 0.85 to 14.88 mg/liter (5 of whom had baseline concentrations of <4.0 mg/liter).
Population pharmacokinetic-pharmacodynamic analysis.
Each patient contributed 1 to 5 plasma samples, resulting in a total of 341 concentration-time samples distributed randomly over the first 24 h of the study. All patients were included in the population pharmacokinetic analysis, and predose quinine concentrations were accommodated by estimating a baseline concentration for these patients (median concentrations of 6.90 mg/liter [range, 0.976 to 14.9 mg/liter]). Postdose plasma concentrations ranged from 0.850 to 33.8 mg/liter. Four patients had very high plasma quinine concentrations (>25 mg/liter), but no serious adverse events or deaths could be attributed to these high plasma quinine concentrations.
A one-compartment disposition model with zero-order absorption resulted in an adequate fit to the observed data. None of the investigated absorption models performed better than the zero-order absorption model. No additional benefit was seen with an additional peripheral disposition compartment (ΔOFV = −4.12; difference of 2 degrees of freedom). There was no substantial between-dose occasion variability in any population parameter (ΔOFV < −0.150), and this was therefore not included in the final model. Michaelis-Menten elimination did not significantly improve the model diagnostics (ΔOFV = −2.75) compared to a first-order elimination model. A Box-Cox transformation of population parameters did not significantly improve the model fit (ΔOFV < −1.88) compared to the usually assumed log-normal distribution. The incorporation of an off-diagonal element in the covariance matrix of elimination clearance and apparent volume of distribution resulted in a significant correlation (99.9%) and an improvement of the model fit. Between-subject variability could not be estimated reliably for the duration of the zero-order absorption (relative standard error [RSE] = 171%) and was therefore not retained in the final model.
Body weight as a fixed allometric function on elimination clearance (exponent of 3/4) and apparent volume of distribution (exponent of 1) resulted in a significant improvement in the model fit (ΔOFV = −39.0) and decreased the between-subject variabilities (CVs from 47.4% to 35.9% and from 61.2% to 51.3%, respectively). The exponents were also estimated, and the bootstrap (n = 500) diagnostics resulted in 95% confidence intervals of 0.484 to 1.51 for elimination clearance and 0.594 to 1.52 for the apparent volume of distribution, respectively. Fixed exponents of 3/4 for elimination clearance and 1 for apparent volume of distribution were selected as the final model, considering the strong biological prior of these relationships supported by the observation that the estimated confidence intervals included these fixed values (35). The following covariate relationships were selected in the forward stepwise addition (P < 0.05) base excess on CL/F, base excess on V/F, hemoglobin on V/F, and age on CL/F as a maturation model. However, none of these covariates could be retained in the backward step with a more stringent statistical criterion (P < 0.001). The final population parameter estimates, variability estimates, and post hoc estimates are summarized in Table 2.
Table 2.
Parameter estimates of the final model describing quinine population pharmacokinetics in children (n = 75) with severe malaria
| Effect and variable | Population estimatea (%RSEb) | 95% CIb |
|---|---|---|
| Fixed PK effects | ||
| CL/F (liters/h) | 0.792 (6.42) | 0.698–0.898 |
| V/F (liters) | 13.7 (6.56) | 12.1–15.6 |
| DUR (h) | 1.42 (21.1) | 0.486–1.70 |
| Fixed PD effects | ||
| Baseline hazard (h−1) | 0.016 (42.7) | 0.00586–0.0342 |
| AUC50 (h · mg/liter) | 64.8 (96.9) | 21.8–387 |
| Random PK effects | ||
| ηCL/F | 0.128 (28.7) | 0.0620–0.201 |
| ηV/F | 0.176 (25.7) | 0.0927–0.262 |
| ηCL/F ∼ ηV/F | 0.15 (21.5) | 0.0835–0.206 |
| σ | 0.0942 (7.76) | 0.0671–0.124 |
| Post hoc PK estimatesc | ||
| CL/F (liters/h/kg) | 0.0741 | 0.0455–0.144 |
| V/F (liters/kg) | 1.24 | 0.645–2.89 |
| t1/2 (h) | 12.1 | 9.63–14.3 |
| Cmax (mg/liter) | 13.4 | 7.20–24.8 |
| AUC0–7.5 h (h · mg/liter) | 78.9 | 42.3–148 |
Computed population mean values from NONMEM are calculated for a typical patient with a body weight of 11 kg.
Assessed by the nonparametric bootstrap method (n = 1,586 successful iterations out of 2,000 for pharmacokinetics [PK] and n = 1,532 successful iterations out of 2,000 for pharmacodynamics [PD]) for the final model. The relative standard error (%RSE) is calculated as 100 × (standard deviation/mean value). The 95% confidence intervals (CIs) are displayed as the 2.5 to 97.5 percentiles of bootstrap estimates.
Post hoc estimates are displayed as median values with 2.5 to 97.5 percentiles of empirical Bayes estimates.
CL/F, elimination clearance; V/F, volume of distribution; F, intramuscular bioavailability; DUR, duration of zero-order absorption; baseline hazard, baseline hazard of death per hour in the time-to-event model; AUC50, cumulative exposure associated with a 50% reduction in the baseline hazard of death; η, interindividual variability; ηCL/F ∼ ηV/F, correlation of random effects on CL/F and V/F; σ, additive residual variance error; t1/2, terminal elimination half-life; AUC0–7.5 h, area under the concentration-time curve from 0 to 7.5 h; Cmax, predicted peak concentration after the first dose.
The final model described the observed data well, with adequate goodness-of-fit diagnostics (Fig. 1) and calculated shrinkages below 15% (CL/F shrinkage, 11.3%; V/F shrinkage, 11.3%; epsilon shrinkage, 14.0%). A prediction-corrected visual predictive check of the final model resulted in no model misspecification with good simulation properties (Fig. 2). The numerical predictive check resulted in 6.92% (95% CI, 2.08 to 9.00%) and 2.08% (95% CI, 1.73 to 9.00%) of observations above and below the 90% prediction interval.
Fig 1.

Goodness-of-fit diagnostics of the final population pharmacokinetic model of quinine in children with severe malaria. Broken line, locally weighted least-squares regression; solid line, line of identity. The observed concentrations, population predictions, and individual predictions were transformed into their logarithms (base 10).
Fig 2.
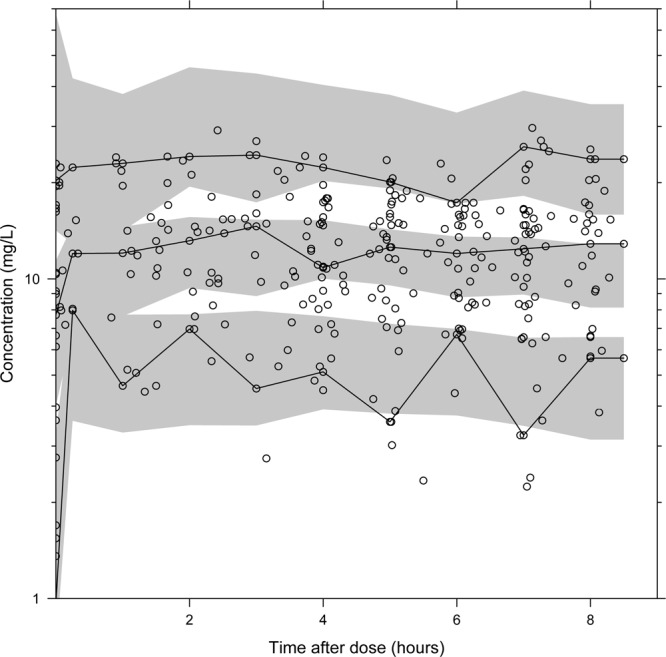
Visual predictive check of the final model describing the population pharmacokinetics of quinine in children with severe malaria. Open circles, observed data points; solid lines, 5th, 50th, and 95th percentiles of the observed data; shaded area, 95% confidence interval of simulated (n = 2,000) 5th, 50th, and 95th percentiles. Venous plasma quinine concentrations were transformed into their logarithms (base 10).
Body weight was the only significant covariate in the final model, with lower body weights being associated with slightly reduced exposures during the first 24 h after the standard weight-based dose. There was a mean reduction of 7.19% in simulated quinine exposure during the first dose (0 to 8 h) when patients weighing 5 kg were compared with patients weighing 20 kg after a 20 mg salt/kg loading dose (data not shown). This reduction in exposure accumulated with repeated maintenance dosing of 10 mg/kg over the first 24 h to a total mean difference of 15.4% (data not shown). Peak concentrations after the first dose were unaffected by body weight but accumulated to a mean difference of 15.9% lower peak concentrations for patients weighing 5 kg than for patients weighing 20 kg after the third dose (Fig. 3A). This difference (<20%) in total exposures and maximum concentrations is not likely to have a significant clinical impact, since more than 85% of patients, irrespective of body weight, reached plasma quinine concentrations over the estimated therapeutic margin of 8 mg/liter after a loading dose of 20 mg/kg, and more than 95% of patients reached the target during the first 24 h with the subsequent maintenance dose of 10 mg/kg. In the absence of a loading dose, the therapeutic range would be reached only in fewer than 30% of patients after the first dose and in 89% of patients during the first 24 h (Fig. 3b).
Fig 3.

(A) Predicted population pharmacokinetic profiles of quinine at different body weights (gray solid profiles). Profiles for body weights of 5 kg (— · —), 10 kg ( ), and 20 kg () are highlighted in black. (B) Predicted population pharmacokinetic profiles of quinine at different body weights (gray solid profiles) after a loading dose of 20 mg/kg (—) versus a routine dose of 10 mg/kg (). Profiles for a body weight of 10 kg are highlighted in black.
), and 20 kg () are highlighted in black. (B) Predicted population pharmacokinetic profiles of quinine at different body weights (gray solid profiles) after a loading dose of 20 mg/kg (—) versus a routine dose of 10 mg/kg (). Profiles for a body weight of 10 kg are highlighted in black.
A constant hazard function model with cumulative exposure implemented as a maximum-effect (Emax) function modulating the hazard described the survival over time well in this study. A simulation-based visual predictive check resulted in the observed survival curve being contained within the 90% prediction interval of the model-simulated survival curve (Fig. 4). The median time to reach a 50% reduction in hazard was approximately 6 h. However, there was no significant difference in post hoc estimates of total exposures (P = 0.1358) or maximum concentrations (P = 0.1786) after the first dose in children who died compared to children who survived (Fig. 5). The exposure-effect relationship is likely to describe the delayed antimalarial effect of quinine, but this approach is biased since many patients had a pretreatment history of quinine, and a larger data set would be necessary for a formal concentration-effect analysis.
Fig 4.

Visual predictive check for the Kaplan-Meier survival curve. The solid black line represents the observed Kaplan-Meier curve, and the shaded gray area represents the corresponding 90% prediction interval derived from model simulations (n = 1,000) of the final pharmacokinetic-pharmacodynamic model.
Fig 5.

Predicted total quinine exposures (area under the concentration-time curve from 0 to 8 h [AUC0–8 h]) and maximum concentrations (Cmax) after the first dose, stratified for outcome. Error bars indicate medians and interquartile ranges.
DISCUSSION
The therapeutic range for quinine in severe malaria has been estimated to be between 8 and 15 mg/liter based on observations in uncomplicated malaria suggesting reduced therapeutic responses when serum or plasma concentrations fall below 5 mg/liter and taking into account the variation in parasite susceptibility and the reduced free fraction observed in severe malaria. Plasma concentrations of up to 20 mg/liter have not been associated with significant toxicity in severe malaria (11, 39–42).
In the present study, intramuscular quinine was rapidly and reliably absorbed, and the currently recommended loading dose regimen resulted in plasma concentrations that rapidly reached the therapeutic range in African children with severe malaria. A wide range of patient covariates did not affect the pharmacokinetic parameters significantly, suggesting that this applied to children of all ages and with all forms of severe malaria. Body weight was the only significant covariate affecting quinine exposure. Monte Carlo simulations resulted in a modest mean reduction of 7.19% in total quinine exposure after the loading dose (0 to 8 h) in children weighing 5 kg compared to that in children weighing 20 kg. This accumulated to a larger difference of 15.4% over 3 doses (0 to 24 h). This is unlikely to have a significant clinical impact, since therapeutic levels of 8 mg/liter were reached rapidly in all weight groups (Fig. 2). Dose adjustment is therefore not recommended for smaller children based on this pharmacokinetic difference. Simulations resulted in median maximum concentrations of 12.6 mg/liter (95% CI, 5.60 to 28.4 mg/liter) after a loading dose of 20 mg/kg, compared to 6.32 mg/liter (95% CI, 2.80 to 14.2 mg/liter) without a loading dose (i.e., after 10 mg/kg). This supports that a loading dose should be used to achieve target concentrations within the first dose interval.
An intramuscular loading dose of quinine was rapidly and reliably absorbed, and patients in this study reached estimated peak median plasma quinine concentrations of 13.4 mg/liter (95% CI, 7.20 to 24.8 mg/liter) within 1.50 h. This is in accordance with previous studies showing similar peak plasma quinine concentrations compared to the intravenous route, with similar efficacies (17, 43, 44). Dilution of the quinine solution to 60 mg/ml has been reported to accelerate the absorption from the intramuscular injection site (8, 12, 21), and this time to peak is shorter than that reported previously for an injection of a more concentrated injectate.
Pharmacokinetic estimates should be extrapolated with caution beyond the range of the studied patients (i.e., elimination clearance of 3.91 liters/h scaled to a 70-kg patient). However, the reported median estimates of a quinine terminal half-life of 12.1 h (95% CI, 9.63 to 14.3 h) and an elimination clearance of 0.0741 liters/h/kg (95% CI, 0.0455 to 0.144 liters/h/kg) are in broad agreement with previously reported estimates from small, conventional, densely sampled pharmacokinetic studies of children with severe falciparum malaria: median half-lives ranging from 8.4 to 23.5 h and clearance rates ranging from 0.027 to 0.0816 liters/h/kg have been reported (reviewed in reference 17). Minor differences between our findings and those reported in a population pharmacokinetic analysis by Krishna et al. (mean half-life of 19.9 h [SD, 4.4 h] and elimination clearance rate of 0.05 liters/h/kg) may be explained by the use of different structural models (a one-compartment model versus a two-compartment model) (7). In this study, we performed sampling during the first 24 h, which could also have contributed to the difference in structural models. Thus, a two-compartment model could prove to be a more appropriate structural model when enough data are collected to support a differentiation between a distribution and a terminal phase. However, the terminal elimination half-life estimate reported previously by Krishna et al. is similar to that for adults with severe malaria, whereas the majority of published data point to a more rapid elimination in children than in adults. Body weight has not been described as a covariate for quinine pharmacokinetics previously, but it was significant in this analysis (7, 10). Physiological processes do not scale linearly with body weight, and consequently, children with a lower body weight will have a higher body weight-normalized elimination clearance rate, which was reported previously for other antimalarials (45, 46).
In accordance with the only previously reported population pharmacokinetic study, we did not find any other covariates explaining the between-subject variability in children with severe malaria despite the different clinical presentations (7). Compared with uncomplicated malaria, patients with severe disease have a small distribution volume and a slow clearance due in part to a high fraction of plasma protein-bound quinine (5).
It is reassuring that intramuscular quinine was reliably absorbed in children with impaired perfusion, shock, and severe anemia, although these were largely corrected for in this study with supportive treatments. In addition, none of the clinical or laboratory parameters with a strong prognostic value, such as coma, impaired renal function (elevated blood urea nitrogen level), or acidosis, affected the pharmacokinetics of quinine (47). Therefore, quinine dosing does not need to be adapted according to the presentation of the disease in children with severe malaria.
Intramuscular quinine treatment is painful, but local toxicity is rare when a sterile injection technique is used and the quinine is diluted to 60 mg/ml (10). In our study site, all concomitant medications, including routinely administered antibiotics, were given by intramuscular injection in the anterior thigh. However, no mobility problems were noted, and all surviving children were well at discharge. The neurological examination at discharge did not disclose any evidence of systemic quinine toxicity such as blindness or hearing problems, even though 4/75 (5%) children had plasma quinine concentrations above 25 mg/liter within the first 24 h of treatment. Peak total plasma concentrations tend to increase during the treatment of severe malaria until clinical resolution (17), so it is possible that some patients might have experienced potentially toxic quinine concentrations later in their treatment course. The levels of quinine associated with toxicity in severe malaria are not well established, since toxicity derives from the free quinine fraction, which depends on the levels of plasma proteins, predominantly AGP, which vary substantially (18, 19). The pharmacokinetic study reported previously by van Hensbroek et al. showed that young children could be more prone to quinine toxicity, as evidenced by a slight prolongation of the QRS interval on the electrocardiogram (intraventricular conduction delay), although this was not related to plasma quinine concentrations (10). The main adverse effect of quinine in severe malaria is hypoglycemia resulting from quinine-stimulated insulin release, and this occurs with therapeutic concentrations (23, 24). Otherwise, given the extensive use of quinine and the widespread and often unreported pretreatment, its use in severe malaria is otherwise remarkably free from serious toxicity.
In our study, children who died did not have higher or lower plasma quinine concentrations than children who survived (Fig. 5) and 12/13 fatal cases had received a loading dose at admission. The one child who died and did not receive a loading dose at admission was reported to have received pretreatment with two quinine injections; however, the baseline quinine concentration was undetectable, suggesting that the child's treatment history was incorrect. Unreliability of the treatment history is commonplace in cases of malaria (48, 49). Administration of a loading dose of 20 mg/kg when the history is uncertain may be safest, as an undertreatment of severe malaria increases the risk of death.
The therapeutic benefit of the loading dose has been widely accepted, but unsubstantiated toxicity concerns have long hindered its implementation in the field (6, 50). Although the potential life-saving benefit of administering a loading dose of quinine has not been tested in large randomized controlled trials, the faster fever and parasite clearance times and an understanding of the basic pathobiology of severe malaria suggest that it is beneficial for the treatment of severe malaria (reviewed in reference 51). Importantly, the loading dose does not alter the risk of hypoglycemia due to quinine-induced hyperinsulinemia (51, 52). One-fifth of the children in our study had already received routine dosing of parenteral quinine prior to admission, none of whom presented with hypoglycemia at admission. Hypoglycemia is an indicator of severe disease, associated with an increased case-fatality rate (52–55). The high incidences of shock and positive blood cultures suggest that concomitant sepsis might also have contributed to the high mortality rate in our study population.
In conclusion, if artemisinin derivatives are unavailable and quinine is used, a loading dose should be given unless there is convincing evidence of adequate pretreatment, since the risk of death from severe malaria is highest and the risk of systemic toxicity is lowest during the first 24 h. Starting with the routine dose is justified only for children who have already definitely received a loading dose within 24 h before admission and those who have received a routine dose within 12 h of admission, but if in doubt, a loading dose should be given.
ACKNOWLEDGMENTS
We are grateful to the patients and their guardians. We thank Ben Amos from Teule Hospital in Muheza for microbiology and laboratory management. We also thank Paul Martine, laboratory technician at AMBRELA, for assisting in the laboratory.
Permission to publish this work was given by the Director General, National Institute for Medical Research, Tanzania.
This work was supported by The Wellcome Trust of Great Britain (grants 076908 and 082541) and was coordinated as part of the Mahidol-Oxford Tropical Medicine Research Programme funded by the Wellcome Trust of Great Britain.
Footnotes
Published ahead of print 26 November 2012
REFERENCES
- 1. WHO 2008. World malaria report 2008. World Health Organization, Geneva, Switzerland: http://whqlibdoc.who.int/publications/2008/9789241563697_eng.pdf [Google Scholar]
- 2. Dondorp AM, Fanello CI, Hendriksen IC, Gomes E, Seni A, Chhaganlal KD, Bojang K, Olaosebikan R, Anunobi N, Maitland K, Kivaya E, Agbenyega T, Nguah SB, Evans J, Gesase S, Kahabuka C, Mtove G, Nadjm B, Deen J, Mwanga-Amumpaire J, Nansumba M, Karema C, Umulisa N, Uwimana A, Mokuolu OA, Adedoyin OT, Johnson WB, Tshefu AK, Onyamboko MA, Sakulthaew T, Ngum WP, Silamut K, Stepniewska K, Woodrow CJ, Bethell D, Wills B, Oneko M, Peto TE, von Seidlein L, Day NP, White NJ. 2010. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet 376:1647–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waller D, Krishna S, Crawley J, Miller K, Nosten F, Chapman D, ter Kuile FO, Craddock C, Berry C, Holloway PA, Brewster D, Greenwood BM, White NJ. 1995. Clinical features and outcome of severe malaria in Gambian children. Clin. Infect. Dis. 21:577–587 [DOI] [PubMed] [Google Scholar]
- 4. WHO 2000. Severe falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 94(Suppl 1):S1–S90 doi:S0035-9203(00)90300-6 [PubMed] [Google Scholar]
- 5. White NJ, Looareesuwan S, Warrell DA, Warrell MJ, Bunnag D, Harinasuta T. 1982. Quinine pharmacokinetics and toxicity in cerebral and uncomplicated falciparum malaria. Am. J. Med. 73:564–572 [DOI] [PubMed] [Google Scholar]
- 6. White NJ, Looareesuwan S, Warrell DA, Warrell MJ, Chanthavanich P, Bunnag D, Harinasuta T. 1983. Quinine loading dose in cerebral malaria. Am. J. Trop. Med. Hyg. 32:1–5 [DOI] [PubMed] [Google Scholar]
- 7. Krishna S, Nagaraja NV, Planche T, Agbenyega T, Bedo-Addo G, Ansong D, Owusu-Ofori A, Shroads AL, Henderson G, Hutson A, Derendorf H, Stacpoole PW. 2001. Population pharmacokinetics of intramuscular quinine in children with severe malaria. Antimicrob. Agents Chemother. 45:1803–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mansor SM, Taylor TE, McGrath CS, Edwards G, Ward SA, Wirima JJ, Molyneux ME. 1990. The safety and kinetics of intramuscular quinine in Malawian children with moderately severe falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 84:482–487 [DOI] [PubMed] [Google Scholar]
- 9. Shann F, Stace J, Edstein M. 1985. Pharmacokinetics of quinine in children. J. Pediatr. 106:506–510 [DOI] [PubMed] [Google Scholar]
- 10. van Hensbroek MB, Kwiatkowski D, van den Berg B, Hoek FJ, van Boxtel CJ, Kager PA. 1996. Quinine pharmacokinetics in young children with severe malaria. Am. J. Trop. Med. Hyg. 54:237–242 [DOI] [PubMed] [Google Scholar]
- 11. White NJ. 1995. Optimal regimens of parenteral quinine. Trans. R. Soc. Trop. Med. Hyg. 89:462–464 [DOI] [PubMed] [Google Scholar]
- 12. Waller D, Krishna S, Craddock C, Brewster D, Jammeh A, Kwiatkowski D, Karbwang J, Molunto P, White NJ. 1990. The pharmacokinetic properties of intramuscular quinine in Gambian children with severe falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 84:488–491 [DOI] [PubMed] [Google Scholar]
- 13. Hall AP, Czerwinski AW, Madonia EC, Evensen KL. 1973. Human plasma and urine quinine levels following tablets, capsules, and intravenous infusion. Clin. Pharmacol. Ther. 14:580–585 [DOI] [PubMed] [Google Scholar]
- 14. Mirghani RA, Yasar U, Zheng T, Cook JM, Gustafsson LL, Tybring G, Ericsson O. 2002. Enzyme kinetics for the formation of 3-hydroxyquinine and three new metabolites of quinine in vitro; 3-hydroxylation by CYP3A4 is indeed the major metabolic pathway. Drug Metab. Dispos. 30:1368–1371 [DOI] [PubMed] [Google Scholar]
- 15. Pukrittayakamee S, Looareesuwan S, Keeratithakul D, Davis TM, Teja-Isavadharm P, Nagachinta B, Weber A, Smith AL, Kyle D, White NJ. 1997. A study of the factors affecting the metabolic clearance of quinine in malaria. Eur. J. Clin. Pharmacol. 52:487–493 [DOI] [PubMed] [Google Scholar]
- 16. Nontprasert A, Pukrittayakamee S, Kyle DE, Vanijanonta S, White NJ. 1996. Antimalarial activity and interactions between quinine, dihydroquinine and 3-hydroxyquinine against Plasmodium falciparum in vitro. Trans. R. Soc. Trop. Med. Hyg. 90:553–555 [DOI] [PubMed] [Google Scholar]
- 17. Krishna S, White NJ. 1996. Pharmacokinetics of quinine, chloroquine and amodiaquine. Clinical implications. Clin. Pharmacokinet. 30:263–299 [DOI] [PubMed] [Google Scholar]
- 18. Mansor SM, Molyneux ME, Taylor TE, Ward SA, Wirima JJ, Edwards G. 1991. Effect of Plasmodium falciparum malaria infection on the plasma concentration of alpha 1-acid glycoprotein and the binding of quinine in Malawian children. Br. J. Clin. Pharmacol. 32:317–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Silamut K, Molunto P, Ho M, Davis TM, White NJ. 1991. Alpha 1-acid glycoprotein (orosomucoid) and plasma protein binding of quinine in falciparum malaria. Br. J. Clin. Pharmacol. 32:311–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wanwimolruk S, Sunbhanich M, Pongmarutai M, Patamasucon P. 1986. Effects of cimetidine and ranitidine on the pharmacokinetics of quinine. Br. J. Clin. Pharmacol. 22:346–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Winstanley P, Newton C, Watkins W, Mberu E, Ward S, Warn P, Mwangi I, Waruiru C, Pasvol G, Warrell D, Marsh K. 1993. Towards optimal regimens of parenteral quinine for young African children with cerebral malaria: the importance of unbound quinine concentration. Trans. R. Soc. Trop. Med. Hyg. 87:201–206 [DOI] [PubMed] [Google Scholar]
- 22. Claessen FA, van Boxtel CJ, Perenboom RM, Tange RA, Wetsteijn JC, Kager PA. 1998. Quinine pharmacokinetics: ototoxic and cardiotoxic effects in healthy Caucasian subjects and in patients with falciparum malaria. Trop. Med. Int. Health 3:482–489 [DOI] [PubMed] [Google Scholar]
- 23. White NJ, Miller KD, Marsh K, Berry CD, Turner RC, Williamson DH, Brown J. 1987. Hypoglycaemia in African children with severe malaria. Lancet i:708–711 [DOI] [PubMed] [Google Scholar]
- 24. White NJ, Warrell DA, Chanthavanich P, Looareesuwan S, Warrell MJ, Krishna S, Williamson DH, Turner RC. 1983. Severe hypoglycemia and hyperinsulinemia in falciparum malaria. N. Engl. J. Med. 309:61–66 [DOI] [PubMed] [Google Scholar]
- 25. Zahn JR, Brinton GF, Norton E. 1981. Ocular quinine toxicity followed by electroretinogram, electro-oculogram, and pattern visually evoked potential. Am. J. Optom. Physiol. Opt. 58:492–498 [DOI] [PubMed] [Google Scholar]
- 26. Maitland K, Kiguli S, Opoka RO, Engoru C, Olupot-Olupot P, Akech SO, Nyeko R, Mtove G, Reyburn H, Lang T, Brent B, Evans JA, Tibenderana JK, Crawley J, Russell EC, Levin M, Babiker AG, Gibb DM. 2011. Mortality after fluid bolus in African children with severe infection. N. Engl. J. Med. 364:2483–2495 [DOI] [PubMed] [Google Scholar]
- 27. Hendriksen IC, Mwanga-Amumpaire J, von Seidlein L, Mtove G, White LJ, Olaosebikan R, Lee SJ, Tshefu AK, Woodrow C, Amos B, Karema C, Saiwaew S, Maitland K, Gomes E, Pan-Ngum W, Gesase S, Silamut K, Reyburn H, Joseph S, Chotivanich K, Fanello CI, Day NP, White NJ, Dondorp AM. 2012. Diagnosing severe falciparum malaria in parasitaemic African children: a prospective evaluation of plasma PfHRP2 measurement. PLoS Med. 9:e1001297 doi:10.1371/journal.pmed.1001297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mirghani RA, Ericsson O, Cook J, Yu P, Gustafsson LL. 2001. Simultaneous determination of quinine and four metabolites in plasma and urine by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 754:57–64 [DOI] [PubMed] [Google Scholar]
- 29. Beal S, Boeckman AJ, Sheiner LB. 1992. NONMEM user's guides, version 4. NONMEM Project Group, University of California, San Francisco, CA [Google Scholar]
- 30. Jonsson EN, Karlsson MO. 1999. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58:51–64 [DOI] [PubMed] [Google Scholar]
- 31. Lindbom L, Pihlgren P, Jonsson EN. 2005. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79:241–257 [DOI] [PubMed] [Google Scholar]
- 32. Lindbom L, Ribbing J, Jonsson EN. 2004. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75:85–94 [DOI] [PubMed] [Google Scholar]
- 33. Savic RM, Jonker DM, Kerbusch T, Karlsson MO. 2007. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 34:711–726 [DOI] [PubMed] [Google Scholar]
- 34. Petersson KJ, Hanze E, Savic RM, Karlsson MO. 2009. Semiparametric distributions with estimated shape parameters. Pharm. Res. 26:2174–2185 [DOI] [PubMed] [Google Scholar]
- 35. McLeay SC, Morrish GA, Kirkpatrick CM, Green B. 2012. The relationship between drug clearance and body size: systematic review and meta-analysis of the literature published from 2000 to 2007. Clin. Pharmacokinet. 51:319–330 [DOI] [PubMed] [Google Scholar]
- 36. Anderson BJ, Holford NH. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab. Pharmacokinet. 24:25–36 [DOI] [PubMed] [Google Scholar]
- 37. de Onis M, Onyango AW, Borghi E, Siyam A, Nishida C, Siekmann J. 2007. Development of a WHO growth reference for school-aged children and adolescents. Bull. World Health Organ. 85:660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. WHO Multicentre Growth Reference Study Group 2006. WHO child growth standards. Length/height-for-age, weight-for-age, weight-for-length, weight-for-height and body mass index-for-age: methods and development. World Health Organization, Geneva, Switzerland: http://www.who.int/childgrowth/publications/technical_report_pub/en/index.html [Google Scholar]
- 39. Earle DP, Jr, Berliner RW, Taggart JV, Welch WJ, Zubrod CG, Wise NB, Chalmers TC, Greif RL, Shannon JA. 1948. Studies on the chemotherapy of the human malarias; method for the quantitative assay of suppressive antimalarial action in falciparum malaria. J. Clin. Invest. 27:75–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pukrittayakamee S, Wanwimolruk S, Stepniewska K, Jantra A, Huyakorn S, Looareesuwan S, White NJ. 2003. Quinine pharmacokinetic-pharmacodynamic relationships in uncomplicated falciparum malaria. Antimicrob. Agents Chemother. 47:3458–3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Silamut K, White NJ, Looareesuwan S, Warrell DA. 1985. Binding of quinine to plasma proteins in falciparum malaria. Am. J. Trop. Med. Hyg. 34:681–686 [DOI] [PubMed] [Google Scholar]
- 42. Taggart JV, Earle DP, Jr, Berliner RW, Zubrod CG, Welch WJ, Wise NB, Schroeder EF, London IM, Shannon JA. 1948. Studies on the chemotherapy of the human malarias; the physiological disposition and antimalarial activity of the cinchona alkaloids. J. Clin. Invest. 27:80–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pasvol G, Newton CR, Winstanley PA, Watkins WM, Peshu NM, Were JB, Marsh K, Warrell DA. 1991. Quinine treatment of severe falciparum malaria in African children: a randomized comparison of three regimens. Am. J. Trop. Med. Hyg. 45:702–713 [DOI] [PubMed] [Google Scholar]
- 44. Schapira A, Solomon T, Julien M, Macome A, Parmar N, Ruas I, Simao F, Streat E, Betschart B. 1993. Comparison of intramuscular and intravenous quinine for the treatment of severe and complicated malaria in children. Trans. R. Soc. Trop. Med. Hyg. 87:299–302 [DOI] [PubMed] [Google Scholar]
- 45. Barnes KI, Little F, Smith PJ, Evans A, Watkins WM, White NJ. 2006. Sulfadoxine-pyrimethamine pharmacokinetics in malaria: pediatric dosing implications. Clin. Pharmacol. Ther. 80:582–596 [DOI] [PubMed] [Google Scholar]
- 46. Tarning J, Zongo I, Some FA, Rouamba N, Parikh S, Rosenthal PJ, Hanpithakpong W, Jongrak N, Day NP, White NJ, Nosten F, Ouedraogo JB, Lindegardh N. 2012. Population pharmacokinetics and pharmacodynamics of piperaquine in children with uncomplicated falciparum malaria. Clin. Pharmacol. Ther. 91:497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. von Seidlein L, Olaosebikan R, Hendriksen IC, Lee SJ, Adedoyin OT, Agbenyega T, Nguah SB, Bojang K, Deen JL, Evans J, Fanello CI, Gomes E, Pedro AJ, Kahabuka C, Karema C, Kivaya E, Maitland K, Mokuolu OA, Mtove G, Mwanga-Amumpaire J, Nadjm B, Nansumba M, Ngum WP, Onyamboko MA, Reyburn H, Sakulthaew T, Silamut K, Tshefu AK, Umulisa N, Gesase S, Day NP, White NJ, Dondorp AM. 2012. Predicting the clinical outcome of severe falciparum malaria in African children: findings from a large randomized trial. Clin. Infect. Dis. 54:1080–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Silamut K, Hough R, Eggelte T, Pukrittayakamee S, Angus B, White NJ. 1995. A simple method for assessing quinine pre-treatment in acute malaria. Trans. R. Soc. Trop. Med. Hyg. 89:665–667 [DOI] [PubMed] [Google Scholar]
- 49. Tran TH, Day NP, Nguyen HP, Nguyen TH, Pham PL, Dinh XS, Ly VC, Ha V, Waller D, Peto TE, White NJ. 1996. A controlled trial of artemether or quinine in Vietnamese adults with severe falciparum malaria. N. Engl. J. Med. 335:76–83 [DOI] [PubMed] [Google Scholar]
- 50. WHO 1990. Severe and complicated malaria. Trans. R. Soc. Trop. Med. Hyg. 84(Suppl 2):1–65 [PubMed] [Google Scholar]
- 51. Lesi A, Meremikwu M. 2004. High first dose quinine regimen for treating severe malaria. Cochrane Database Syst. Rev. 2004:CD003341 doi:10.1002/14651858.CD003341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ogetii GN, Akech S, Jemutai J, Boga M, Kivaya E, Fegan G, Maitland K. 2010. Hypoglycaemia in severe malaria, clinical associations and relationship to quinine dosage. BMC Infect. Dis. 10:334 doi:10.1186/1471-2334-10-334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. English M, Wale S, Binns G, Mwangi I, Sauerwein H, Marsh K. 1998. Hypoglycaemia on and after admission in Kenyan children with severe malaria. Q. J. Med. 91:191–197 [DOI] [PubMed] [Google Scholar]
- 54. Marsh K, Forster D, Waruiru C, Mwangi I, Winstanley M, Marsh V, Newton C, Winstanley P, Warn P, Peshu N, Pasvol G, Snow R. 1995. Indicators of life-threatening malaria in African children. N. Engl. J. Med. 332:1399–1404 [DOI] [PubMed] [Google Scholar]
- 55. Taylor TE, Molyneux ME, Wirima JJ, Fletcher KA, Morris K. 1988. Blood glucose levels in Malawian children before and during the administration of intravenous quinine for severe falciparum malaria. N. Engl. J. Med. 319:1040–1047 [DOI] [PubMed] [Google Scholar]