

Abstract
Escherichia coli is implicated in the pathogenesis of inflammatory bowel disease (IBD). Rifaximin, a nonabsorbable derivative of rifampin effective against E. coli, improves symptoms in mild-to-moderate IBD. However, rifaximin resistance can develop in a single step in vitro. We examined the prevalence and mechanisms of rifaximin resistance in 62 strains of E. coli isolated from the ileal mucosa of 50 patients (19 with ileal Crohn's disease [L1+L3], 6 with colonic Crohn's disease [L2], 13 with ulcerative colitis [UC], 4 with symptomatic non-IBD diagnoses [NI], and 8 healthy [H]). Resistance (MIC > 1,024 mg/liter) was present in 12/48 IBD-associated ileal E. coli strains. Resistance correlated with prior rifaximin treatment (P < 0.00000001) but not with the presence of ileal inflammation (P = 0.73) or E. coli phylogroup. Mutations in a 1,057-bp region of rpoB, which encodes the bacterial target of rifaximin, were identified in 10/12 resistant strains versus 0/50 sensitive strains (P < 0.000000001) and consisted of seven amino acid substitutions. The efflux pump inhibitor Phe-Arg-β-naphthylamide (PAβN) lowered the MIC of 9/12 resistant strains 8- to 128-fold. Resistance was stable in the absence of rifaximin in 10/12 resistant strains after 30 passages. We conclude that IBD-associated ileal E. coli frequently manifest resistance to rifaximin that correlates with prior rifaximin use, amino acid substitutions in rpoB, and activity of PAβN-inhibitable efflux pumps, but not with the presence of ileal inflammation or E. coli phylogroup. These findings have significant implications for treatment trials targeting IBD-associated E. coli.
INTRODUCTION
Inflammatory bowel disease (IBD) is considered a consequence of uncontrolled intestinal inflammation in response to a combination of environmental, microbial, and immunological factors in a genetically susceptible individual, although its exact etiology remains unclear (1–3). The gut microbiota is increasingly implicated in the pathogenesis of both Crohn's disease (CD) and ulcerative colitis (UC) (4, 5). In particular, the detection of Escherichia coli antigens and DNA in granulomas and peri-ulcerative lesions in CD (6), and circulating antibodies against E. coli outer membrane porin C (OmpC) in patients with CD suggests the involvement of E. coli in the pathogenesis of CD (7). Studies have found selective enrichment of E. coli relative to Firmicutes in patients with Crohn's ileitis (ICD), termed dysbiosis, and a correlation between the severity of ileal inflammation and the density of E. coli colonization (8). Dysbiosis has also been implicated in the pathogenesis of UC, with an increase in Escherichia spp., Rhodococcus spp., and Shigella spp., relative to a decrease in anti-inflammatory Lactobacillus spp. and Pediococcus spp. in UC patients (9).
The potential role of gut microbes in IBD provides a rationale for antibiotic treatment, and drugs such as metronidazole, ciprofloxacin, and clofazimine have been used in the treatment of CD (10, 11), UC (12), and pouchitis (4). However, the success rate of antibiotic treatment varies dramatically with the different forms of IBD, and there is a lack of precise therapeutic guidelines (13). Moreover, IBD treatment involves long-term use of these antibiotics, which promotes emergence of resistance and leads to a poor patient tolerability profile due to adverse systemic side effects such as peripheral neuropathy, nausea, and diarrhea (13, 14). In light of such concerns, the gut-selective antibiotic rifaximin has recently garnered much attention as a therapeutic option in IBD. Rifaximin is a semisynthetic derivative of rifampin with broad-spectrum in vitro activity against aerobic and anaerobic, Gram-positive and Gram-negative bacteria. It is virtually unabsorbed in the intestinal tract after oral administration (<0.4%) due to its pyridoimidazole ring, and remains largely localized in the gastrointestinal tract, avoiding systemic circulation (15, 16). In the United States, it was originally approved for the treatment of uncomplicated Travelers' diarrhea (TD) and is currently used for treating disorders such as hepatic encephalopathy, small bowel bacterial overgrowth, irritable bowel syndrome, and IBD (15).
A growing number of studies have found rifaximin to be effective in the treatment of IBD. A multicenter, double-blind, placebo-controlled trial involving 83 patients with mild-to-moderate CD found that monotherapy with rifaximin at 800 mg for 12 weeks was superior to placebo in promoting clinical remission, only in patients with baseline concentrations of C-reactive protein above the upper reference limit (4, 17). A recent study involving 402 patients with moderately active CD found that administration of 800 mg of rifaximin-EIR (extended intestinal release) formulation twice daily for 12 weeks induced remission with few adverse events (18). Other studies have found rifaximin to be effective in the treatment of UC and antibiotic-dependent pouchitis (19–21). In light of such positive results, rifaximin is increasingly being prescribed for IBD treatment. However, the specific factors that determine positive and negative responses of a patient with IBD to rifaximin remain to be determined.
One factor that is likely to impact the ability of a patient to respond to rifaximin is the presence of antimicrobial resistance. Rifaximin, like other rifamycins, exerts its antimicrobial effects by binding to the β-subunit of bacterial DNA-dependent RNA polymerase and blocking the path of the elongating RNA transcript at the 5′ end, thus inhibiting transcription (22). Point mutations in the rpoB gene, which encodes for the β subunit, have been implicated in resistance to rifaximin and its parent compound rifampin (23). Ninety-five percent of mutations associated with resistance to rifamycins map to four regions of rpoB: the N-terminal cluster (codons 143 to 148) and clusters I (codons 505 to 537), II (codons 563 to 575), and III (codons 684 to 690) (16, 22, 24, 25). Another potential mechanism implicated in resistance to rifaximin is activity of bacterial multidrug resistance (MDR) efflux pump systems that are capable of extruding chemically unrelated compounds, thus lowering the intracellular drug concentration (26). Phe-Arg-β-naphthylamide (PAβN) is a known inhibitor of MDR efflux pumps and has been shown to lower the MIC of rifaximin and rifampin in both resistant and sensitive E. coli strains in vitro (16, 27, 28).
We have previously reported that resistance to antimicrobials, including rifaximin, is common (71% in ICD patients) in E. coli associated with Crohn's ileitis (ICD) (1). In the present study, we investigated the molecular mechanisms of rifaximin resistance, and the association of resistance with E. coli phylogenetic group, prior rifaximin use, and the presence of ileal inflammation in E. coli isolated from the ileal mucosa of ICD, colonic CD (CCD), UC, symptomatic non-IBD (NI), and healthy (H) patients. The CCD, UC, and NI patients were included as a control group that did not present ileal inflammation but were likely to have received rifaximin treatment. The healthy patients were included as a control group that had neither ileal inflammation nor prior rifaximin treatment. Using this diverse group of patients, we found that resistance to rifaximin correlates with prior rifaximin use, amino acid substitutions in rpoB, and the activity of PAβN-inhibitable efflux pumps but not with the presence of ileal inflammation or E. coli phylogroup. Additionally, we found that resistance remains stable in the absence of rifaximin in 10/12 resistant strains over 30 passages. These findings have significant implications for treatment trials targeting IBD-associated E. coli.
MATERIALS AND METHODS
Patients and bacterial strains.
We examined resistance to rifaximin in 62 E. coli strains isolated from the ileal mucosa of 50 patients. Patients with CD were categorized according to the Montreal Classification (29) as L1 (disease restricted to ileum), L2 (colonic involvement only), and L3 (ileal and colonic involvement). In the study group (patients showing ileal inflammation), 25 strains were previously obtained from 19 ICD (L1+L3) patients (6 with a history of rifaximin treatment) (1). In the control group (patients without ileal inflammation), 9 strains were isolated from the ileum of 6 CCD (L2) patients (1 with history of rifaximin treatment), 14 strains were isolated from the ileum of 13 patients with UC (3 with history of rifaximin treatment), 4 strains were previously obtained from 4 patients with non-IBD (NI) related conditions (1 with history of rifaximin treatment) (1), and 10 strains were previously isolated from 8 healthy (H) patients (no known history of rifaximin use) (1). The 4 NI patients were investigated for signs of gastrointestinal disease with a final diagnosis not consistent with IBD (celiac disease [n = 1], H. pylori [n = 1], tubular adenoma [n = 1], lactose intolerance [n = 1]). The 10 H patients were undergoing surveillance endoscopy (with a history of polyps [n = 3], colorectal cancer [n = 3], and age-based surveillance endoscopy [n = 4]).
In all patients, the terminal ileum was intubated as part of the T1 standard of care. A single ileal biopsy was taken from each patient with standard single-use sterile endoscopic forceps into a sterile cryovial containing Luria-Bertani (LB) broth and glycerol (20%) on ice and stored at −80°C until processing. Ten E. coli colonies from each biopsy were screened by random amplified polymorphic DNA (RAPD)-PCR with informative primer 1283, and representative isolates from each individual that differed in overall genotype were selected for subsequent analyses (30). The major E. coli phylogenetic groups (A, B1, B2, and D) were determined by triplex PCR (1, 31). This study was approved by the Cornell University Committee on Human Subjects (protocol 05-05008). All patients gave signed informed consent to participate and to provide mucosal biopsies to the Tissue Bank (protocol 0603-859).
Determination of MIC of rifaximin.
The MIC of rifaximin (Sigma-Aldrich, St. Louis, MO, USA) was determined by the agar dilution method using a breakpoint of 32 mg/liter between sensitive (≤32 mg/liter) and resistant (>32 mg/liter) strains (32).
Mutations in rpoB.
The presence of mutations in rpoB was determined by PCR amplification of a 1,057-bp long region, which includes clusters I, II, and III. The following primers and reaction conditions were used: 5′-AAG CTC ATC GAT ATC CGT AAC G-3′ and 5′-GCA CGT CGC CAC GTT CAA CC-3′; 30 cycles at 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, followed by elongation at 72°C for 10 min (16). The N-terminal cluster (codons 143 to 148) of rpoB was sequenced using the following primers: 5′-CTG CGC GTT AAA CTG CGT CTG GTG-3′ and 5′-CAA CCG GGA CTT CGA TCA GTT TGA-3′. PCR was carried out in a MasterCycler Gradient (Eppendorf, Germany), and the products obtained were purified using a QIAquick PCR purification kit (Qiagen, Germany). PCR products were sequenced at the Cornell University Life Sciences Core Laboratories Center (Ithaca, NY, USA) and aligned with a reference sequence (E. coli K-12, substrain MG1655; GenBank accession no. NC_000913.2), using MegAlign (DNASTAR, Madison, WI, USA).
Determination of the role of bacterial efflux pumps.
To determine the role of MDR efflux pumps in the development of resistance, the MIC of rifaximin in the presence or absence of the efflux pump inhibitor PAβN (Sigma-Aldrich, St. Louis, MO, USA) was measured using the agar dilution method (32). Cultures were grown in two sets of eight Mueller-Hinton agar plates each, one with rifaximin and the other with rifaximin plus PAβN (20 mg/liter). Rifaximin concentrations ranged from 8 to 1,024 mg/liter. A rifaximin stock solution of 50 mg/ml in methanol was used.
Stability of resistance to rifaximin.
Resistant strains were streaked daily on LB agar plates in the absence of rifaximin selective pressure for 30 consecutive passages, representing 2,160 bacterial generations. Every 10 days, the MIC of rifaximin was determined for all passaged strains using the agar dilution method (32).
Statistical analysis.
To identify differences between groups, the Fisher exact test (two-tailed) was used. P < 0.05 was considered statistically significant.
RESULTS
Resistance patterns.
Resistance to rifaximin was present in 12/62 E. coli strains isolated from the ilea of 11/50 patients examined in the study (Table 1). All resistant strains displayed an MIC of >1,024 mg/liter, while sensitive strains displayed an MIC of 0.125 to 16 mg/liter. A total of 13/50 patients had received prior rifaximin treatment, 11 of whom showed presence of resistant strains; in the 2/13 patients who did not show presence of resistant strains, the treatment duration was <2 weeks, which may not be sufficient time to select for resistant strains. Overall, resistance showed a strong correlation with prior rifaximin treatment (P < 0.00000001), but not with the presence of ileal inflammation (P = 0.73) (Table 1). In patients without ileal inflammation (L2, UC, and NI), resistance correlated strongly with prior rifaximin treatment (P < 0.000001) as well. Among patients with rifaximin-resistant E. coli, treatment duration ranged from 13 days to 26 months. Rifaximin-resistant strain 60CM-1 was isolated 131 days after the last rifaximin dose, suggesting that resistance is persistent without continued selection pressure. The E. coli phylogenetic group was not associated with resistance (A, 4/15; B1, 4/17; B2, 2/17; D, 2/11).
Table 1.
Patterns of rifaximin resistancea
| Parameterb | Ileal inflammation present |
Ileal inflammation absent |
Total | |||
|---|---|---|---|---|---|---|
| CD (L1+L3) | CD (L2) | UC | NI | H | ||
| Prior rifaximin treatment | Y N | Y N | Y N | Y N | Y N | |
| No. of patients with resistant E. coli/total patients | 5/6 0/13 | 2/2 0/4 | 4/4 0/9 | 0/1 0/3 | 0/0 0/8 | 11/50 |
| No. of resistant E. coli strains/total no. of strains | 6/7 0/18 | 2/2 0/7 | 4/4 0/10 | 0/1 0/3 | 0/0 0/10 | 12/62 |
Diagnoses: CD, Crohn's disease; L1+L3, ileal+ileocolonic; L2, colonic; UC, ulcerative colitis; NI, symptomatic non-IBD; H, healthy. Y, yes; N, no.
Resistance correlates with prior rifaximin treatment (P < 0.00000001) but not with the presence of ileal inflammation (P = 0.73).
Mutations in rpoB.
Mutations in rpoB were identified in 10/12 resistant strains versus 0/50 sensitive strains (P < 0.000000001) (Table 2). In all but one strain, mutations were encoded by single amino acid substitutions at residues 513, 516, 518, 526, and 574 of the β-subunit of RNA polymerases; strain 58PP-1 was found to have two substitutions, at positions 513 and 574. Overall, seven different amino acid substitutions were identified: Q513H (one strain), D516N (two strains), N518D (one strain), H526N (three strains), H526L (one strain), S574F (two strains), and S574Y (one strain) (Fig. 1). All polymorphisms were present in clusters I and II of rpoB; none were detected in the N-terminal cluster and cluster III.
Table 2.
Molecular mechanisms of rifaximin resistancea
| Diagnosis | Patient ID | E. coli strain ID | Rifx treatment |
Strain type | MIC Rifx (mg/liter) | MIC Rifx + PAβN (mg/liter) | Mutations in rpoB |
||
|---|---|---|---|---|---|---|---|---|---|
| Duration (mo) | Time between last Rifx dose and ileal biopsy (days) | Codon | Amino acid substitution | ||||||
| CD (L2) | 524 | 524-2 | 2.6 | 27 | R | >1,024 | 128 | 526 | H→N |
| 14 | 14GK-1 | 24.5 | 0 | R | >1,024 | 64 | 526 | H→N | |
| CD (L1+L3) | 75 | 75TI-1 | 5 | 6 | R | >1,024 | 8 | 518 | N→D |
| 552 | 552-P4 | 5 | 0 | R | >1,024 | 8 | None | None | |
| 552 | 552-2 | 5 | 0 | R | >1,024 | 8 | None | None | |
| 24 | 24LW-1 | 16 | 0 | R | >1,024 | >1,024 | 516 | D→N | |
| 32 | 32SY-1 | 26 | 0 | R | >1,024 | 32 | 574 | S→F | |
| 578 | 578-1 | 0.6 | 0 | R | >1,024 | 128 | 516 | D→N | |
| UC | 40 | 40EM-1 | 0.4 | 0 | R | >1,024 | >1,024 | 526 | H→L |
| 48 | 48JD-1 | Unknown | 0 | R | >1,024 | 32 | 526 | H→N | |
| 58 | 58PP-1 | 7 | 0 | R | >1,024 | >1,024 | 513, 574 | Q→H, S→F | |
| 60 | 60CM-1 | 13 | 131 | R | >1,024 | 8 | 574 | S→Y | |
| CD (L1+L3) | 541 | 541-1 | NT | NT | S | 32 | 8 | None | None |
| 541 | 541-15 | NT | NT | S | 32 | 8 | None | None | |
| 41 | 41CB-1 | NT | NT | S | 16 | 8 | None | None | |
Rifx, rifaximin; NT, no rifaximin treatment; R, resistant; S, sensitive; rifaximin resistant, MIC > 32 mg/liter; rifaximin sensitive, MIC ≤ 32 mg/liter.
Fig 1.
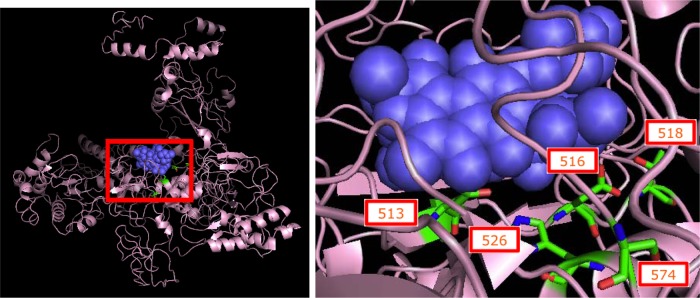
Mutated residues in the RNA polymerase β-subunit. (Left panel) Thermus aquaticus RNA polymerase β-subunit (pink diagram) with bound rifampin (purple spheres), the parent compound of rifaximin. (Right panel) A magnified view of bound rifampin and the T. aquaticus equivalents of the 5 amino acid codons (shown as RBG stick models) that were found to be mutated in our rifaximin resistant E. coli strains. All five codons occur close to the binding pocket of rifamycins; 3/5 codons (513, 516, and 526) are known to interact directly with the antibiotic and affect its binding (22). Image of 1I6V (22) created using The PyMOL Molecular Graphics System (version 1.5.0.4; Schrödinger, LLC).
Role of efflux pumps.
The efflux pump inhibitor PAβN lowered the MIC of 75% (9/12) of resistant strains between 8- and 128-fold, rendering six strains sensitive with MIC of 8 to 32 mg/liter (Table 2). PAβN did not display any effect on the MIC of three resistant strains. In two of the nine strains affected by PAβN, no mutations in rpoB were detected.
Stability of resistant strains.
After 30 passages in culture, 10/12 strains remained resistant (an MIC of >1,024 mg/liter) to rifaximin. The exceptions were strain 75TI-1, in which the MIC dropped from >1,024 to 128 mg/liter after 20 passages, and 58PP-1, in which the MIC dropped from >1,024 to 16 mg/liter after 10 passages. The basis of this change in MIC was investigated by sequencing the same 1,057-bp region of rpoB, which revealed that the Q513H and S574F substitutions in 58PP-1 had reverted back to wild type. In contrast, the N518D substitution was still present in 75TI-1, and the strain had maintained an active efflux mechanism with PAβN lowering the MIC of passaged 75TI-1 from 128 to <8 mg/liter, suggesting that a decrease in the activity or expression level of PAβN-inhibitable efflux pumps was responsible for the decreased MIC of passaged 75TI-1. RAPD-PCR analysis of both passaged and nonpassaged parent strains with informative primer 1283 yielded identical banding patterns indicating the absence of substantial mutations and contamination during passage (30).
DISCUSSION
A growing number of studies have found rifaximin to be effective in the treatment of IBD, and it is increasingly prescribed for the treatment of CD and UC (4). However, the specific factors that determine the clinical response of a patient with IBD to rifaximin remain to be determined. One factor that can impact the ability of an individual to respond to rifaximin is the presence of antimicrobial resistance. The results of the present study indicate that E. coli isolated from the ileum of 11/38 (29%) patients with IBD (5/19 = 26% of L1+L3 patients, 2/6 = 33% of L2 patients, and 4/13 = 31% of UC patients) exhibit resistance to rifaximin with an MIC of >1,024 mg/liter. The presence of resistance correlates strongly with prior rifaximin treatment but not with the presence of ileal inflammation or with E. coli phylogroup. The observation of similar levels of resistance in the study group (with ileal inflammation: L1+L3) and the control group (without ileal inflammation: L2, UC, and NI) suggests that ileal inflammation does not play a role in selecting for rifaximin-resistant E. coli. Conversely, the strong correlation between resistance and prior rifaximin treatment in both the study group and the control group suggests that exposure to rifaximin selects for resistant E. coli independent of ileal inflammation.
Mutations in rpoB alone accounted for resistance in 3/12 resistant strains, activity of PAβN-inhibitable efflux pumps alone accounted for resistance in 2/12 strains, while a combined effect of both mechanisms accounted for resistance in the majority (7/12) of resistant strains. Thus, our findings suggest that both mutations in rpoB and the activity of PAβN-inhibitable efflux pumps, occurring together or independently, contribute to the development of rifaximin resistance. These results are consistent with a previous study that examined in vitro-selected rifaximin-resistant E. coli mutants associated with traveler's diarrhea (16).
Mutations in rpoB have been widely reported in the development of resistance to rifamycins. We identified seven distinct polymorphisms occurring at five residues of the β-subunit of RNA polymerase in 10/12 resistant strains versus 0/50 sensitive strains. Four of these polymorphisms (Q513H, D516N, H526N, and H526L) occur at residues that are known to interact directly with rifampin, and substitutions at these have been shown to confer resistance to rifamycins (Fig. 1) (22, 33). Among these four polymorphisms, D516N (in E. coli) and H526L (in Mycobacterium tuberculosis) are known to be involved in the development of rifampin resistance but, to our knowledge, the specific Q513H and H526N changes have not previously been reported in the literature (22, 24). Of the three polymorphisms that do not directly interact with the drug, N518D has previously been identified in rifampin-resistant E. coli. Although residue 518 does not interact directly with the antibiotic, it is present in the drug's binding pocket and may indirectly affect binding to the β-subunit (22, 24). S574Y has previously been implicated in the development of rifaximin resistance in vitro in E. coli, even though this residue is not known to affect rifamycin binding directly or indirectly (16, 22, 34). S574F has been reported in the literature as a secondary mutation in a serially passaged rifampin-resistant strain carrying a primary mutation at another rpoB residue (34).
In addition to these well-characterized mutations in rpoB, our findings suggest that the activity of PAβN-inhibitable efflux pumps contributes to the development and maintenance of rifaximin resistance. PAβN is a broad-spectrum inhibitor of MDR efflux pumps in a variety of Gram-negative bacteria and is believed to exert its effects by inhibiting resistance nodulation-cell division (RND) type efflux pumps, although this proposition has been called into question by several studies (28, 35, 36). Our results replicate previous observations that PAβN lowers the MIC of rifampin and rifaximin in vitro in both resistant and sensitive E. coli strains (16, 27). PAβN lowered the MIC of 9/12 resistant strains 8 to 128 fold, rendering six strains sensitive to rifaximin (MIC, 8 to 32 mg/liter), while the other three strains exhibited low-level resistance (64 to 128 mg/liter); PAβN lowered the MIC of three sensitive strains 1- to 2-fold. In the two resistant strains with no mutations in rpoB (552-P4 and 552-2), PAβN lowered the MIC from >1,024 to 8 mg/liter, indicating that activity of PAβN-inhibitable efflux pumps can independently induce high-level rifaximin resistance. Intriguingly, both these strains were isolated from the same patient.
In 7/12 resistant strains both mechanisms of resistance were observed, raising questions about the extent to which each mechanism contributes to the development of resistance. Since PAβN lowers the MIC of sensitive strains as well, it appears that efflux pump mechanisms are constitutively active in all E. coli strains. It seems likely that resistant strains overexpress these efflux pumps, which may partially confer their resistant phenotype, together with rpoB polymorphisms that reduce the binding affinity of rifaximin for the β-subunit of RNA polymerase.
The finding that resistance remained stable in 10/12 strains after 30 consecutive passages in the absence of rifaximin selective pressure, and the observation that rifaximin resistant E. coli were isolated from a patient even after 131 days from the last rifaximin dose are in agreement with previous studies that have found rifaximin resistance to be generally stable (15, 23). However, a study evaluating the effect of rifaximin on commensal flora, including E. coli found that the majority of resistant strains regained sensitivity after discontinuing treatment (37). Of the two strains in our study where resistance did not remain stable, 75TI-1 showed a drop in MIC from >1,024 to 128 mg/liter after 20 passages even though the N518D mutation remained intact. However, since PAβN lowered the MIC of this strain from 128 to <8 mg/liter, it appears that activity of PAβN-inhibitable efflux pumps, rather than the N518D substitution, is the primary mechanism of resistance in this strain. In contrast, the drop in MIC of strain 58PP-1, from >1,024 to 16 mg/liter after 10 passages, was associated with reversion of the Q513H and S574F substitutions to wild type and not with the inhibitory effect of PAβN, suggesting that these mutations independently confer the high-level rifaximin resistance observed in this strain. To determine the functional effect of mutations in rpoB and distinguish it from the effect of PAβN-inhibitable efflux pumps, future experiments could examine the transcriptional activity of RNA polymerases isolated from resistant strains in the presence of various concentrations of rifaximin using a cell-free transcription assay.
Although the present study has focused on polymorphisms in rpoB and activity of PAβN-inhibitable efflux pumps, it is possible that there are other mechanisms contributing to resistance such as efflux pumps not inhibited by PAβN, structural modifications of rifaximin by bacteria, or a reduction in membrane permeability to the antibiotic. Studies on rifaximin resistance in in vitro-selected E. coli associated with Traveler's diarrhea (TD) have found no difference in the expression levels of outer membrane porin (Omp) proteins between resistant and sensitive strains (16). On the other hand, studies in Bifidobacterium have implicated reduction in membrane permeability due to specific changes in fatty acid composition, but not structural modifications of the antibiotic moiety by bacterial enzymatic activities in the development of rifaximin resistance (25, 38).
Although development of in vivo resistance to rifaximin during therapy has been reported in Clostridium difficile (39), studies investigating rifaximin resistance in TD-associated E. coli have generally found no clinically significant resistance (15, 40–42). However, one must note that therapy for TD is usually short-term, lasting 3 to 5 days, while therapy for IBD is long-term and may last for months or even years, creating a substrate for resistance development. Another important consideration in interpreting the results of this study is determining the clinical significance of the findings as rifaximin reaches concentrations of up to 8 mg/g of feces after 3 days of treatment with 800 mg daily (43). For most antibiotics, resistance breakpoints are set by CLSI guidelines based on the drug's plasma concentration. Since, rifaximin is largely unabsorbed and remains localized in the gut, there is no formal resistance breakpoint for it. Most studies, including ours, use a breakpoint of 32 mg/liter between sensitive (≤32 mg/liter) and resistant (>32 mg/liter) strains, although the manufacturers Alfa Wasserman recommend a resistance breakpoint of 64 mg/liter (15). Using either breakpoint, the strains described in this study are highly resistant with an MIC of >1,024 mg/liter and were isolated from patients in whom we would anticipate luminal rifaximin concentrations were in the range of 8 mg/g of feces and can therefore be considered clinically resistant.
The present study reveals valuable insights about the mechanisms and patterns of rifaximin resistance in a patient group with diverse disease phenotypes, although the broad applicability of our results is limited by the absence of information on temporal associations and dosing practices that may promote rifaximin resistance. In our study, resistance emerged in one patient after just 13 days of rifaximin treatment and was found to be stable in another patient even after 131 days from the last rifaximin dose. Since observations from two patients do not allow us to reach any broadly applicable conclusions, future studies should investigate rifaximin resistance in clinical settings where bacterial samples are obtained from IBD patients before, during, and after commencing antibiotic therapy in order to determine the temporal association between rifaximin therapy and resistance development.
Although the present study informs our knowledge of rifaximin resistance, it is clear that the mechanisms of action and clinical pharmacology of rifaximin in patients with IBD remain to be fully elucidated. Despite the high enteric concentrations rifaximin achieves, its bioavailability may be greatly limited by its hydrophobic nature and consequent insolubility in aqueous media (10, 11). This situation is further complicated by the increased solubility of rifaximin in the presence of bile acids, suggesting that it may have greater bioavailability and antimicrobial activity in the bile acid-rich small bowel than in the largely aqueous media of the colon (44). This may explain the absence of changes in the composition of the colonic microbiota, despite positive therapeutic responses in IBD patients (15, 45), and suggests differential responses to rifaximin may occur in patients with ileal versus colonic inflammation.
In conclusion, the present study clearly demonstrates that resistance to rifaximin in ileal IBD-associated E. coli correlates with prior rifaximin treatment, amino acid substitutions in rpoB, and activity of PAβN-inhibitable efflux pumps, but not with the presence of ileal inflammation and E. coli phylogroup. These findings have significant implications for treatment trials targeting IBD-associated E. coli and suggest that antimicrobial trials in IBD that are informed by knowledge of disease phenotype and antimicrobial susceptibility are more likely to demonstrate valid outcomes than those based on nonstratified empirical approaches.
ACKNOWLEDGMENTS
This study was supported by a grant from the Jill Roberts Center for Inflammatory Bowel Disease, Weill Medical College of Cornell University, New York, NY.
We thank Fatiha Chabouni for technical assistance.
Footnotes
Published ahead of print 26 November 2012
REFERENCES
- 1. Dogan B, Scherl E, Bosworth B, Yantiss R, Altier C, McDonough PL, Jiang ZD, Dupont HL, Garneau P, Harel J, Rishniw M, Simpson KW. 16 April 2012. Multidrug resistance is common in Escherichia coli associated with ileal Crohn's disease. Inflamm. Bowel Dis. [Epub ahead of print.] doi:10.1002/ibd.22971 [DOI] [PubMed] [Google Scholar]
- 2. Hanauer SB. 2006. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm. Bowel Dis. 12(Suppl 1):S3–S9 [DOI] [PubMed] [Google Scholar]
- 3. Sartor RB. 2006. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 3:390–407 [DOI] [PubMed] [Google Scholar]
- 4. Guslandi M. 2011. Rifaximin in the treatment of inflammatory bowel disease. World J. Gastroenterol. 17:4643–4646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, Weber J, Hoffmann U, Schreiber S, Dietel M, Lochs H. 2002. Mucosal flora in inflammatory bowel disease. Gastroenterology 122:44–54 [DOI] [PubMed] [Google Scholar]
- 6. Liu Y, van Kruiningen HJ, West AB, Cartun RW, Cortot A, Colombel JF. 1995. Immunocytochemical evidence of Listeria, Escherichia coli, and Streptococcus antigens in Crohn's disease. Gastroenterology 108:1396–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arnott ID, Landers CJ, Nimmo EJ, Drummond HE, Smith BK, Targan SR, Satsangi J. 2004. Sero-reactivity to microbial components in Crohn's disease is associated with disease severity and progression, but not NOD2/CARD15 genotype. Am. J. Gastroenterol. 99:2376–2384 [DOI] [PubMed] [Google Scholar]
- 8. Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, Orsi RH, Wiedmann M, McDonough P, Kim SG, Berg D, Schukken Y, Scherl E, Simpson KW. 2007. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn's disease involving the ileum. ISME J. 1:403–418 [DOI] [PubMed] [Google Scholar]
- 9. Sasaki M, Klapproth JM. 2012. The role of bacteria in the pathogenesis of ulcerative colitis. J. Signal. Transduct. 2012:704953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feller M, Huwiler K, Schoepfer A, Shang A, Furrer H, Egger M. 2010. Long-term antibiotic treatment for Crohn's disease: systematic review and meta-analysis of placebo-controlled trials. Clin. Infect. Dis. 50:473–480 [DOI] [PubMed] [Google Scholar]
- 11. Nikfar S, Mirfazaelian H, Abdollahi M. 2010. Efficacy and tolerability of immunoregulators and antibiotics in fistulizing Crohn's disease: a systematic review and meta-analysis of placebo-controlled trials. Curr. Pharm. Des. 16:3684–3698 [DOI] [PubMed] [Google Scholar]
- 12. Rahimi R, Nikfar S, Rezaie A, Abdollahi M. 2007. A meta-analysis of antibiotic therapy for active ulcerative colitis. Dig. Dis. Sci. 52:2920–2925 [DOI] [PubMed] [Google Scholar]
- 13. Guslandi M. 2005. Antibiotics for inflammatory bowel disease: do they work? Eur. J. Gastroenterol. Hepatol. 17:145–147 [DOI] [PubMed] [Google Scholar]
- 14. Shafran I, Burgunder P. 2010. Adjunctive antibiotic therapy with rifaximin may help reduce Crohn's disease activity. Dig. Dis. Sci. 55:1079–1084 [DOI] [PubMed] [Google Scholar]
- 15. DuPont HL. 2011. Biologic properties and clinical uses of rifaximin. Expert Opin. Pharmacother. 12:293–302 [DOI] [PubMed] [Google Scholar]
- 16. Pons MJ, Mensa L, Gascon J, Ruiz J. 2012. Fitness and molecular mechanisms of resistance to rifaximin in in vitro selected Escherichia coli mutants. Microb. Drug Resist. 18:376–379 [DOI] [PubMed] [Google Scholar]
- 17. Prantera C, Lochs H, Campieri M, Scribano ML, Sturniolo GC, Castiglione F, Cottone M. 2006. Antibiotic treatment of Crohn's disease: results of a multicentre, double blind, randomized, placebo-controlled trial with rifaximin. Aliment. Pharmacol. Ther. 23:1117–1125 [DOI] [PubMed] [Google Scholar]
- 18. Prantera C, Lochs H, Grimaldi M, Danese S, Scribano ML, Gionchetti P, and Retic Study Group (Rifaximin-EIR Treatment in Crohn'S Disease) 2012. Rifaximin-extended intestinal release induces remission in patients with moderately active Crohn's disease. Gastroenterology 142:473–481.e4 [DOI] [PubMed] [Google Scholar]
- 19. Gionchetti P, Rizzello F, Ferrieri A, Venturi A, Brignola C, Ferretti M, Peruzzo S, Miglioli M, Campieri M. 1999. Rifaximin in patients with moderate or severe ulcerative colitis refractory to steroid-treatment: a double-blind, placebo-controlled trial. Dig. Dis. Sci. 44:1220–1221 [DOI] [PubMed] [Google Scholar]
- 20. Guslandi M, Petrone MC, Testoni PA. 2006. Rifaximin for active ulcerative colitis. Inflamm. Bowel Dis. 12:335. [DOI] [PubMed] [Google Scholar]
- 21. Shen B, Remzi FH, Lopez AR, Queener E. 2008. Rifaximin for maintenance therapy in antibiotic-dependent pouchitis. BMC Gastroenterol. 8:26 doi:10.1186/1471-230X-8-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. 2001. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104:901–912 [DOI] [PubMed] [Google Scholar]
- 23. Ruiz J, Mensa L, Pons MJ, Vila J, Gascon J. 2008. Development of Escherichia coli rifaximin-resistant mutants: frequency of selection and stability. J. Antimicrob. Chemother. 61:1016–1019 [DOI] [PubMed] [Google Scholar]
- 24. Tupin A, Gualtieri M, Roquet-Baneres F, Morichaud Z, Brodolin K, Leonetti JP. 2010. Resistance to rifampicin: at the crossroads between ecological, genomic and medical concerns. Int. J. Antimicrob. Agents 35:519–523 [DOI] [PubMed] [Google Scholar]
- 25. Vitali B, Turroni S, Dal Piaz F, Candela M, Wasinger V, Brigidi P. 2007. Genetic and proteomic characterization of rifaximin resistance in Bifidobacterium infantis BI07. Res. Microbiol. 158:355–362 [DOI] [PubMed] [Google Scholar]
- 26. Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, Shimer G. 2001. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob. Agents Chemother. 45:1126–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kern WV, Steinke P, Schumacher A, Schuster S, von Baum H, Bohnert JA. 2006. Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Escherichia coli. J. Antimicrob. Chemother. 57:339–343 [DOI] [PubMed] [Google Scholar]
- 28. Saenz Y, Ruiz J, Zarazaga M, Teixido M, Torres C, Vila J. 2004. Effect of the efflux pump inhibitor Phe-Arg-beta-naphthylamide on the MIC values of the quinolones, tetracycline and chloramphenicol, in Escherichia coli isolates of different origin. J. Antimicrob. Chemother. 53:544–545 [DOI] [PubMed] [Google Scholar]
- 29. Silverberg MS, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, Caprilli R, Colombel JF, Gasche C, Geboes K, Jewell DP, Karban A, Loftus EV, Jr, Pena AS, Riddell RH, Sachar DB, Schreiber S, Steinhart AH, Targan SR, Vermeire S, Warren BF. 2005. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology Can. J. Gastroenterol. 19(Suppl A):5–36 [DOI] [PubMed] [Google Scholar]
- 30. Wang G, Whittam TS, Berg CM, Berg DE. 1993. RAPD (arbitrary primer) PCR is more sensitive than multilocus enzyme electrophoresis for distinguishing related bacterial strains. Nucleic Acids Res. 21:5930–5933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clermont O, Bonacorsi S, Bingen E. 2000. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl. Environ. Microbiol. 66:4555–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clinical and Laboratory Standards Institute 2007. Methods for antimicrobial susceptibility testing of anaerobic bacteria. Approved standard M11-A7, 7th ed Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 33. Villain-Guillot P, Bastide L, Gualtieri M, Leonetti JP. 2007. Progress in targeting bacterial transcription. Drug Discov. Today. 12:200–208 [DOI] [PubMed] [Google Scholar]
- 34. Reynolds MG. 2000. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156:1471–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bohnert JA, Kern WV. 2005. Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob. Agents Chemother. 49:849–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pages JM, Masi M, Barbe J. 2005. Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med. 11:382–389 [DOI] [PubMed] [Google Scholar]
- 37. De Leo C, Eftimiadi C, Schito GC. 1986. Rapid disappearance from the intestinal tract of bacteria resistant to rifaximin. Drugs Exp. Clin. Res. 12:979–981 [PubMed] [Google Scholar]
- 38. Vitali B, Turroni S, Serina S, Sosio M, Vannini L, Candela M, Guerzoni ME, Brigidi P. 2008. Molecular and phenotypic traits of in-vitro-selected mutants of Bifidobacterium resistant to rifaximin. Int. J. Antimicrob. Agents. 31:555–560 [DOI] [PubMed] [Google Scholar]
- 39. Liao CH, Ko WC, Lu JJ, Hsueh PR. 2012. Characterizations of clinical isolates of Clostridium difficile by toxin genotypes and by susceptibility to 12 antimicrobial agents, including fidaxomicin (OPT-80) and rifaximin: a multicenter study in Taiwan. Antimicrob. Agents Chemother. 56:3943–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DuPont HL, Jiang ZD, Ericsson CD, Adachi JA, Mathewson JJ, DuPont MW, Palazzini E, Riopel LM, Ashley D, Martinez-Sandoval F. 2001. Rifaximin versus ciprofloxacin for the treatment of traveler's diarrhea: a randomized, double-blind clinical trial. Clin. Infect. Dis. 33:1807–1815 [DOI] [PubMed] [Google Scholar]
- 41. DuPont HL, Ericsson CD, Mathewson JJ, Palazzini E, DuPont MW, Jiang ZD, Mosavi A, de la Cabada FJ. 1998. Rifaximin: a nonabsorbed antimicrobial in the therapy of travelers' diarrhea. Digestion 59:708–714 [DOI] [PubMed] [Google Scholar]
- 42. DuPont HL, Jiang ZD. 2004. Influence of rifaximin treatment on the susceptibility of intestinal Gram-negative flora and enterococci. Clin. Microbiol. Infect. 10:1009–1011 [DOI] [PubMed] [Google Scholar]
- 43. Jiang ZD, Ke S, Palazzini E, Riopel L, Dupont H. 2000. In vitro activity and fecal concentration of rifaximin after oral administration. Antimicrob. Agents Chemother. 44:2205–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Darkoh C, Lichtenberger LM, Ajami N, Dial EJ, Jiang ZD, DuPont HL. 2010. Bile acids improve the antimicrobial effect of rifaximin. Antimicrob. Agents Chemother. 54:3618–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maccaferri S, Vitali B, Klinder A, Kolida S, Ndagijimana M, Laghi L, Calanni F, Brigidi P, Gibson GR, Costabile A. 2010. Rifaximin modulates the colonic microbiota of patients with Crohn's disease: an in vitro approach using a continuous culture colonic model system. J. Antimicrob. Chemother. 65:2556–2565 [DOI] [PubMed] [Google Scholar]