

Abstract
GS-9669 is a highly optimized thumb site II nonnucleoside inhibitor of the hepatitis C virus (HCV) RNA polymerase, with a binding affinity of 1.35 nM for the genotype (GT) 1b protein. It is a selective inhibitor of HCV RNA replication, with a mean 50% effective concentration (EC50) of ≤11 nM in genotype 1 and 5 replicon assays, but lacks useful activity against genotypes 2 to 4. The M423T mutation is readily generated clinically upon monotherapy with the thumb site II inhibitors filibuvir and lomibuvir, and it is notable that GS-9669 exhibited only a 3-fold loss in potency against this variant in the genotype 1b replicon. Rather than M423T, resistance predominantly tracks to residues R422K and L419M and residue I482L in GT 1b and 1a replicons, respectively. GS-9669 exhibited at least additive activity in combination with agents encompassing four other direct modes of action (NS3 protease, NS5A, NS5B via an alternative allosteric binding site, and NS5B nucleotide) as well as with alpha interferon or ribavirin in replicon assays. It exhibited high metabolic stability in in vitro human liver microsomal assays, which, in combination with its pharmacokinetic profiles in rat, dog, and two monkey species, is predictive of good human pharmacokinetics. GS-9669 is well suited for combination with other orally active, direct-acting antiviral agents in the treatment of genotype 1 chronic HCV infection. (This study has been registered at ClinicalTrials.gov under registration number NCT01431898.)
INTRODUCTION
Chronic hepatitis C virus (HCV) infection is a global health problem with an estimated prevalence of 2.2 to 3.3% worldwide (1). In up to 30% of those infected, the disease progresses over the course of 10 to 20 years to liver fibrosis, cirrhosis, and, ultimately, hepatocellular carcinoma (2). In the United States, where genotype (GT) 1 HCV predominates, HCV infection is the leading cause of liver transplants, and mortality rates associated with HCV overtook HIV mortality rates in 2007 (3). Treatment with pegylated alpha interferon (IFN-α) and ribavirin (RBV) is poorly tolerated and of limited efficacy in patients infected with GT 1 (4).
HCV is a small, single-stranded RNA virus whose genome encodes a single polyprotein that is processed by host and viral proteases to generate four structural proteins and six nonstructural proteins. Of the latter, NS3-NS4A (the viral protease), NS5A (an essential component of the cellular replicase complex, although its precise function is unknown), and NS5B (the viral RNA-dependent RNA polymerase) have proven particularly fruitful as targets for the discovery of direct-acting anti-HCV agents. Two protease inhibitors (boceprevir and telaprevir) received regulatory approval in 2011, and a burgeoning group of potential drugs acting via all three viral targets are currently in clinical development. Because of the genetic diversity of HCV due to the high rate and error-prone nature of viral replication, it is anticipated that a combination of agents may be necessary to provide effective eradication in patients (4).
Like several other polymerases, NS5B adopts a topology similar to that of a right hand, with “palm,” “fingers,” and “thumb” subdomains. Inhibitors may be divided into two classes: nucleos(t)ide analogs that serve as false substrates for the enzyme and result in a defective elongation of the nascent RNA chain and nonnucleoside analogs that inhibit the initiation or elongation phases of replication, depending upon the allosteric site to which they bind (5). The nucleotide analog sofosbuvir (GS-7977) (6) is currently in phase 3 clinical studies. Examples of nonnucleoside inhibitors (NNIs) currently in phase 2 clinical studies include BI-207127 and BMS-791325 (binding to thumb site I); filibuvir and lomibuvir (binding to thumb site II) (Fig. 1); setrobuvir, ABT-072, and ABT-333 (binding to palm site I); and tegobuvir (also binding in the palm). While the nucleos(t)ide sofosbuvir exhibits activity against all GTs of the virus, the NNIs mentioned above are active only against GT 1 (7).
Fig 1.
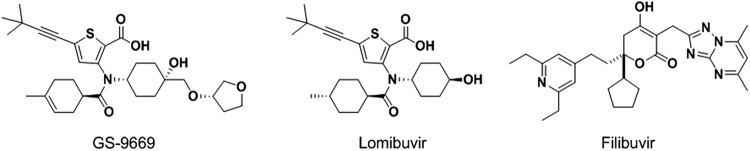
Structures of NS5B thumb site II inhibitors.
Among the nonnucleoside inhibitors of NS5B, clinical efficacy following 3 to 7 days of monotherapy varies from 1.5 to 3.7 log10 declines in viral RNA levels in serum, with the greatest reduction being achieved by lomibuvir (previously known as VX-222 and VCH-222) (7). This encouraging level of clinical validation led to a program in our laboratories directed at the inhibition of NS5B via binding to thumb site II, culminating in the identification of GS-9669, whose preclinical profile is described here.
MATERIALS AND METHODS
Inhibitors.
GS-9669, lomibuvir, filibuvir, the benzimidazole thumb site I inhibitor JT-16 [1H-benzimidazole-5-carboxylic acid, 2-(4-{[4-(acetylamino)-4′-chloro(1,1′-biphenyl)2-yl]methoxy}phenyl)-1-cyclohexyl-], GS-9256, GS-9451, GS-5885, GS-6620, tegobuvir, and daclatasvir were synthesized at Gilead Sciences according to procedures reported previously (8–12; E. Canales, M. O. H. Clarke, S. E. Lazerwith, W. Lew, P. A. Morganelli, and W. J. Watkins, 14 January 2011, International patent application WO 2011088345; C. C. Kong, S. D. Kumar, C. Poisson, C. G. Yannopoulos, G. Falardeau, L. Vaillancourt, and R. Denis, 15 November 2007, International patent application WO 2008058393; A. Cho, C. U. Kim, A. S. Ray, and L. Zhang, 26 May 2011, International patent application WO 2011150288; C. Bachand, M. Belema, D. H. Deon, A. C. Good, J. Goodrich, C. A. James, R. Lavoie, O. D. Lopez, A. Martel, N. A. Meanwell, V. N. Nguyen, J. L. Romine, E. H. Ruediger, L. B. Snyder, D. R. St. Laurent, F. Yang, D. R. Langley, G. Wang, and L. G. Hamann, 9 August 2007, International patent application WO 2008021927). The NS3 protease inhibitor BILN-2061 {14-cyclopentyloxycarbonylamino-18[2-(2-isopropylaminothiazol-4-yl)-7-methoxy-quniolin-4-yloxy]-2,15-dioxo-3,16-diaza-tricyclo(14.3.0.0)nonadec-7-ene-4-carboxylic acid} and the nucleoside 2′-C-methyl adenosine (2′-C-Me-A) [2-(6-amino-purin-9-yl)-5-hydroxymethyl-3-methyl-tetrahydro-furan-3,4-diol] were purchased from Acme Bioscience (Palo Alto, CA). The benzothiadiazine palm site I inhibitor 2-{3-[1-(cyclopropylmethyl-amino)-4-hydroxy-2-oxo-1,2-dihydro-quinolin-3-yl]-1,1-dioxo-1,4-dihydro-1λ6-benzo(1,2,4)thiadiazin-7-yloxy}-acetamide was purchased from ChemALong Laboratories (Lemont, IL). IFN-α and ribavirin were purchased from Sigma-Aldrich. The compounds were dissolved in dimethyl sulfoxide (DMSO) at a final concentration of 10 mM and stored at −80°C before use.
NS5B protein.
NS5BΔ21 GT1b, con-1 was expressed in Escherichia coli and purified to homogeneity as described previously (13).
Surface plasmon resonance assay.
All experiments were performed with a Biacore T200 instrument (GE Healthcare, Piscataway, NJ), using CM5 sensor chips. Binding studies employed the C-terminally truncated soluble form of the NS5B protein (amino acids 1 to 570; GT 1b; Δ21). Standard amine coupling was used to immobilize the protein to the surfaces of preconditioned sensor chips. An immobilization density of 4,000 to 5,000 response units was achieved with a coupling buffer containing 10 mM sodium acetate at pH 5.5. Experiments were run at 25°C in buffer containing 50 mM HEPES (pH 7.5), 5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 1 mM tris(2-carboxyethyl)phosphine, 0.01% Tween 20, and 5% dimethyl sulfoxide. Compounds were tested in a 3-fold concentration dilution series at 6 concentrations (0.823 nM to 200 nM) at a flow rate of 100 μl/min. At the end of each binding cycle, a 5-s injection of a buffer containing 10 mM disodium tetraborate and 1 M NaCl (pH 8.5) at 50 μl/min was used as a regeneration step to remove any remaining compounds bound to the NS5B surfaces. Response data were double referenced using Scrubber 2.0c software and fitted with the 1:1 binding model accounting for mass transport.
Biochemical assay.
RNA-dependent HCV NS5B polymerase activity was determined by the incorporation of nucleotides into secondary-structure-free heteropolymorphic RNA (sshRNA), as described previously (14). The polymerase assay reaction mixture contained 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 10 mM KCl, 1 mM dithiothreitol, 1 mM EDTA, and 0.2 U/μl of RNasin (Promega, Madison, WI). Compounds were serially diluted in 100% (vol/vol) DMSO at a ratio of 1:3, and 10 of these dilutions starting from 25.0 μM to 1.2 nM were further diluted in the polymerase assay reaction mixture at a final DMSO concentration of 0.25%. Compound dilutions were preincubated with 4 ng/μl sshRNA template and 25 nM purified NS5BΔ21 enzyme for 15 min at room temperature. The reaction was started by the addition of 0.2 μM CTP and 250 μM the remaining nucleoside triphosphates, followed by 0.005 μM [33P]CTP (3,300 Ci/mmol) (PerkinElmer, Waltham, MA). Following incubation for 60 min at 30°C, the assay mixtures were transferred into 96-well DE81 filter plates (PerkinElmer, Waltham, MA). Plates were then washed three times with 125 mM Na2HPO4, once with water, and once with 100% ethanol and dried. Microscint-20 (PerkinElmer, Waltham, MA) scintillation cocktail was added, and radioactivity was measured in a TopCount microscintillation reader (PerkinElmer, Waltham, MA). To determine 50% inhibitory concentrations (IC50s), a nonlinear regression sigmoidal dose-response model equation, fit = A + ([B − A]/{1 + [(C/x)^D]}), where A is the lowest percent activity (set at 0), B is the highest percent activity (set at 100), C is the IC50, and D is the Hill slope, was used. Ten-point dose-response curves were generated in XLFit (IDBS, Guildford, Surrey, United Kingdom), using model 205.
Cell culture assays.
Huh7 and Huh7-Lunet cells were obtained from ReBLikon GmbH (Mainz, Germany) (15). Stable replicon cell lines for HCV GTs 1a-H77, 1b-con-1, and 2a-JFH-1 were described previously (16, 17). All Huh7-Lunet cells and derived replicon cell lines were propagated in Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX-I (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 1 unit/ml penicillin, 1 μg/ml streptomycin (Invitrogen), and 0.1 mM nonessential amino acids (Invitrogen). Replicon cell lines were selected and maintained in 0.5 mg/ml G418 (Invitrogen). MT-4 cells, obtained from the AIDS Research and Reference Reagent program, were maintained in RPMI 1640 medium (Irvine Scientific, Santa Ana, CA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 1 unit/ml penicillin, 1 μg/ml streptomycin (Invitrogen), and 0.1 mM HEPES (Invitrogen).
Replicon assays.
HCV GT 1a-H77, 1b-con-1, and 2a-JFH-1 replicon cells were seeded into 96-well plates at a density of 5,000 cells/well in 100 μl of DMEM culture medium without G418. Compounds were serially diluted 1:3 in 100% DMSO and added to cells at a 1:200 dilution, for a final concentration of 0.5% DMSO. Alternately, in a 384-well plate, replicon cells were seeded at 2,000 cells/well. Similarly, compounds were serially diluted 1:3 in 100% DMSO but were added to cells at a 1:225 dilution, for a final concentration of 0.44% DMSO. Assay plates were incubated for 72 h at 37°C in an incubator with 5% CO2 and 85% humidity, after which cell culture medium was removed and cells were assayed for luciferase activity as markers for replicon levels. Luciferase activity was quantified by using a commercial kit (Promega). For the HCV NS5B chimeric replicons, consensus sequence chimeras for GTs 2b, 3a, 4a, and 5a were described previously (18, 19). For the HCV GT 1b NS3/4A, NS5A, and NS5B mutant replicons, mutations were introduced into the GT 1b wild-type replicon by using a QuikChange II XL mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. After confirming the mutations by DNA sequencing, replicon RNA was transcribed in vitro from replicon-encoding plasmids using a MEGAscript kit (Ambion, Austin, TX). RNA for the GT 1b mutants, as well as the HCV NS5B chimeric replicons, was transfected into Huh7-Lunet cells as previously described (17). Briefly, Huh7-Lunet cells were trypsinized and washed twice with phosphate-buffered saline (PBS). A suspension of 4 × 106 cells in 400 μl of PBS was mixed with 10 μg RNA and electroporated (960 μF and 270 V). Cells were transferred into prewarmed cell culture medium at a density of 2 × 105 cells/ml, and 100-μl aliquots were then seeded into 96-well plates. Compound dilution and assay methods were performed as described above. The 50% effective concentration (EC50) was defined as the concentration at which luciferase activity was decreased by 50%. Data were analyzed using GraphPad Prism 5.0 (GraphPad, La Jolla, CA). EC50s were calculated by nonlinear regression analysis using a sigmoidal dose-response (variable-slope) equation (four-parameter logistic equation), Y = bottom + (top − bottom)/{1 + 10^[(log CC50 − X) × Hill slope]}, where the bottom and top values were fixed at 0 and 100.
Cell toxicity assays.
For Huh7 and MT-4 cell cytotoxicity assays, compound serial dilutions were performed in 100% DMSO in a 384-well plate for 10 cycles of 3-fold dilution using a Biomek FX workstation (Beckman Coulter, Brea, CA). DMSO-treated wells were used as the no-compound cytotoxicity control, and 40 μM puromycin-treated (catalog number P9620; Sigma, St. Louis, MO) Huh7 cells or 10 μM puromycin-treated MT-4 cells were used as the 100% cytotoxicity control. Two thousand cells/well were added into a 384-well plate with a Biotek μFlow workstation (Biotek, Winooski, VT). A volume of 0.4 μl of the compound solution was transferred from the serial dilution plate to the cell culture plate on a Biomek FX workstation. The final DMSO concentrations in the assay mixtures were 0.44% for Huh7 cells and 0.5% for MT-4 cells. The plates were incubated for 3 days for Huh7 cells or 5 days for MT-4 cells at 37°C in an incubator with 5% CO2 and 85% humidity. For Huh7 cell viability determinations, the medium in the 384-well cell culture plate was first aspirated, and the plate was then washed with four cycles of 100 μl PBS per well using a Biotek EL405 plate washer. A volume of 50 μl of a solution containing 400 nM calcein-AM (catalog number 25200-056; Anaspec, Fremont, CA) in PBS was added to each well of the plate with a Biotek μFlow workstation. The plate was incubated for 30 min at room temperature before the fluorescence signal (emission at 490 nm and excitation at 520 nm) was quantified with a PerkinElmer Envision plate reader. The percent cell viability was measured by calcein-AM conversion to a fluorescent product. For MT-4 cell viability determinations, 22 μl Cell Titer Glo (catalog number G7573; Promega, Madison, WI) was added to the assay plates with a Biotek μFlow workstation. Plates were subsequently placed onto a PerkinElmer Envision plate reader for 5 min before the luminescence signal was read. CC50 values were calculated from the compound concentration that caused a 50% decrease in signal, a measure of toxicity, and calculated by nonlinear regression using Pipeline Pilot software (Accelrys, San Diego, CA).
In vitro combination experiments.
For combination studies, one compound was serially diluted in nine steps of 1:2 dilutions toward the horizontal direction, with the other compound serially diluted in seven steps of 1:2 dilutions toward the vertical direction. This achieved a defined set of different drug concentrations and ratios. For each individual drug, the EC50 was selected as the midpoint for the concentration range tested. To each well of a black polystyrene 384-well plate (cell culture treated) (catalog number 781086; Greiner Bio-one, Monroe, NC), 90 μl of cell culture medium (without G418) containing 2,000 suspended HCV replicon cells was added with a Biotek μFlow workstation. For compound transfer into cell culture plates, 0.4 μl of compound solution from the combined serial dilution plate was transferred to the cell culture plate on a Biomek FX workstation. The DMSO concentration in the final assay wells was 0.44%. The plates were incubated for 3 days at 37°C with 5% CO2 and 85% humidity. The antiviral activity of the combined drugs was tested as described above for replicon assays. Combination data were analyzed based on the Bliss independence model using MacSynergy II software (version 1.0; University of Michigan, Ann Arbor, MI). Results of the combination assays were expressed as mean synergy and antagonism volumes (nM2) calculated at 95% confidence from two independent experiments performed in four replicates. Combination effects are defined (20) as follows:
Strong synergy if volumes are >100 nM2; this amount of synergy is probably important in vivo.
Moderate synergy if volumes are >50 and ≤100 nM2; this amount of synergy may be important in vivo.
Minor synergy if volumes are >25 and <50 nM2.
Additivity if volumes are >−25 and ≤25 nM2.
Minor antagonism if volumes are <−25 and >−50 nM2.
Moderate antagonism if volumes are >−100 nM2 and ≤−50 nM2; this amount of antagonism may be important in vivo.
Strong antagonism if volumes are ≤−100 nM2; this amount of antagonism is probably important in vivo.
Protein binding.
The unbound fraction in human plasma and cell culture medium (CCM) was determined by equilibrium dialysis against isotonic phosphate buffer (pH 7.4) in an HTDialysis HTD96b unit. The membrane had a nominal cutoff of 10 kDa and was pretreated in accordance with the manufacturer's instructions. The final concentration of test compound added to the protein-containing matrix was 2 μM. Dialysis was done at 37°C overnight with gentle agitation. Samples were then treated by the addition of equal volumes of either buffer (to the protein-containing samples) or drug-free plasma or CCM (to the buffer samples), followed by protein precipitation with an organic solvent containing the internal standard (IS) for liquid chromatography-tandem quadrupole mass spectrometry (LC-MS/MS) and centrifugation. Supernatants were analyzed on a Waters Micromass Quattro Premier XE MS/MS instrument coupled to an Agilent 1200 series liquid chromatograph through an electrospray interface. Quantification was done through analyte/IS peak area ratios.
Pharmacokinetics.
Studies were performed with male Sprague-Dawley rats, beagle dogs, cynomolgus monkeys, and rhesus monkeys (3 animals per dosing route) and complied fully with all federal and Institutional Animal Care and Use Committee (IACUC) guidelines. Intravenous administration was performed at a dose of 0.5 mg/kg of body weight by infusion over 30 min, formulated as a solution in a 1:1:18 (vol/vol/vol) mixture of ethanol, polyethylene glycol 400, and 5% (wt/vol) aqueous dextrose, adjusted to pH 7. Oral dosing was done by gavage at a dose of 2 mg/kg in solution in a 1:11:8 (vol/vol/vol) mixture of ethanol, polyethylene glycol 400, and water, adjusted to pH 7. Blood samples were collected through a jugular vein catheter, and plasma was prepared by centrifugation. After precipitation of plasma proteins, concentrations of analyte were determined by using a Thermo-Finnigan TSQ Quantum Ultra MS/MS coupled to an Agilent 1200 series LC instrument. The limit of quantification was 2 nM. Pharmacokinetic parameters were determined by noncompartmental analysis.
Metabolic stability.
Stability was determined with pooled hepatic microsomal fractions (final protein concentration, 1 mg/ml; BD Biosciences, Woburn, MA) at a substrate concentration of 3 μM. Microsomal membranes were permeabilized by the addition of alamethicin, and cofactors for oxidation (NADPH regenerating system) and glucuronidation (UDP-glucuronic acid) were supplied (21). Reactions were performed for 1 h at 37°C, and after deproteination of the samples, quantification of analytes was performed as described above for protein binding. Hepatic intrinsic clearance was calculated as described previously by Obach (22), and the predicted clearance was calculated using the well-stirred liver model without protein restriction.
RESULTS
NS5B binding and polymerase assays.
Direct binding of GS-9669 to NS5b polymerase was established through surface plasmon resonance (SPR) studies with the Δ21 C-terminally truncated protein (Table 1). Direct measurement of binding affinity is informative in assessing potency and comparing compounds because the format of the biochemical assay necessitates use of the protein at 25 nM; as a result, potent compounds such as GS-9669 and lomibuvir yield IC50s close to this value, and their relative potency is not well discriminated. Furthermore, in cells, the enzyme functions as part of a multimolecular replicase complex; kinetic and inhibitory studies with the isolated enzyme are therefore of limited utility. Nonetheless, the loss in binding affinity conferred by the clinically relevant M423T mutation is reflected in the biochemical assay and is consistent with data from other studies (23, 24); it is apparent that the impact of the mutation in these assays on the potency of GS-9669 and lomibuvir is less than for filibuvir.
Table 1.
Binding to and inhibition of HCV NS5B GT 1b-con-1 polymerase by GS-9669 and other reference NNIs
| Compound | Avg wild-type KD (nM) ± SDa | M423T KD fold shiftb | Mean wild-type IC50 (nM) ± SDc | M423T IC50 fold shiftd |
|---|---|---|---|---|
| GS-9669 | 1.35 ± 0.49 | 10.5 | 40 ± 17 | 4 |
| Lomibuvir | 2.21 ± 0.66 | 10.3 | 55 ± 17 | 4 |
| Filibuvir | 31.5 ± 14.4 | 950 | 73 ± 4 | >94 |
Average kinetic parameters across three surfaces extracted by using a global fit of a 1:1 binding model.
Ratio of mutant KD (equilibrium dissociation constant) over wild-type KD values.
Arithmetic mean of data from at least two experiments.
Ratio of mutant IC50 over wild-type IC50 values.
Activity in cellular assays.
Activity in cell lines, in conjunction with assessment of free-drug levels, has proven a useful predictor of clinical efficacy (25). The activities of GS-9669 and relevant reference inhibitors were compared across HCV GTs, using 3-day assays against subgenomic replicons encoding luciferase genes (for GTs 1a, 1b, and 2a), and against chimeric replicons (for GTs 2b, 3a, 4a, and 5a) in which the relevant NS5B genes, using sequences derived from patient-derived isolates (18, 19), were synthesized and cloned into GT 1b Rluc-neo replicons (thereby replacing the parent NS5B gene) (26). The cytotoxicity of the compounds in the replicon cell line and in MT4 cells was also assessed (Table 2).
Table 2.
Activities of GS-9669 and other reference NS5B inhibitors across GTs and in cytotoxicity assays
| Inhibitor | Mean EC50 (nM) ± SD for GTa,b |
Mean CC50 (nM) ± SD for cell lineb |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 2a | 2b | 3a | 4a | 5a | Huh7 | MT4 | |
| GS-9669 | 11 ± 2.2 | 2.8 ± 0.8 | 79,102 ± 906 | 30,000 ± 8,042 | >50,000 | 348 ± 124 | 8.8 ± 1.0 | >44,000 | 42,829 ± 9,065 |
| Lomibuvir | 13 ± 3.8 | 6.0 ± 1.7 | 13,906 ± 2,146 | >50,000 | >50,000 | 2,275 ± 727 | 13 ± 1.9 | >44,000 | 23,108 ± 3,822 |
| Filibuvir | 447 ± 155 | 170 ± 27 | 16,083 ± 3,627 | 15,323 ± 4,814 | 8,007 ± 5,935 | 2,461 ± 1,250 | 9,128 ± 4,964 | >44,000 | 58,972 ± 6,181 |
| 2′-C-Me-A | 625 ± 290 | 561 ± 197 | 672 ± 185 | 350 ± 71 | 192 ± 11 | 240 ± 57 | 95 ± 7.1 | >44,000 | NT |
GT 1a, 1b, and 2a subgenomic replicons; GT 2b, 3a, 4a, and 5a NS5B chimeric replicons.
Arithmetic mean of data from at least two experiments. NT, not tested.
Collectively, these cell-based data indicate that GS-9669 is active against HCV GT 1a, GT 1b, and GT 5a (EC50s of ≤15 nM) but, like other thumb site II inhibitors, lacks potency against other GTs (GTs 2a, 2b, 3a, and 4a). No cytotoxicity was observed at the highest concentration tested.
Resistance profile of NS5B thumb site II resistance mutations.
M423T, M423I, M423V, I482L, R422K, and L419M mutations have all been generated in replicon-based resistance selection experiments with thumb site II inhibitors (23, 26–28). The binding of both GS-9669 and lomibuvir to the NS5B M423T mutant was reduced 10-fold compared to the wild-type, and for filibuvir, the reduction in binding affinity was much greater (Table 1). Similarly, the inhibitory potency of GS-9669 and lomibuvir in the M423T biochemical assay was reduced by 4-fold, with no activity detectable for filibuvir. Results of transient-transfection replicon assays revealed that GS-9669 is more active against the M423T and M423I mutants than lomibuvir (Table 3). The fold resistance of the I482L and R422K mutants against GS-9669 is higher than that of M423 mutants, although, even against these mutations, GS-9669 remains more potent than lomibuvir. In vitro resistance selection with GS-9669 was reported previously (29): at 10× to 20× the EC50, the dominant mutations were R422K and L419M, and I482L, in GT 1b and 1a replicons, respectively. These data provide strong evidence for the inhibitory effect in the replicon arising from binding to NS5B thumb site II.
Table 3.
Susceptibility of wild-type and NS5b site II mutant HCV GT 1b replicons to GS-9669 and other reference NS5B inhibitors
| Compound | Mean EC50 (nM) for WT (GT 1b) ± SDa | Fold resistanceb against NS5B mutant |
|||||
|---|---|---|---|---|---|---|---|
| M423T | M423I | M423V | I482L | R422K | L419M | ||
| GS-9669 | 11 ± 1.9 | 3.4 | 7.2 | 12 | 38 | 36 | 19 |
| Lomibuvir | 11 ± 3.0 | 81 | 40 | 33 | 105 | 105 | 24 |
| Filibuvir | 80 ± 28 | 37 | 31 | 37 | 17 | NT | NT |
| 2′-C-Me-A | 206 ± 86 | 0.8 | 0.7 | 0.4 | 0.6 | 0.7 | 1.2 |
Arithmetic means of data from at least two experiments Values were generated in a 96-well assay manual format. WT, wild type.
Fold resistance is calculated as the ratio of mutant EC50 to wild-type EC50 values. NT, not tested.
Cross-resistance to non-thumb site II NS5B polymerase, NS3, and NS5A resistance mutations.
The phenotype of drug-resistant NS5B mutants is associated with the binding sites of different inhibitory classes (7). S282T is a resistance mutation in the active site that is selected by 2′-C-methyl-modified ribonucleosides (30). The P495L mutation is in the thumb domain at the site of interaction with the loops extending from the finger domain (thumb site I), selected by a series of benzimidazoles (9). M414T is a mutation selected in the palm region (palm site I) by a series of allosteric benzothiadiazine inhibitors (7). The double mutation of Y448H and Y452H has been selected by and confers resistance to tegobuvir (31, 32). Results of previously reported transient-replicon-transfection assays (19) indicated that GS-9669 and lomibuvir retain full activity against these resistance mutations, in contrast to the relevant controls (see Table S1 in the supplemental material). Similarly, the activity against the major NS3 protease inhibitor (R155K and D168V) (33) and NS5A (Y93H) (34) resistance-associated mutations was assessed. As expected, GS-9669 and the other NS5B inhibitors retained full activity against these resistance mutations (see Table S2 in the supplemental material).
In vitro combination studies.
The activity of GS-9669 was tested in the GT 1b replicon in combination with tegobuvir, GS-9256, and GS-9451 (NS3 protease inhibitors); GS-5885 (NS5A inhibitor); GS-6620 (nucleoside NS5B inhibitor); IFN-α; and RBV (Table 4). The combination of the allosteric NS5B inhibitors tegobuvir and GS-9669 resulted in moderate synergy in the replicon assay. Studies with other HCV inhibitors, including IFN-α and RBV, revealed additive to minor synergistic interactions.
Table 4.
Antiviral effects of GS-9669 in combination with other anti-HCV drugs in GT 1b replicon cells
| Drug combined with GS-9669 | Antiviral mechanism | Mean synergy volume (nM2) ± SDa | Mean antagonism volume (nM2) ± SDa | Interaction |
|---|---|---|---|---|
| GS-5885 | NS5A | 34 ± 19 | 0 ± 0 | Minor synergy |
| GS-9451 | NS3 protease | 19 ± 13 | −2 ± 2 | Additive |
| GS-9256 | NS3 protease | 22 ± 12 | −7 ± 7 | Additive |
| Tegobuvir | NS5B NNI | 70 ± 26 | 0 ± 0 | Moderate synergy |
| GS-6620 | NS5B nucleotide | 26 ± 28 | −1 ± 3 | Minor synergy |
| IFN-α | 31 ± 23 | −2 ± 4 | Minor synergy | |
| RBV | 12 ± 8 | −12 ± 9 | Additive |
Values represent the means ± standard deviations of data from two independent experiments performed in triplicate.
Plasma and CCM binding.
Antiviral effects in vivo and in vitro are dependent upon unbound drug concentrations. Equilibrium dialysis studies were undertaken to assess the impact of binding in cell culture medium (CCM) (containing 10% fetal bovine serum) in replicon assays and to take into account plasma binding for pharmacokinetic-pharmacodynamic (PK-PD) predictions in patients. While the degree of binding in CCM is similar for the three compounds listed in Table 5, the plasma binding differs, with filibuvir less bound than lomibuvir and GS-9669 intermediate between them. When these levels and observed potencies in the replicon (Table 2) are taken into account, a 2- to 4-fold-lower total plasma concentration is predicted to be necessary for GS-9669 to produce an antiviral effect equivalent to that of lomibuvir in patients with wild-type GT 1 infection, and this was corroborated by direct equilibrium dialysis of CCM versus plasma (data not shown).
Table 5.
Binding to human plasma and cell culture medium and calculated EC50s
| Compound | Mean % unbound in cell culture medium ± SDa | Calculated unbound EC50 (nM)b |
Mean % unbound in plasma ± SD | Calculated plasma-adjusted EC50 (nM)c |
||
|---|---|---|---|---|---|---|
| GT 1a | GT 1b | GT 1a | GT 1b | |||
| GS-9669 | 26.5 ± 0.7 | 2.9 | 0.7 | 1.50 ± 0.08 | 194 | 50 |
| Lomibuvir | 22.0 ± 7.6 | 2.9 | 1.3 | 0.64 ± 0.22 | 447 | 206 |
| Filibuvir | 30.3 ± 0.5 | 136 | 52 | 2.12 ± 0.59 | 6,395 | 2,432 |
By equilibrium dialysis versus phosphate buffer.
Unbound drug concentration at the observed EC50.
Total plasma concentration at the unbound EC50.
In vitro metabolism and pharmacokinetics in nonclinical species.
Metabolic stability in vitro was determined using microsomal fractions with mixed cofactors (NADPH for oxidation and UDP-glucuronic acid for conjugation) and in primary hepatocytes, for which the results were very similar (data not shown). The microsomal stability of GS-9669 is compared with that of lomibuvir, and also with observed clearance values in vivo, in Table 6. There was excellent prediction of clearance in the dog from hepatic microsomal data. GS-9669 is stable in rat hepatic microsomal fractions, and this represents a correct characterization of the compound, as low clearance was observed for this species (in vivo hepatic extraction ratio of ca. 4%). Similarly, microsomal stability studies correctly characterized GS-9669 as having moderate to high clearance in both cynomolgus and rhesus monkeys.
Table 6.
Predicted and observed clearance across species
| Species | Compound | Clearance rate (liters/h/kg) |
|
|---|---|---|---|
| Predicted from microsomal assays | Observed in vivo | ||
| Rat | GS-9669 | <0.20 | 0.15 |
| Lomibuvir | 0.26 | 0.23 | |
| Dog | GS-9669 | 0.34 | 0.46 |
| Lomibuvir | 0.63 | 0.63 | |
| Cynomolgus monkey | GS-9669 | 0.24 | 0.93 |
| Lomibuvir | 0.12 | 1.06 | |
| Rhesus monkey | GS-9669 | 0.20 | 1.09 |
| Lomibuvir | 0.10 | 1.12 | |
| Human | GS-9669 | <0.09 | |
| Lomibuvir | 0.12 | ||
Other pharmacokinetic parameters for GS-9669 following dosing intravenously and as an oral solution are summarized in Table 7, with rat results depicted graphically in Fig. 2.
Table 7.
Pharmacokinetic parameters for GS-9669 across species
| Dosage route and parameterc | Mean value ± SD for species |
|||
|---|---|---|---|---|
| Sprague-Dawley rat | Beagle dog | Cynomolgus monkey | Rhesus monkey | |
| Intravenousa | ||||
| Vss (liters/kg) | 0.31 ± 0.09 | 0.68 ± 0.48 | 0.58 ± 0.10 | 0.70 ± 0.52 |
| CL (liters/h/kg) | 0.15 ± 0.11 | 0.46 ± 0.10 | 0.93 ± 0.04 | 1.09 ± 0.43 |
| t1/2 (h) | 3.33 ± 0.53 | 2.35 ± 1.13 | 2.33 ± 0.28 | 2.46 ± 1.60 |
| Oralb | ||||
| Tmax (h) | 0.83 ± 0.29 | 0.5 ± 0.0 | ||
| t1/2 (h) | 2.69 ± 0.79 | 4.51 ± 1.41 | ||
| F (%) | 34 ± 32 | 49 ± 15 | ||
Infusion of 0.5 mg/kg.
Oral gavage of a solution of 2 mg/kg.
Vss, volume of distribution at steady state; CL, clearance; t1/2, half-life; Tmax, time to maximum concentration of drug in serum.
Fig 2.
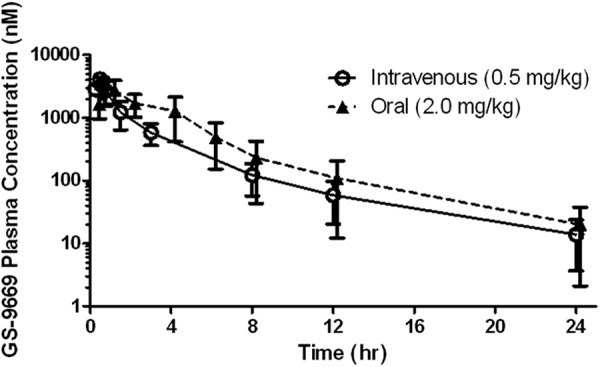
Rat pharmacokinetic profile of GS-9669. Values are means and standard deviations of data for samples from three animals, analyzed separately.
Across species, steady-state volumes of distribution are typical of an acidic drug and approximate extracellular fluid. The compound was rapidly absorbed, and bioavailability was good.
DISCUSSION
Paradigms for the treatment of chronic HCV infection are changing rapidly. The introduction of direct-acting antiviral agents brings with it the prospect of oral, IFN-free regimens of a greatly shortened duration, which will enable viral eradication in a greater proportion of patients and alleviate the significant societal burden of the sequelae of infection (3). Clinical data, however, clearly demonstrate the need for complementary combinations of agents in order to suppress the emergence of resistant virus and ensure maximal sustained virologic response (SVR) (4), and a need remains for the identification of compounds with potency, selectivity, pharmacokinetic profile, and physical properties suitable for convenient administration with other drugs, ideally as part of a single-tablet regimen.
GS-9669 is an inhibitor of HCV replication with low-nanomolar antiviral potency in GT 1a and 1b replicon assays and a selectivity index of >4,400-fold with respect to cytotoxicity. The compound also displays very low potential for cytotoxicity in MT-4 cells as well as in a further panel of diverse immortalized cell lines (data not shown). GS-9669 inhibits NS5B polymerase activity by binding to the thumb site II pocket, as determined by its profile against known resistance mutations. Structure-activity relationships in conjunction with X-ray crystallographic studies adding further insight into the determinants for binding have been presented (W. J. Watkins, presented at the 244th ACS National Meeting, Philadelphia, PA, 19 to 23 August 2012). The M423T mutation is readily generated clinically upon monotherapy with either filibuvir (23) or lomibuvir (M. Jiang, A. Ardzinski, M. Nelson, C. Lin, O. Nicolas, J. Spanks, D. Bartels, E. Zhang, A. Tiggers, J. Sullivan, J. Dorrian, J. Bedard, A. Kwong, and T. Kieffer, presented at the 17th International Meeting on Hepatitis C Virus and Related Viruses, Yokohama, Japan, 10 to 14 September 2010), and it is notable that GS-9669 exhibits only a 3-fold loss in potency against this variant in the GT 1b replicon.
Consistent with the profile of other NS5B thumb site II inhibitors, decreased potency was observed for GS-9669 against replicons containing NS5B from GTs 2a, 2b, 3a, and 4a, but the compound retains activity against GT 5a. The reduced potency in GTs 2 to 4 is attributable to changes in the shape of the binding site as a result of the complementary amino acid residue changes I482L and L419I.
The activity of GS-9669 against diverse resistance mutations beyond NS5B thumb site II in GT 1 HCV makes it a promising candidate for use in combination with other agents. No significant loss of activity was observed against three mutants indicative of other known NNI binding sites or against the S282T mutant, which can be observed in vitro upon selection against nucleoside inhibitors. Full activity is also retained against two major NS3 protease resistance-associated mutations, R155K and D168V, as well as the NS5A Y93H mutant. No antagonism has been observed in combination studies with prototypical compounds encompassing four other direct modes of action or with IFN-α or RBV.
For the treatment of chronic hepatitis C infection, the unbound drug concentration in the liver is of primary importance. However, determination of unbound concentration in the liver is difficult, and measurement of total concentrations can be misleading. We therefore adopted the conservative approach of optimizing unbound plasma concentrations, recognizing that any facilitated uptake into hepatocytes would further enhance potency. The plasma binding of GS-9669 (98.5%) is typical of a moderately lipophilic carboxylic acid, and when this and the extent of binding in CCM are accounted for, the predicted EC50 versus wild-type GT 1 HCV in human plasma is <200 nM, 2- to 4-fold lower than that of lomibuvir.
From in vitro studies, the metabolic stability of GS-9669 in humans is predicted to be high, suggesting that the predominant determinants of plasma exposure following oral dosing will be absorption, distribution, and excretion. The bioavailability of GS-9669 following oral gavage dosing in rat and dog is good. Across nonclinical species, there was good agreement in general between predicted and observed clearance, and steady-state volumes of distribution approximated extracellular fluid; together, these findings suggest that the half-life in human will be suitable for oral dosing no more frequently than twice daily.
GS-9669 is a selective inhibitor of GT 1 and GT 5 HCV with low-nanomolar potency and with an unusually low-potency shift against NS5B M423 mutants. It retains full activity against resistance mutations associated with other mechanisms for inhibition of replication. Its preclinical pharmacokinetic profile suggests that it is well suited for use in combination with other oral agents in IFN-free regimens. Exploratory clinical studies are under way (ClinicalTrials.gov registration number NCT01431898).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Gilead Sciences.
We acknowledge all members of the discovery project team and those providing assay support, especially Lee Chong, Carmen Ip, Christine Zhu, Gene Eisenberg, Jennifer Chau, Kelly Wang, Megan Tessler, Sara Eng, Yujin Wang, Uli Schmitz, Xiaowu Chen, Kati e Dashner, John Ward, Brian Schultz, Anita Niedziela-Majka, Joy Feng, George Stepan, Yili Xu, Weidong Zhong, and Melanie Wong. We also gratefully acknowledge the assistance of Christian Voitenleitner in compiling the manuscript.
Footnotes
Published ahead of print 26 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02052-12.
REFERENCES
- 1. Ghany MG, Strader DB, Thomas DL, Seeff LB, Shuhart MC, Davis GL, Bambha K, Cardenas A, Davern TJ, Franco J, Han SH, Harrison SA, Howell CD, Ling SC, Liu LU, Martin P, O'Shea RS, Reau N, Runyon BA, Talwalkar JA, Wong JB, Yim C. 2009. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 49:1335–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558–567 [DOI] [PubMed] [Google Scholar]
- 3. Ly KN, Xing J, Klevens M, Jiles R, Ward JW, Holmberg SD. 2012. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann. Intern. Med. 156:271–278 [DOI] [PubMed] [Google Scholar]
- 4. Manns MP, Foster GR, Rockstroh JK, Zeuzem S, Zoulim F, Houghton M. 2007. The way forward in HCV treatment—finding the right path. Nat. Rev. Drug Discov. 6:991–1000 [DOI] [PubMed] [Google Scholar]
- 5. Beaulieu PL. 2007. Non-nucleoside inhibitors of the HCV NS5B polymerase: progress in the discovery and development of novel agents for the treatment of HCV infections. Curr. Opin. Investig. Drugs 8:614–634 [PubMed] [Google Scholar]
- 6. Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. 2010. Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53:7202–7218 [DOI] [PubMed] [Google Scholar]
- 7. Watkins WJ, Ray AS, Chong LS. 2010. HCV NS5B polymerase inhibitors. Curr. Opin. Drug Discov. Devel. 13:441–446 [PubMed] [Google Scholar]
- 8. Li H, Tatlock J, Linton A, Gonzalez J, Jewell T, Patel L, Ludlum S, Drowns M, Rahavendran SV, Skor H, Hunter R, Shi ST, Herlihy KJ, Parge H, Hickey M, Yu X, Chau F, Nonomiya J, Lewis C. 2009. Discovery of (R)-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (PF-00868554) as a potent and orally available hepatitis C virus polymerase inhibitor. J. Med. Chem. 52:1255–1258 [DOI] [PubMed] [Google Scholar]
- 9. Hirashima S, Suzuki T, Ishida T, Noji S, Yata S, Ando I, Komatsu M, Ikeda S, Hashimoto H. 2006. Benzimidazole derivatives bearing substituted biphenyls as hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors: structure-activity relationship studies and identification of a potent and highly selective inhibitor JTK-109. J. Med. Chem. 49:4721–4736 [DOI] [PubMed] [Google Scholar]
- 10. Sheng XC, Appleby T, Butler T, Cai R, Chen X, Cho A, Clarke MO, Cottell J, Delaney WE, IV, Doerffler E, Link J, Ji M, Pakdaman R, Pyun HJ, Wu Q, Xu J, Kim CU. 2012. Discovery of GS-9451: an acid inhibitor of the hepatitis C virus NS3/4A protease. Bioorg. Med. Chem. Lett. 22:2629–2634 [DOI] [PubMed] [Google Scholar]
- 11. Guo H, Kato D, Kirschberg TA, Liu H, Link JO, Mitchell ML, Parrish JP, Sun J, Taylor J, Bacon EM, Canales E, Cho A, Kim CU, Cottell JJ, Desai MC, Halcomb RL, Lazerwith SE, Mackman RL, Pyun H-J, Saugier JH, Trenkle JD, Tse WC, Watkins WJ, Xu L. January 2012. Antiviral compounds. US patent 8,088,368
- 12. Bondy SS, Dahl TC, Oare DA, Oliyai R, Tse WC, Zia V. June 2011. Novel pyridazine compound and use thereof. US patent 7,956,184
- 13. Hung M, Wang R, Liu X. 2011. Preparation of HCV NS3 and NS5B proteins to support small-molecule drug discovery. Curr. Protoc. Pharmacol. 13:Unit13B.6. doi:10.1002/0471141755.ph13b06s54 [DOI] [PubMed] [Google Scholar]
- 14. Hung M, Gibbs CS, Tsiang M. 2002. Biochemical characterization of rhinovirus RNA-dependent RNA polymerase. Antiviral Res. 56:99–114 [DOI] [PubMed] [Google Scholar]
- 15. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robinson M, Yang H, Sun SC, Peng B, Tian Y, Pagratis N, Greenstein AE, Delaney WE, IV 2010. Novel hepatitis C virus reporter replicon cell lines enable efficient antiviral screening against genotype 1a. Antimicrob. Agents Chemother. 54:3099–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lohmann VF, Korner JK, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 18. Wong KA, Xu S, Martin R, Miller MD, Mo H. 2012. Tegobuvir (GS-9190) potency against HCV chimeric replicons derived from consensus NS5B sequences from genotypes 2b, 3a, 4a, 5a, and 6a. Virology 429:57–62 [DOI] [PubMed] [Google Scholar]
- 19. Herlihy KJ, Graham JP, Kumpf R, Patick AK, Duggal R, Shi ST. 2008. Development of intergenotypic chimeric replicons to determine the broad-spectrum antiviral activities of hepatitis C virus polymerase inhibitors. Antimicrob. Agents Chemother. 52:3523–3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prichard MN, Shipman C., Jr 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 14:181–205 [DOI] [PubMed] [Google Scholar]
- 21. Fisher MB, Campanale K, Ackerman BL, VandenBranden M, Wrighton SA. 2000. In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab. Dispos. 28:560–566 [PubMed] [Google Scholar]
- 22. Obach RS. 1999. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 27:1350–1359 [PubMed] [Google Scholar]
- 23. Wagner F, Thompson R, Kantaridis C, Simpson P, Troke PJ, Jagannatha S, Neelakantan S, Purohit VS, Hammond JL. 2011. Antiviral activity of the hepatitis C virus polymerase inhibitor filibuvir in genotype 1-infected patients. Hepatology 54:50–59 [DOI] [PubMed] [Google Scholar]
- 24. Yi G, Deval J, Fan B, Cai H, Soulard C, Ranjith-Kumar CT, Smith DB, Blatt L, Beigelman L, Kao CC. 2012. Biochemical study of the comparative inhibition of hepatitis C virus RNA polymerase by VX-222 and filibuvir. Antimicrob. Agents Chemother. 56:830–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith DA, Di I, Kerns EH. 2010. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discov. 9:929–939 [DOI] [PubMed] [Google Scholar]
- 26. Nicolas O, Boivin I, Bedard J. 2008. Genotypic analysis of HCV NS5b variants selected from patients treated with VCH-759, and viral load declines. J. Hepatol. 48(Suppl 2):S317 doi:10.1016/S0168-8278(08)60846-6 [Google Scholar]
- 27. Nicolas O, Boivin I, D'Amours AB, Fex P, Denis F, Selliah S, Bedard J. 2009. Activity & genotypic and phenotypic analysis of HCV NS5b variants selected from patients treated with VCH-916. J. Hepatol. 50(Suppl 1):S349 doi:10.1016/S0168-8278(09)60963-6 [Google Scholar]
- 28. Le Pogam S, Kang H, Harris SF, Leveque V, Giannetti AM, Ali S, Jiang WR, Rajyaguru S, Tavares G, Oshiro C, Hendricks T, Klumpp K, Symons J, Browner MF, Cammack N, Nájera I. 2006. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J. Virol. 80:6146–6154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mabery E, Eng S, Fenaux M, Watkins W, Delaney W. 2012. In vitro resistance profile of GS-9669, a non-nucleoside NS5B inhibitor of genotype 1 HCV. Rev. Antivir. Ther. Infect. Dis. 6:36 [Google Scholar]
- 30. Migliaccio G, Tomassini JE, Carroll SS, Tomei L, Altamura S, Bhat B, Bartholomew L, Bosserman MR, Ceccacci A, Colwell LF, Cortese R, De Francesco R, Eldrup AB, Getty KL, Hou XS, LaFemina RL, Ludmerer SW, MacCoss M, McMasters DR, Stahlhut MW, Olsen DB, Hazuda DJ, Flores OA. 2003. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 278:49164–49170 [DOI] [PubMed] [Google Scholar]
- 31. Shih I, Vliegen I, Peng B, Yang H, Hebner C, Paeshuyse J, Pürstinger G, Fenaux M, Tian Y, Mabery E, Qi X, Bahador G, Paulson M, Lehman LS, Bondy S, Tse W, Reiser H, Lee WA, Schmitz U, Neyts J, Zhong W. 2011. Mechanistic characterization of GS-9190 (tegobuvir), a novel nonnucleoside inhibitor of hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 55:4196–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeuzem S, Buggisch P, Agarwal K, Marcellin P, Sereni D, Klinker H, Moreno C, Zarski J-P, Horsmans Y, Mo H, Arterburn S, Knox S, Oldach D, McHutchison JG, Manns MP, Foster GR. 2012. The protease inhibitor, GS-9256, and non-nucleoside polymerase inhibitor tegobuvir alone, with ribavirin, or pegylated interferon plus ribavirin in hepatitis C. Hepatology 55:749–758 [DOI] [PubMed] [Google Scholar]
- 33. Lin C, Gates CA, Rao BG, Brennan DL, Fulghum JR, Luong YP, Frantz JD, Lin K, Ma S, Wei YY, Perni RB, Kwong AD. 2005. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J. Biol. Chem. 280:36784–36791 [DOI] [PubMed] [Google Scholar]
- 34. Fridell RA, Qiu D, Wang C, Valera L, Gao M. 2010. Resistance analysis of the hepatitis C virus NS5A inhibitor BMS-790052 in an in vitro replicon system. Antimicrob. Agents Chemother. 54:3641–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.