

Abstract
There is evidence that HIV-1 evolution under maraviroc (MVC) pressure can lead to the selection of either X4-tropic variants and/or R5-tropic, MVC-resistant isolates. However, the viral dynamics of HIV-1 variants in patients with virological failure (VF) on MVC-containing regimens remain poorly studied. Here, we investigated the V3 loop evolution of HIV-1 on MVC in relation to coreceptor usage and the nature of HIV-1 quasispecies before MVC therapy using bulk population sequences and ultradeep sequencing. The majority of patients had no detectable minority X4 variant at baseline. The evolution of tropism was followed up until VF and showed three possibilities for viral evolution in these patients: emergence of preexisting X4 variants, de novo selection of R5 variants presenting V3 loop mutations, or replication of R5 variants without selection of known mutations.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) infection involves an interaction between the viral envelope protein and the CD4 molecule. The V3 loop is then exposed and engages the coreceptor (CCR5 or CXCR4), which mediates membrane fusion. In the early stages of HIV-1 infection, CCR5 usage is predominant. The receptor switch to CXCR4 occurs often in infected individuals (around 40%) and is associated with faster disease progression (1). CCR5 inhibitors inhibit HIV-1 entry by blocking the CCR5 coreceptor. The binding of these small molecules to a cavity of the membrane domain of CCR5 stabilizes the coreceptor in a conformation which can no longer be recognized by the HIV-1 gp120 (2, 3). Maraviroc (MVC) is the first CCR5 inhibitor used for the treatment of CCR5-tropic HIV-1 infection (4). Two mechanisms of escape to MVC have been so far described in vitro and in vivo (5, 6). The first mechanism includes the selection of minority variants using the CXCR4 coreceptor; the second possibility is the emergence of CCR5-tropic resistant isolates which can still use the CCR5 coreceptor in the presence of the inhibitor. However, few studies have described the relative importance of the two mechanisms in MVC-treated patients (7, 8).
In this study, we described that the coreceptor switch occurred in only 30% of the patients, and we also characterized the genotypic evolution of HIV-1 isolates in patients with virological failure on MVC-based regimens.
MATERIALS AND METHODS
Study population.
Included patients were screened for the maraviroc expanded-access protocol (MVC EAP) in France between January 2007 and August 2008 and received MVC associated with an optimized background therapy if the result of the phenotypic assay for coreceptor use determination was CCR5, using a previously validated assay (Trofile; Monogram Biosciences) (9). For some patients, a modified version with an optimized sensitivity of the assay was performed (ESTA) once it became available (10). Inclusion criteria for the MVC EAP were HIV-1 infection, age of ≥18 years, with previous antiretroviral therapy and virological failure with plasma HIV-1 RNA of >1,000 copies/ml. Patients from 18 centers in France were included in the virological GenoTropism study (11). Sociodemographic data, clinical data, and treatment histories were collected for all enrolled patients at the screening date. MVC-treated patients were followed up at baseline (M0) and at months 1, 3, and 6 on MVC-containing regimens (M1, M3, and M6). The patients had signed the MVC EAP informed consent form and were specifically informed about their participation in the GenoTropism study. The study was approved by the Comité Consultatif de Traitement de l'Information dans la Recherche Scientifique et Médicale and the Commission Nationale Informatique et Libertés. Only patients treated by MVC and with virological failure (VF; defined as plasma viral load [VL] above 50 copies/ml at month 3 or month 6) and available plasma samples at baseline and follow-up were studied here.
Virological methods. (i) Population sequencing.
The sequence analysis comprising the complete V3 loop sequence was performed from plasma sampled at baseline MVC and at the time of follow-up with VL of >50 copies/ml. PCR primers and conditions and sequencing primers are described in the ANRS consensus techniques (http://www.hivfrenchresistance.org). PCR products were sequenced as described in the previous publication (11).
(ii) Ultradeep sequencing.
Env C2V3 quasispecies were determined at M0 by 454 ultradeep sequencing (UDS; Roche). A 415-nucleotide fragment encompassing the V3 env region was generated by nested reverse transcription (RT)-PCR. The nested PCR was performed with the Expand high-fidelity Plus PCR system (Roche Diagnostics), with the following conditions: 1 cycle of 94°C for 2 min; 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 3 min; followed by a final extension at 72°C for 7 min. The amplified PCR products were purified by using Agencourt Ampure PCR purification beads (Beckman Coulter, Brea, CA) and quantified with the Quant-iT Picogreen double-stranded DNA (dsDNA) assay kit (Invitrogen) on a LightCycler 480 (Roche). Pooled PCR products were clonally amplified on capture beads in water-in-oil emulsion microreactors, and pyrosequencing was performed by using PicoTiterPlate, following the standard approach for PCR amplicon sequencing. A total of 500,000 enriched DNA beads were deposited in the wells of a full GS Junior Titanium PicoTiterPlate device and pyrosequenced in both forward and reverse directions. The 200 nucleotide cycles were performed in a 10-h sequencing run. For each sample, a fasta file containing nucleotide sequence data was obtained.
(iii) Phylogenetic analysis.
Phylogenetic analyses were performed on V3 sequences, including both 454 sequences and population sequencing data, using the neighbor-joining method on Clustal W2 software and trees designed with the iTOL website (12).
(iv) Genotypic and phenotypic prediction of coreceptor usage.
Coreceptor usage was predicted from V3 sequences using the Geno2pheno algorithm (http://coreceptor.bioinf.mpi-inf.mpg.de/) by selecting a false-positive rate (FPR) at 10% for sequences issued from population sequencing or a false-positive rate at 5.75% or 3.5% (13, 14) for the calculation of the X4 virus frequency for the 454 sequences.
Coreceptor usage was also determined at M0 and at follow-up by phenotypic analysis using a recombinant assay (Toulouse tropism test) (15).
RESULTS
Patients'characteristics.
Seventeen patients were included in this study. Patient characteristics are detailed in Table 1. At baseline, before maraviroc-containing therapy, the median plasma HIV-1 RNA was 5.2 log10 copies/ml (range, 2.1 to 5.8), and at VF (M6), the median plasma HIV-1 RNA was 3 log10 copies/ml (range, 1.6 to 5.6). The median CD4+ cell count was 131 cells/mm3 (range, 3 to 607) at baseline MVC and 320 cells/mm3 (range, 8 to 642) at M6. The antiretroviral drugs coadministered with MVC included nucleoside or nucleotide RT inhibitors for 9 patients, ritonavir-boosted protease inhibitors in 12 patients, a nonnucleoside RT inhibitor (etravirine) in 4 patients, an integrase inhibitor (raltegravir) in 11 patients, and a fusion inhibitor (enfuvirtide) in 3 patients.
Table 1.
Patient characteristicsa
| Patient no. | Time of follow-up | OBT | Viral load (log10 copies/ml) | CD4+ cell count (cells/μl) | Tropism (population sequencing) | Tropism 454 (% X4) | Tropism TTT (pheno) |
|---|---|---|---|---|---|---|---|
| 1 | M0 | DRV RAL ENF | 5.8 | 20 | R5 | 2.5 | R5 |
| M1 | 5.5 | 12 | X4 | ND | R5X4 | ||
| M6 | 5.6 | 39 | X4 | ND | R5X4 | ||
| 2 | M0 | ETR RAL | 5.2 | 336 | R5 | 0 | R5 |
| M3 | 2.8 | 376 | R5 | ND | ND | ||
| 3 | M0 | 3TC ETR DRV RAL | 5.2 | 3 | R5 | 0 | R5 |
| M3 | 4.4 | 6 | R5 | ND | ND | ||
| M6 | 4.6 | 8 | R5 | ND | ND | ||
| 4 | M0 | TDF FTC RAL | 5.4 | 130 | R5 | 0 | R5 |
| M6 | 2.1 | 342 | R5 | ND | ND | ||
| 5 | M0 | AZT 3TC ABC ETR DRV RAL | 5.2 | 15 | R5 | 0.74 | ND |
| M1 | 5 | 70 | X4 | ND | X4 | ||
| M3 | 4.5 | 93 | X4 | ND | ND | ||
| M6 | 3.2 | 193 | X4 | ND | ND | ||
| 6 | M0 | DRV RAL | 4.7 | 607 | R5 | 0 | R5 |
| M1 | 5 | 374 | R5 | ND | R5 | ||
| M3 | 3 | 460 | R5 | ND | R5 | ||
| M6 | 2.5 | 642 | R5 | ND | ND | ||
| 7 | M0 | DRV RAL | 5.2 | 607 | R5 | ND | ND |
| M3 | 2.9 | 460 | X4 | ND | ND | ||
| 8 | M0 | TDF FTC DRV | 4.5 | 45 | R5 | 0 | R5 |
| M6 | 3 | 124 | R5 | ND | R5 | ||
| 9 | M0 | fAPV RAL ENF | 4.5 | 131 | R5 | 0 | R5 |
| M3 | 3.3 | 132 | X4 | ND | X4 | ||
| M9 | 3.6 | 206 | R5 | ND | ND | ||
| 10 | M0 | TDF FTC DRV | 5.6 | 27 | R5 | 1.3 | R5 |
| M6 | 5.1 | 58 | R5 | ND | R5 | ||
| 11 | M0 | ABC 3TC DRV ENF | 4.6 | 316 | R5 | 0.3 | R5 |
| M6 | 2.2 | 400 | R5 | ND | ND | ||
| 12 | M0 | 3TC ABC TDF ETR DRV ATV | 5.2 | 82 | R5 | 0 | R5 |
| M3 | 3.5 | 383 | R5 | ND | R5 | ||
| 13 | M0 | 3TC ETR DRV ATV RAL | 2.4 | 276 | R5 | 76.1 | R5 |
| M3 | 2.3 | 457 | R5 | ND | R5 | ||
| 14 | M0 | DRV RAL | 2.1 | 541 | R5 | ND | ND |
| M3 | 2.4 | 390 | R5 | ND | ND | ||
| M6 | 2.7 | 243 | R5 | ND | ND | ||
| 15 | M0 | RAL | 3 | 323 | R5 | 0 | R5 |
| M6 | 2.1 | 360 | R5 | ND | ND | ||
| 16 | M0 | DRV | 3.9 | 434 | R5 | 0 | R5 |
| M6 | 5.1 | 642 | R5 | ND | R5 | ||
| 17 | M0 | TDF FTC TPV RAL | 5.5 | 131 | X4 | 100 | R5X4 |
| M1 | 4.3 | 121 | X4 | ND | ND | ||
| M3 | 4.5 | 160 | X4 | ND | ND |
OBT, optimized background therapy; TTT, Toulouse tropism test; M0, follow-up at baseline; M1, M3, and M6, follow-up at months 1, 3, and 6 on MVC-containing regimens; DRV, darunavir; RAL, raltegravir; ENF, enfuvirtide; ETR, etravirine; 3TC, lamivudine; TDF, tenofovir; FTC, emtricitabine; AZT, zidovudine; ABC, abacavir; fAPV, fosamprenavir; ATV, atazanavir; R5, CCR5; X4, CXCR4; ND, no data.
Baseline MVC HIV-1 coreceptor usage.
The inferred HIV-1 coreceptor usage from genotype was determined at baseline MVC from bulk HIV-1 plasma RNA. The genotypic analysis of the V3 loop, using the Geno2pheno (FPR, 10%) algorithm, showed an R5 tropism for all patients but one (Table 1). HIV-1 tropism using a phenotypic test (15) was determined at baseline MVC for 14 patients. These results were concordant with the genotypic determination, with R5 tropism for 13 patients and an R5X4 result for the patient with X4 genotypic tropism at baseline (and determined as R5 by Trofile assay).
The percentage of X4 variants could be determined by ultradeep sequencing of the V3 loop in 15 patients at baseline. The median number of analyzed sequences for each patient was 3,240 (range, 1,034 to 7,585). Out of 14 patients with R5 tropism from bulk population analysis, 9 had 0% of X4 isolates with UDS; 4 patients had minority X4 variants, ranging between 0.3% to 2.5% of the global population; and one patient had 76.1% of X4 variants. The patient with the X4 bulk population tropism was shown to have 100% of X4 variants by UDS.
Evolution of coreceptor usage at virological failure.
Both population sequencing and phenotypic analysis showed a switch from R5 isolates at baseline to X4 isolates at VF in 4 patients, a stable R5 coreceptor use in 12 patients, and a stable X4 tropism in one patient. Of note, one patient (patient 9) with a switch from R5 isolate at baseline to X4 isolate at M3 reversed to R5 tropism at M6. Three (patients 1, 5, and 17) of 6 patients with minority X4 variants and 1/9 patient (patient 9) with 0% of X4 variants at baseline harbored X4 viruses at virological failure (Fisher's exact test, P = 0.23).
Evolution of V3 quasispecies on MVC.
Phylogenetic trees could be constructed for 15 patients, with each tree including V3 loop sequences obtained from population sequencing at baseline MVC and at virological failure and sequences obtained by ultradeep sequencing at baseline MVC. This enabled us to define the most proximal UDS sequences to bulk sequences at baseline and at failure. Four examples of phylogenetic trees are shown in Fig. 1, showing different dynamics: a switch from the R5 isolate to a stable X4 population for patient 5, an R5 population for patient 6 with a low sequence variability, a selection of the X4 variant followed by a return to the baseline R5 population in patient 9, and an evolving X4 population in patient 17. In order to define specific mutations appearing at virological failure on MVC, we aligned the V3 loop amino acid sequences obtained at baseline and at failure and the proximal baseline UDS sequences for each patient (Fig. 2). For the three patients (1, 5, and 9) with selection of X4 variants, multiple mutations were selected at failure, including changes at critical amino acids 11 and 25. For patient 1 and 5, 2.5% and 0.74% of X4 variants were detected at baseline, including most proximal sequences to VF. For patient 9, no X4 minority variant was detected at baseline.
Fig 1.
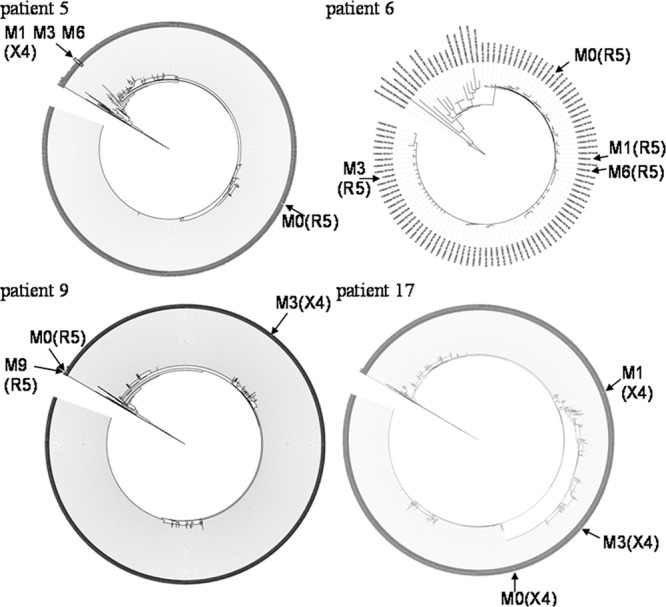
V3 quasispecies at baseline and at failure. Phylogenetic analysis, based on the neighbor-joining method, of the HIV-1 V3 region using 454 sequences at M0 and population sequences at times indicated for 4 patients. Corresponding tropisms are indicated between brackets. Only consensus sequences are represented for patient 6.
Fig 2.

Virological evolution of the V3 region in the patients failing on MVC. The number of 454 sequences used for the analysis is determined in the first column. Prox M indicated the 454 sequence found to be the most closely related to the bulk population sequence at the studied time. For patient 10, no proximal sequence was identified to M6 bulk sequence. No 454 data were available for patients 7 and 14. Columns for amino acids at positions 11 and 25 are in gray. Black arrows indicate positions of known mutations. Black circles indicate positions of mutations observed in this study.
In a majority of patients, R5 sequences were found at virological failure. In 8 patients (patients 2, 4, 6, 8, 11, 12, 13, 16), a stable genotype was found over time. In three patients, the presence of multiple mutations was found in the V3 loop: mutations 13S, 22T, and 24A for patient 3; mutations 13P and 25Q for patient 10; and mutations 13S, 25G, 29D, and 34Y for patient 15. The corresponding mutations were not found in the most proximal UDS sequences at M0, suggesting that these mutations could have been selected later during MVC exposure.
DISCUSSION
In this study, we could characterize the evolution of HIV-1 quasispecies in patients enrolled in the GenoTropism study and having VF on MVC-containing regimens. The risk of VF has been shown to be independently associated with an X4 tropism at baseline MVC (determined from V3 sequences and the Geno2pheno algorithm), with a lower weighed genotypic sensitivity score (GSS), a higher baseline viral load, and a lower nadir of CD4+ cells (11). Here, we focused on the evolution of coreceptor usage and the genetic evolution of V3 sequences in 17 patients with VF. Both genotypic and phenotypic tests showed that in all patients but one, the majority viral population used the CCR5 coreceptor at baseline. The diversity of env quasispecies at baseline MVC was studied by UDS. In a majority of patients (9/14) with R5 tropism from bulk population analysis, no minority X4 variant could be found. However, 4 patients had minority X4 variants, and one was found to have majority X4 variants by UDS. The evolution of tropism could be followed up at VF, showing a switch from R5 isolates to X4 isolates in 4 patients, a stable R5 tropism in 12 patients, and a stable X4 tropism in one patient.
To investigate the dynamics of evolution of the env V3 loop between baseline MVC and VF, we constructed phylogenetic trees comprising all env baseline quasispecies and the bulk variants selected at VF. The switch from R5 to X4 tropism occurred in only 30% of the patients. In this case, multiple V3 mutations were selected at VF, including typical changes at amino acids 11 and 25 associated with X4 tropism. In 2/3 patients, preexisting X4 variants carrying all or most of these mutations were detected as minority variants at baseline, in agreement with a previous report showing that minority X4 variants could be selected by MVC (8). In patients with R5 tropism at VF, the picture was more complex. A majority of patients showed no evolution of V3 sequence between baseline MVC and VF. At least three mechanisms could be involved in these patients: (i) a nonoptimal adherence to antiretroviral therapy (ART) could have led to VF; however, most patients showed a decrease in plasma HIV-1 RNA between M0 and VF, which is not in favor of this possibility; (ii) a suboptimal efficacy of the whole ART regimen, despite partial efficacy due to MVC; (iii) resistance to MVC or CXCR4 usage could be encoded by genetic changes occurring out of the V3 loop, since changes in gp41 have been previously shown to influence coreceptor tropism (16, 17) and mutations in the gp120 V4 loop have been shown to modulate resistance to MVC (18). As an additional means of resistance, we could not exclude that MVC-resistant viruses utilized the drug-bound form of the receptors as previously described in vitro (5, 6, 19).
Three patients presented R5 isolates at VF with the selection of multiple mutations in the V3 loop. Because these mutations were absent from baseline sequences, they are likely to correspond to de novo viral evolution to use MVC-bound CCR5, as previously suggested (18). In the latter report, mutations P/T308H, T320H, and I322V in HXB2 gp120 (corresponding to amino acids 13, 23, and 25 in the V3 loop) were described in clinical isolates from patients with VF on MVC and were shown to code for resistance to MVC. In a previous report, mutations A316T, A319S, and I323V (V3 loop positions 19, 20, and 26), in vitro selected in the presence of MVC, were also shown to promote MVC resistance. Other patterns have been described in patients with VF in the MOTIVATE trial (20, 21) but without full molecular characterization. In our three patients, changes at position 13 were selected or present at baseline and VF, suggesting that this position may be critical for resistance to MVC. However, more data will be necessary to establish correlations between genotype and phenotype (and/or VF) for resistance to MVC, since mutation patterns seem to vary importantly among patients, suggesting a role of env variability background for establishment of resistance.
These V3 loop mutations should be further investigated to understand how they mediate MVC antiviral effects. We cannot also exclude a relationship between the background antiviral regimens and the likelihood of development of resistance against MVC. The numbers of patients were, however, too small to assess a significant relationship between the GSS and the different patterns observed at VF.
In conclusion, our study confirms two possibilities for viral evolution in patients with VF on MVC: emergence of preexisting X4 variants or de novo selection of R5 variants presenting V3 loop mutations. Besides these two patterns, the replication of R5 variants without selection of mutations warrants further pharmacokinetical and/or virological studies outside the V3 loop.
ACKNOWLEDGMENTS
We thank all patients included in the study.
The GenoTropism study was funded by the Agence Nationale de Recherches sur le SIDA et les Hépatites Virales (ANRS; Paris, France) and Pfizer. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under the project “Collaborative HIV and Anti-HIV Drug Resistance Network (CHAIN)” grant agreement no. 223131.
The members of the ANRS AC11 Study Group are given by location as follows. The following members represent the virology laboratories: Bordeaux, P. Recordon-Pinson, H. Fleury, and B. Masquelier; Caen, A. Vabret; Kremlin-Bicetre, C. Pallier; Lille, M. Lazrek; Lyon, P. Andre, J. C. Tardy, and M. A. Trabaud; Marseille, C. Tamalet; Montpellier, B. Montes and M. Segondy; Nantes, V. Ferre; Nice, J. Cottalorda; Orleans, M. Mace; Paris Bichat Claude Bernard, D. Descamps and F. Brun-Vezinet; Paris HEGP, A. Si-Mohammed and C. Charpentier; Paris Paul Brousse, D. Desbois and E. Dussaix; Paris Pitie Salpetriere, A. G. Marcelin, C. Soulie, V. Calvez, and F. Flandre; Paris Saint Antoine, L. Morand-Joubert; Paris Tenon, C. Amiel, and V. Schneider; Rennes, A. Maillard and A. Ruffault; and Toulouse, J. Izopet, F. Nicot, P. Delobel, and S. Raymond. The following members represent the clinical centers: Bordeaux, P. Morlat, I. Louis, J. M. Ragnaud, D. Neau, M. Dupon, and I. Raymond; Caen, R. Verdon; Kremlin-Bicetre, J. F. Delfraissy; Lille, Y. Yazdanpanah; Lyon, C. Chidiac and L. Cotte; Marseille, I. Poizot-Martin and I. Ravaut; Montpellier, J. Reynes; Nantes, F. Raffi; Nice, J. Durant; Orleans, T. Prazuck; Paris Bichat Claude Bernard, P. Yeni; Paris HEGP, L. Weiss; Paris Paul Brousse, D. Vittecocq; Paris Pitie Salpetriere, C. Katlama; Paris Saint Antoine, P. M. Girard; Paris Tenon, G. Pialoux; Rennes, C. Michelet; Toulouse, B. Marchou and Theresa Mo, BC Centre for Excellence in HIV/AIDS.
Footnotes
Published ahead of print 3 December 2012
REFERENCES
- 1. Cicala C, Arthos J, Fauci AS. 2011. HIV-1 envelope, integrins and coreceptor use in mucosal transmission of HIV. J. Transl. Med. 9:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacArthur RD, Novak RM. 2008. Reviews of anti-infective agents: maraviroc: the first of a new class of antiretroviral agents. Clin. Infect. Dis. 47:236–241 [DOI] [PubMed] [Google Scholar]
- 3. Safarian D, Carnec X, Tsamis F, Kajumo F, Dragic T. 2006. An anti-CCR5 monoclonal antibody and small molecule CCR5 antagonists synergize by inhibiting different stages of human immunodeficiency virus type 1 entry. Virology 352:477–484 [DOI] [PubMed] [Google Scholar]
- 4. Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, Montana JB, McHale M, Sullivan J, Ridgway C, Felstead S, Dunne MW, van der Ryst E, Mayer H. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359:1429–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berro R, Klasse PJ, Lascano D, Flegler A, Nagashima KA, Sanders RW, Sakmar TP, Hope TJ, Moore JP. 2011. Multiple CCR5 conformations on the cell surface are used differentially by human immunodeficiency viruses resistant or sensitive to CCR5 inhibitors. J. Virol. 85:8227–8240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moore JP, Kuritzkes DR. 2009. A pièce de resistance: how HIV-1 escapes small molecule CCR5 inhibitors. Curr. Opin. HIV AIDS 4:118–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wirden M, Soulié C, Fourati S, Valantin MA, Simon A, Ktorza N, Tubiana R, Bonmarchand M, Schneider L, Calvez V, Katlama C, Marcelin AG. 6 September 2012. Pitfalls of HIV genotypic tropism testing after treatment interruption. J. Antimicrob. Chemother. [Epub ahead of print.] doi:10.1093/jac/dks362 [DOI] [PubMed] [Google Scholar]
- 8. Westby M, Lewis M, Whitcomb J, Youle M, Pozniak AL, James IT, Jenkins TM, Perros M, van der Ryst E. 2006. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 80:4909–4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whitcomb JM, Huang W, Fransen S, Limoli K, Toma J, Wrin T, Chappey C, Kiss LD, Paxinos EE, Petropoulos CJ. 2007. Development and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob. Agents Chemother. 51:566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Su Z, Gulick RM, Krambrink A, Coakley E, Hughes MD, Han D, Flexner C, Wilkin TJ, Skolnik PR, Greaves WL, Kuritzkes DR, Reeves JD. 2009. Response to vicriviroc in treatment-experienced subjects, as determined by an enhanced-sensitivity coreceptor tropism assay: reanalysis of AIDS Clinical Trials Group A5211. J. Infect. Dis. 200:1724–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Recordon-Pinson P, Soulié C, Flandre P, Descamps D, Lazrek M, Charpentier C, Montes B, Trabaud MA, Cottalorda J, Schneider V, Morand-Joubert L, Tamalet C, Desbois D, Macé M, Ferré V, Vabret A, Ruffault A, Pallier C, Raymond S, Izopet J, Reynes J, Marcelin AG, Masquelier B; ANRS AC11 Resistance Study Group 2010. Evaluation of the genotypic prediction of HIV-1 coreceptor use versus a phenotypic assay and correlation with the virological response to maraviroc: the ANRS GenoTropism Study. Antimicrob. Agents. Chemother. 54:3335–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Letunic I, Bork P. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128 [DOI] [PubMed] [Google Scholar]
- 13. Saliou A, Delobel P, Dubois M, Nicot F, Raymond S, Calvez V, Masquelier B, Izopet J; ANRS AC11 Resistance Study Group 2011. Concordance between two phenotypic assays and ultradeep pyrosequencing for determining HIV-1 tropism. Antimicrob. Agents. Chemother. 55:2831–2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swenson LC, Mo T, Dong WW, Zhong X, Woods CK, Jensen MA, Thielen A, Chapman D, Lewis M, James I, Heera J, Valdez H, Harrigan PR. 2011. Deep sequencing to infer HIV-1 co-receptor usage: application to three clinical trials of maraviroc in treatment-experienced patients. J. Infect. Dis. 203:237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raymond S, Delobel P, Mavigner M, Cazabat M, Souyris C, Encinas S, Bruel P, Sandres-Sauné K, Marchou B, Massip P, Izopet J. 2010. Development and performance of a new recombinant virus phenotypic entry assay to determine HIV-1 coreceptor usage. J. Clin. Virol. 47:126–130 [DOI] [PubMed] [Google Scholar]
- 16. Huang W, Toma J, Fransen S, Stawiski E, Reeves JD, Whitcomb JM, Parkin N, Petropoulos CJ. 2008. Coreceptor tropism can be influenced by amino acid substitutions in the gp41 transmembrane subunit of human immunodeficiency virus type 1 envelope protein. J. Virol. 82:5584–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taylor BM, Foulke JS, Flinko R, Heredia A, DeVico A, Reitz M. 2008. An alteration of human immunodeficiency virus gp41 leads to reduced CCR5 dependence and CD4 independence. J. Virol. 82:5460–5471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tilton JC, Wilen CB, Didigu CA, Sinha R, Harrison JE, Agrawal-Gamse C, Henning EA, Bushman FD, Martin JN, Deeks SG, Doms RW. 2010. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J. Virol. 84:10863–10876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, Stockdale M, Dorr P, Ciaramella G, Perros M. 2007. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J. Virol. 81:2359–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lewis M, Mori J, Simpson P. 2008. Changes in V3 loop sequence associated with failure of maraviroc treatment in patients enrolled in the MOTIVATE 1 and 2 trials, abstr 871. Fifteenth Conference on Retroviruses and Opportunistic Infections, Boston, MA [Google Scholar]
- 21. Lewis M, Simpson P, Delogne C. 2010. Genotypic analysis of the HIV-1 gp120 V3 loop for treatment-experienced patients enrolled into the MOTIVATE studies and who received maraviroc+optimized background therapy, abstr 539. Seventeenth Conference on Retroviruses and Opportunistic Infections, San Francisco, CA [Google Scholar]