

Abstract
The genome of the coprophilic ascomycete Podospora anserina encodes 33 different genes encoding copper-dependent lytic polysaccharide monooxygenases (LPMOs) from glycoside hydrolase family 61 (GH61). In this study, two of these enzymes (P. anserina GH61A [PaGH61A] and PaGH61B), which both harbored a family 1 carbohydrate binding module, were successfully produced in Pichia pastoris. Synergistic cooperation between PaGH61A or PaGH61B with the cellobiose dehydrogenase (CDH) of Pycnoporus cinnabarinus on cellulose resulted in the formation of oxidized and nonoxidized cello-oligosaccharides. A striking difference between PaGH61A and PaGH61B was observed through the identification of the products, among which were doubly and triply oxidized cellodextrins, which were released only by the combination of PaGH61B with CDH. The mass spectrometry fragmentation patterns of these oxidized products could be consistent with oxidation at the C-6 position with a geminal diol group. The different properties of PaGH61A and PaGH61B and their effect on the interaction with CDH are discussed in regard to the proposed in vivo function of the CDH/GH61 enzyme system in oxidative cellulose hydrolysis.
INTRODUCTION
In response to environmental concerns, industrial processes such as second-generation bioethanol production are emerging. On the basis of enzymatic cellulose conversion, these processes are confronted with a major problem: the recalcitrance of lignocellulosic biomass (1–3). To solve the problem caused by substrate recalcitrance and the high cost of cellulase cocktails, research has focused on various methods to enhance cellulose conversion (4). Fungi are known to be natural degraders of wood and consequently are used in derived biotechnological applications (5, 6). Oxidation was already known to be an important reaction in the fungal degradation of cellulose (7), and it is only recently that several studies have revealed the role played by lytic polysaccharide monooxygenases (LPMOs), formerly known as glycoside hydrolase family 61 (GH61), in the oxidative degradation of lignocellulose (8–14).
Carbohydrate-Active EnZymes database (CAZy) family GH61 (15; www.cazy.org) comprises fungal enzymes that are known for their weak endoglucanase activity (16). Harris et al. (9) revealed that GH61 exhibited a boosting effect on enzymatic cellulose conversion, thus reducing the enzyme loading of cellulase cocktails. More recently, ascorbate, gallate, and even lignin were shown to potentiate GH61 activity on biomass by acting as reductants (13, 14, 17). These LPMOs are believed to act on the surfaces of the insoluble substrate without the need of first extracting individual chains from their crystalline matrix (18). The three-dimensional structure of GH61 shows the presence of highly conserved histidine residues implicated in a type 2 copper center (12, 13, 19) and the presence of a unique N-methylated histidine motif in the binding site (13, 19).
Cellobiose dehydrogenases (CDHs; EC 1.1.99.18; cellobiose:[acceptor] 1-oxidoreductase) are extracellular fungal hemoflavoenzymes that belong to the glucose-methanol-choline (GMC) oxidoreductase superfamily. CDHs are monomeric enzymes carrying two prosthetic groups, a heme b and a flavin adenine dinucleotide (FAD) (20). The flavoprotein domain of CDH catalyzes the two-electron oxidation of cellobiose and, more generally, cellodextrins to the corresponding lactones (21) using electron acceptors such as dioxygen, quinones, and phenoxy radicals (22, 23). The heme is involved in intramolecular electron transfer from FAD to the heme and from the heme to another electron acceptor, such as Fe3+ (24, 25). It is now established that CDHs are secreted by fungi under cellulolytic conditions and are involved in cellulose/lignin degradation (26–30).
Recent studies demonstrated that LPMOs act in concert with CDH since their association resulted in an increase in the conversion of cellulose, assuming a key role of this oxidative system in fungi (8, 11, 12, 31). The effectiveness of LPMO/CDH synergy seems to depend on enzyme concentrations and the type of substrate used. Since oxidized sugars are the major products resulting from cellulose degradation, we wished to obtain more insights into the nature of the products formed. For this purpose, we cloned and heterologously expressed two family GH61 enzymes from the coprophilic ascomycete Podospora anserina. The comparative action of the two GH61 enzymes was evaluated through the identification of oxidized products generated in combination with the CDH of Pycnoporus cinnabarinus that we have recently characterized (28).
MATERIALS AND METHODS
Biological material.
P. anserina strain S mat+ was provided by P. Silar (UMR 8621 CNRS, Orsay, France). Heterologous expression of CDH from P. cinnabarinus ss3 monokaryotic strain BRFM 137 (CIRM-CF, UMR1163, INRA Marseille, France) was described by Bey et al. (28). Pichia pastoris yeast strain X33 and the pPICZαA vector are components of the Pichia pastoris Easy Select expression system (Invitrogen, Cergy-Pontoise, France).
Media and culture conditions.
P. anserina S mat+ was grown at 27°C on M2 plates (KH2PO4, 0.25 g · liter−1; K2HPO4, 0.3 g · liter−1; MgSO4 · 7H2O, 0.25 g · liter−1; urea, 0.5 g · liter−1; thiamine, 0.05 g · liter−1; biotin, 0.25 μg · liter−1; citric acid, 2.5 mg · liter−1; ZnSO4, 2.5 mg · liter−1; CuSO4, 0.5 mg · liter−1; MnSO4, 125 μg · liter−1; boric acid, 25 μg · liter−1; sodium molybdate, 25 μg · liter−1; iron alum, 25 μg · liter−1; dextrin, 5 g · liter−1; yeast extract, 10 g · liter−1; agar, 12.5 g · liter−1; the pH was adjusted to 7 with KH2PO4). Precultures in Roux flasks containing 200 ml of M2 medium without agar supplementation were inoculated by five disks (diameter, 0.5 cm) of P. anserina grown in M2 medium plates. Inoculum was obtained from 5-day-old static precultures incubated at 27°C. An inoculum suspension was obtained from seven mycelial mats ground with an Ultra-Turrax disperser tool in 200 ml of sterile water. Ten milliliters was used to inoculate 500-ml baffled conical flasks containing 100 ml of M2 medium.
All media and protocols for the heterologous expression of P. anserina GH61 (PaGH61) in P. pastoris are described in the Pichia expression manual (Invitrogen).
Isolation of mRNA and cloning of gh61 cDNAs.
From the M2 culture plate of P. anserina, a 1-mm square of agar was placed in 500 μl of sterile water and ground using a FastPrep system (MP Biomedicals, Santa Ana, CA) for 30 s. The suspension obtained was used to inoculate Roux flasks containing 200 ml of M2 medium. The mycelium was harvested after 7 days of culture at 27°C, frozen in liquid nitrogen, and ground with a mortar. Isolation of total RNA was performed on a 3- to 8-day-old culture of P. anserina on microcrystalline cellulose (Avicel) medium using a total RNA purification kit from Plant (Macherey-Nagel, Düren, Germany), as recommended by the manufacturer. The mRNA of P. anserina was purified from the total RNA using an mRNA isolation kit from Roche (Basel, Switzerland) following the standard protocol. Contaminant DNA was digested by Turbo DNase (Ambion Inc., Austin, TX) according to the manufacturer's instructions. First-strand cDNA synthesis was performed using SuperScript reverse transcriptase (Invitrogen) and oligo(dT)18 primer following the manufacturer's instructions. The amplification of the full-length Pagh61A and Pagh61B cDNAs was performed using specific primers (in the sequences below, the vector-specific part is underlined) using a Clontech In-Fusion PCR cloning system (TaKaRa Bio Inc., Japan) to fuse the ends of the PCR fragment to the homologous ends of linearized pPICZαA (Invitrogen). Primer designs were performed using the In-Fusion primer design tool (Clontech). Forward primer PaGH61AF (5′-AGGGGTATCTCTCGAGAAAAGACACGGCCACGTCTCCC-3′) and reverse primer PaGH61AR (5′-GAGTTTTTGTTCTAGACCGATGCACTGGCTGTAGTAAG-3′) were designed from the P. anserina S mat+ gh61A gene (GenBank accession number CAP73254.1). Forward primer PaGH61BF (5′-AGGGGTATCTCTCGAGAAAAGACATTCCACCTTCCAACAGC-3′) and reverse primer PaGH61BR (5′-GAGTTTTTGTTCTAGACCCACGCACTGGTGATACCA-3′) were designed from the P. anserina S mat+ gh61B gene (GenBank accession number CAP68375.1). Amplification was carried out using an Expand High Fidelity PCR system (Roche) following the amplification program of 1 cycle at 94°C for 2 min, 30 cycles composed of three steps for each cycle (94°C for 15 s, 58°C for 30 s, and 72°C for 55 s), and a final step of 72°C for 7 min. The PCR product was fused to the pPICZα-A vector linearized using EcoRI and XbaI. Recombinant expression plasmids were further sequenced to check the integrity of the cDNA sequences (GATC Biotech, Mulhouse, France).
Transformation and screening.
Transformation of competent P. pastoris X33 was performed by electroporation with PmeI-linearized pPICZαA recombinant plasmids, as described by Couturier et al. (32). The pPICZαA vector without an insert was used as a control. Transformants were first screened on yeast extract-peptone-dextrose-sorbitol (YPDS) plates with different concentrations of zeocin (100 to 1,000 μg · ml−1). After incubation at 30°C, transformants were picked from minimal dextrose (MD) plates and transferred to minimal methanol (MM) plates. Zeocin-resistant P. pastoris transformants were then screened for protein expression in 10 ml of BMGY (10 g · liter−1 of yeast extract, 20 g · liter−1 of peptone, 10 g · liter−1 of glycerol in 50-ml tubes) at 30°C in an orbital shaker (200 rpm) for 16 h to an optical density at 600 nm (OD600) of 2 to 6, expression was induced by transferring cells into 2 ml of buffered complex methanol medium (BMMY), and the cells were grown for another 3 days. Each day the medium was supplemented with 3% (vol/vol) methanol. The supernatant was then analyzed by SDS-PAGE to determine the transformant with the best secretion yield.
Production of recombinant enzymes.
The best-producing transformant was grown in 1 liter of BMGY in shaken flasks as described above. The cells were then transferred to 200 ml of BMMY and stirred at 200 rpm and 30°C for 4 days. Bioreactor production of the best-producing transformant was carried out in a 2-liter bioreactor (Tryton; Pierre Guerin, Mauze, France) according to the P. pastoris fermentation process guidelines (Invitrogen). Cultures were first performed in a 0.5-liter shake flask containing 100 ml of BMGY medium. After 60 h of growth at 28°C under shaking at 130 rpm, 50 ml of culture was used to inoculate the bioreactor to reach an OD600 of 1.0.
The basal salts medium used for the batch phase (phase I) in the bioreactor was composed of 40 g · liter−1 glycerol, 26.7 ml · liter−1 H3PO4, 14.9 g · liter−1 MgSO4 · 7H2O, 0.93 g · liter−1 CaSO4, 7.7 g · liter−1 KCl, 4.13 g · liter−1 KOH, 4.35 ml · liter−1 PTM1 and trace salt solution (see Invitrogen's Pichia fermentation process guidelines). Batch phase was performed at 30°C with agitation of 600 rpm, and the pH was controlled at 5.0 with ammonium hydroxide (28%, vol/vol). The total O2 flow was kept constant at 0.3 volume of oxygen per volume of fermentation culture per minute.
After 20 to 24 h on batch phase, the second phase consisted of the simultaneous addition of 50 g of sorbitol and 0.5% (vol/vol) methanol to the bioreactor until the yeast cells switched to methanol metabolism (i.e., 3 h later). The induction phase (phase III) was carried out for 72 h. During this phase, a solution of methanol containing 12 ml · liter−1 of PTM1 salts was added at a rate of 5 ml · h−1 · liter−1 of culture under agitation at 800 rpm. Dissolved oxygen was maintained at 20% in the bioreactor. For CDH production, the induction phase was conducted at 30°C. The temperature was decreased to 25°C for the production of PaGH61A and PaGH61B to ensure correct maturation of proteins.
Enzyme purification.
Culture supernatant was concentrated at least 10-fold using Amicon centrifugal units with a 30-kDa cutoff at 4,000 × g or an Amicon Vivaflow unit (Millipore, Bedford, MA) with a 10-kDa cutoff, depending on the culture volume. The concentrated supernatant was dialyzed against buffer A (Tris-HCl, 50 mM, pH 7.8; NaCl, 150 mM; imidazole, 10 mM) and loaded onto a nickel chelate His-bind resin (GE Healthcare, Buc, France) column (0.7 by 5 cm) equilibrated with buffer A that was connected to an Äkta fast-performance liquid chromatograph (GE Healthcare). Recombinant enzymes that displayed His6 tags at the C terminus were eluted with buffer B (Tris-HCl, 50 mM, pH 7.7; imidazole, 500 mM; NaCl, 150 mM). Fractions containing recombinant enzymes were pooled, concentrated, and dialyzed against HEPES-NaCl buffer (HEPES, 20 mM; NaCl, 150 mM; pH 5) and further purified onto a Sephacryl 200 HR column (GE Healthcare) using the same buffer. Fractions corresponding to the core of the unique peak were pooled and dialyzed against sodium acetate buffer (50 mM, pH 4.8).
Protein analysis.
SDS-polyacrylamide gels (12%) were prepared as described by Laemmli (33). Proteins were stained with Coomassie blue G-250. The molecular mass under denaturing conditions was determined with reference standard proteins (PageRuler prestained protein ladder; Thermo Fisher Scientific, IL).
Zymograms were performed in polyacrylamide gels under native and denaturing conditions. Gels were copolymerized with 0.4% carboxymethyl cellulose (CMC). Under native conditions, 50 μg of purified PaGH61A or PaGH61B was mixed with the loading buffer (without heating and a reducing agent such as β-mercaptoethanol) before separation onto the 12% polyacrylamide gel. After electrophoresis, the polyacrylamide gel was soaked in 50 mM sodium phosphate buffer (pH 5) containing 10 mM ascorbic acid and the mixture was incubated overnight at 45°C under shaking. Under denaturing condition, classical SDS-PAGE was performed with 150 μg of purified PaGH61A or PaGH61B mixed with the loading buffer (without reducing agent). After electrophoresis, the SDS-polyacrylamide gel was washed with deionized water and soaked in 2.5% (vol/vol) Triton X-100. After 1 h of incubation at 4°C, the gel was soaked in 50 mM sodium phosphate buffer (pH 5) containing 10 mM ascorbic acid and the mixture was incubated overnight at 45°C under shaking. After overnight incubation, gels were stained with 0.1% Congo red solution under gentle shaking for 1 h and destained with 1 M NaCl for 1 h. Protein bands exhibiting cellulolytic activity were observed as clear halos on a red background.
Protein concentration was determined using a Bio-Rad protein assay (Bio-Rad, Marnes-la-Coquette, France), based on the Bradford procedure, using bovine serum albumin as the standard (34).
Enzyme assays.
The activity of CDH was determined by monitoring the reduction of 0.2 mM 2,6-dichlorophenol indophenol (DCPIP) in 100 mM sodium acetate buffer (pH 4.8) containing 10 mM cellobiose, as described by Bey et al. (28).
Cellulose cleavage assays.
Cleavage assays were performed in 50-ml Falcon tubes (BD Bioscience) containing 1% (wt/vol) phosphoric acid-swollen cellulose (PASC) in 50 mM sodium phosphate buffer (pH 4.8) in a final reaction volume of 10 ml. PASC was prepared as described by Wood (35). Purified enzymes (CDH, PaGH61A, and PaGH61B) were added to the reaction mixture described above at different concentrations: 500 μg · g−1 of CDH and 5 to 50 mg · g−1 of PaGH61 enzymes. Assays were performed at 45°C in an orbital shaker (Infors AG, Switzerland) for 48 h at 140 rpm. After 48 h of incubation, all the samples were centrifuged at 3,500 rpm for 15 min. The supernatants were filtered through a 0.22-μm-pore-size membrane before carbohydrate determination. Saccharification assays were performed as triplicate independent experiments.
Carbohydrate determination.
Monosaccharides, oligosaccharides, and their corresponding aldonic acid forms generated after PASC cleavage were analyzed by high-performance anion-exchange chromatography (HPAEC) as described by Forsberg et al. (36) using nonoxidized cello-oligosaccharides (Megazyme) as standards. Corresponding oxidized standards were produced from nonoxidized cello-oligosaccharides by CDH treatment. All assays were carried out in triplicate. Products were also analyzed by mass spectrometry (MS) with an Ultraflex II matrix-assisted laser desorption ionization–time of flight/time of flight (MALDI-TOF/TOF) instrument (Bruker Daltonic GmbH, Bremen, Germany) with a 200-Hz smart-beam laser as described by Vaaje-Kolstad et al. (18).
RESULTS
Heterologous expression of P. anserina GH61A and GH61B in P. pastoris.
Genome analysis of P. anserina revealed 33 genes encoding putative GH61 enzymes (37). Of those genes, 8 showed the presence of a carbohydrate binding module from family 1 (CBM1), which are known to display cellulose-binding function. Three amino acids (two histidines, with one at the first position, and one tyrosine) exposed at enzyme surfaces are known to be required for GH61 activity because they are involved in metal coordination. In PaGH61, these key amino acids were all conserved. In order to investigate the specificities of P. anserina GH61, we selected two members, PaGH61A (GenBank accession number CAP73254.1) and PaGH61B (GenBank accession number CAP68375.1), for further cloning and heterologous expression. These two GH61 proteins (i) harbored a CBM1 module, (ii) belonged to two distinct subgroups of GH61 (not shown), and (iii) displayed only 34% amino acid identity with each other.
The two cDNAs encoding PaGH61A and PaGH61B were cloned by reverse transcription-PCR using mRNA purified from P. anserina grown on cellulose. After sequencing, both coding sequences were in agreement with those predicted from genomic data. The two cDNAs encoding PaGH61A and PaGH61B were inserted into pPICZαA for subsequent expression in the yeast P. pastoris. Following induction of selected P. pastoris transformants, PaGH61A and PaGH61B were successfully expressed and further produced in a bioreactor to high yield, as we obtained 150 mg and 1 g of each protein per liter of culture, respectively. Electrophoretic analysis revealed that purified PaGH61A and PaGH61B displayed apparent molecular masses of 60 and 50 kDa, respectively (see Fig. 2, lanes 2 and 3), which were higher than the theoretical ones (39 and 36 kDa, respectively). This might be due to glycosylation of the linker regions between the catalytic and the CBM1 modules that contain multiple glycosylation sites. In parallel, CDH was produced in a bioreactor as described by Bey et al. (28).
Fig 2.

Products generated following the synergistic action of PaGH61 and CDH on cellulose. Analysis was performed using HPAEC-PAD after 48 h of incubation at 45°C under shaking of 1% (wt/vol) PASC in 50 mM sodium acetate (pH 5) with CDH/PaGH61. (A, B) Action of PaGH61A at 5 mg · g−1 (A) and PaGH61B at 5 mg · g−1 (B) in association with 500 μg · g−1 of CDH; (C) products formed by combination of PaGH61B (50 mg · g−1) with 500 μg · g−1 of CDH. All experiments were carried out in triplicate. nC, nanocoulomb.
Zymogram analysis of PaGH61A and PaGH61B.
In order to observe the cellulose-cleaving activity of PaGH61A and PaGH61B, we performed zymogram analysis in the presence of CMC (Fig. 1). When gel electrophoresis was performed under native conditions, clear halos were visualized at the top of the zymogram in the presence of ascorbic acid, indicating that cellulose was depolymerized (Fig. 1, lanes 4 and 5). Under denaturing conditions, halos were visualized at molecular masses corresponding to those of PaGH61A and PaGH61B (Fig. 1, lanes 6 and 7).
Fig 1.
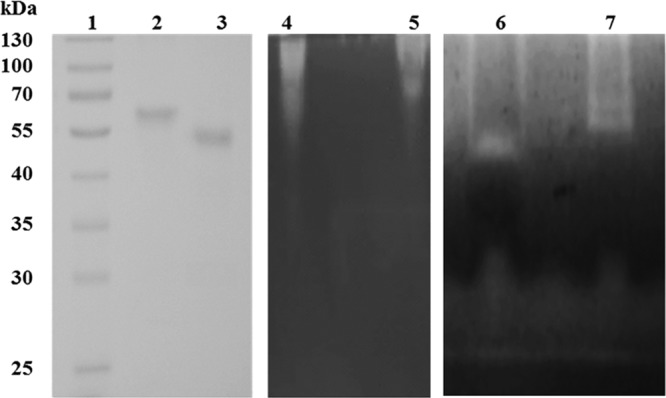
SDS-PAGE (lanes 1 to 3) and zymogram analysis (lanes 4 to 7) of PaGH61 enzymes. For SDS-PAGE, lane 1, prestained molecular mass marker; lane 2, 10 μg of purified PaGH61A; lane 3, 10 μg of purified PaGH61B; for a CMC zymogram under native conditions incubated overnight at 45°C under shaking with addition of 10 mM ascorbic acid, lane 4, 50 μg of rPaGH61B; lane 5, 50 μg of PaGH61A; for a CMC zymogram under denaturing conditions, lane 6, 150 μg of purified PaGH61B; lane 7, 150 μg of purified PaGH61A.
Synergistic action of PaGH61A and PaGH61B with CDH.
To investigate the synergy between each of P. anserina GH61 and CDH, cellulose cleavage assays were performed. The products formed following cellulose cleavage were analyzed by HPAEC-pulsed amperometric detection (PAD) (Fig. 2) and mass spectrometry (Fig. 3). In control reaction mixtures containing P. anserina GH61 enzymes or CDH only without any reductant, only traces of nonoxidized (for PaGH61) or oxidized (for CDH) cellodextrins were observed.
Fig 3.


MALDI TOF/TOF spectra of products following the combined action of PaGH61 with CDH. Analysis was performed after 48 h of incubation at 45°C under shaking of 1% (wt/vol) PASC in 50 mM sodium acetate (pH 5). (A) PaGH61A (5 mg · g−1) combined with CDH (500 μg · g−1); (B) PaGH61B (5 mg · g−1) combined with CDH (500 μg · g−1); (C) PaGH61B (50 mg.g−1) combined with CDH (500 μg · g−1). MS/MS fragmentation was performed on products released at m/z 1,045 (D) and m/z 1,245 (E) under the same conditions described for panel C. Identified compounds are labeled in black for native cellodextrins and in red when the products are oxidized. aa, aldonic acid; al, aldonolactone/lactone; OI, oxygen incorporation; *, compounds are subject to hypothesis. The numbers in different-colored boxes refer to the masses of compounds identified during MS/MS fragmentation.
Figure 3 illustrates the type of compounds formed when each PaGH61 enzyme was incubated with CDH on amorphous cellulose. Using HPAEC, it was possible to discriminate a ladder of oxidized cello-oligosaccharides (from DP2ox [i.e., oxidized cello-oligosaccharides with a degree of polymerization of 2] to DP5ox) and nonoxidized cello-oligosaccharides (from DP2 to DP6) (Fig. 2). The peaks close to nonoxidized cello-oligosaccharides appearing as shoulders in Fig. 2 were identified as the corresponding lactones (m/z −2) using mass spectrometry (Fig. 3). The presence of cellodextrins presumably oxidized at the C-1 position (m/z +16), as well as doubly oxidized cellodextrins at the C-1 and C-4 positions (m/z +14), was confirmed by mass spectrometry. Mass spectrometry analysis revealed that oxidized compounds are underestimated compared to the estimate obtained by HPAEC-PAD analysis due to the presence of cations from the acetate buffer (association of oxidized compounds with Na+ or 2Na+) (Fig. 3), although the relative quantities are unclear without further analysis. Comparison of the two PaGH61 enzymes revealed that PaGH61A yielded significantly larger amounts of lactone intermediates than PaGH61B (Fig. 2A and B and 3A and B). The main difference between the two enzymes is the presence among oxidized compounds of two additional peaks with PaGH61B at m/z 1,045 and m/z 1,245 (Fig. 3B) that were absent in the PaGH61A experiment (Fig. 3A). The product corresponding to m/z 1,045 could be explained by combined oxidations such as doubly oxidized DP6 (m/z +32) with an Na+ adduct (m/z +23). The product at m/z 1,245 could correspond to triple oxidation of DP7 (m/z +48) with 2Na+-H+ (m/z +47). Since the identification of such compounds could represent novel types of oxidations, we increased the loading of PaGH61B to enhance this phenomenon (Fig. 2C). It led almost exclusively to the formation of oxidized cellodextrins (Fig. 2C and 3C). As expected, mass spectrometry revealed an enhancement of peaks corresponding to m/z 1,045 and m/z 1,245 (Fig. 3C). MS/MS fragmentation of m/z 1,045 and m/z 1,245 peaks revealed the presence of DP6 (Fig. 3D) and DP7 (Fig. 3E) oxidized cellodextrins, respectively. In both cases, fragmentation patterns could be consistent with oxidation at the C-6 position (geminal diol form) at the reducing end (Fig. 3D and E).
DISCUSSION
Podospora anserina represents an interesting model to study oxidative deconstruction of lignocellulose since this coprophilic fungus displays an impressive array of genes encoding putative GH61 enzymes (37). The reason for the existence of multiple enzymes in fungi is not yet known, and the question arises whether each enzyme has a different role in fungi by targeting different substrates and/or generating different products, as suggested by Phillips et al. (12). Among the set of P. anserina GH61 enzymes, we successfully produced PaGH61A and PaGH61B to high levels in a bioreactor (up to 1 g · liter−1 for PaGH61B). As GH61 activity is not easily detectable, we set up a simple method based on zymogram analysis using ascorbate as a reductant to evaluate GH61 activity. The presence of activity halos demonstrated that PaGH61A and PaGH61B were functionally produced by P. pastoris with a correct processing of the signal peptide that is required for activity since the N-terminal residue is one of the residues that coordinates copper.
The synergistic action between P. anserina GH61 monooxygenase and CDH was revealed by the identification of nonoxidized, singly oxidized, and multiply oxidized cello-oligosaccharides (Fig. 2A to C and 3A to C). It has been suggested that apparition of native cellodextrins could take origins in oxidative cleavages near the reducing ends releasing an intact nonreducing moiety (12). When PaGH61A and PaGH61B were associated with CDH, different behaviors were observed. PaGH61A released more lactone products than PaGH61B. These lactone products are spontaneously hydrolyzed to their corresponding C-1 oxidized products (Fig. 4). In the PaGH61A experiment, doubly oxidized compounds were detected; they displayed masses that presumably correspond to C-4 oxidation (4-keto sugar) and C-1 oxidation (aldonic acid form). PaGH61B mainly yielded peaks corresponding to oxidized cello-oligosaccharides, and this phenomenon was enhanced when PaGH61B was added to high concentrations. Another striking difference between PaGH61A and PaGH61B was observed by the identification of two products (m/z 1,045 and m/z 1,245) released by the combination of PaGH61B with CDH. This suggests that PaGH61A and PaGH61B display different specificities with respect to the oxidation position. It has been proposed that the m/z 1.245 product could result in ketal formation from the 4-keto sugar (12). However, a 6-ketal sugar (i.e., geminal diol) is more likely because (i) it is more stable than a 4-ketal sugar, (ii) it would be further promoted under acidic conditions, and (iii) it could be stabilized by the environment. An equivalent form has already been identified following oxidation of sugars by galactose oxidase. Indeed, fungal galactose oxidases (EC 1.1.3.9) are single copper metalloenzymes that catalyze the oxidation of primary alcohols to their corresponding aldehydes with strict regioselectivity, and a galactose oxidase from Fusarium spp. was shown to target the C-6 position of galactose to produce aldehyde. The aldehyde was identified, using nuclear magnetic resonance, as a 6-ketal sugar (i.e., geminal diol) in water (38). Our hypothesis is supported by the MS/MS fragmentation patterns of the m/z 1,245 and the m/z 1,045 products that would correspond to triple oxidation for DP7 and double oxidation for DP6 at the reducing end, respectively (Fig. 3D and E). As hydrogen abstraction at the C-4 or the C-1 position would lead to the formation of a copper hydroperoxo intermediate with subsequent cleavage of the glycosidic bond, the most probable explanation for the observed fragmentation pattern of the m/z 1,245 and the m/z 1,045 products would be a triple oxidation (m/z +48) with a C-1 aldonic acid together with two 6-ketal sugar groups and a double oxidation (m/z +32) with a C-1 aldonic acid together with one 6-ketal sugar group (Fig. 3D and E). The increase of these oxidized species upon high-level loading of PaGH61B could be interpreted in two ways: the additional oxidation takes place directly either onto cellulose or onto soluble cello-oligosaccharides. Such C-6 oxidations could occur along the cellulose chain during hydrogen abstraction without subsequent cleavage.
Fig 4.

Schematic representation of products formed by the synergistic action of PaGH61 and CDH.
To date, three classes of LPMOs have been described. Type 1 LPMOs generate products oxidized at C-1, probably initially in the lactone form, which is then spontaneously hydrolyzed to yield an aldonic acid. Type 2 LPMOs generate products oxidized at the nonreducing end. Formation of a 4-ketoaldose could result from oxidative cleavage proceeding by the same general mechanism proposed for oxidation at C-1. It has been suggested that LPMO-3 catalyzed oxidation of cellulose with less specificity than LPMO-1 (oxidation in the C-1 position) and LPMO-2 (oxidation in the C-4 position) (8). The regiospecificities of PaGH61A and PaGH61B observed in the present study do not allow us to assign them to these proposed LPMO classes since experiments were performed in the presence of CDH.
Analysis of the genome sequence of P. anserina demonstrated an unexpected enzymatic arsenal (32, 37, 39). This coprophilic fungus displays a large array of GMC oxidoreductases, including two CDHs, a pyranose oxidase, a galactose oxidase, a copper radical oxidase, and several laccases, that could be relevant for in vivo synergy with LPMOs upon lignocellulose oxidative reactions. We are only at the beginning of the characterization of the enzymatic cocktail of P. anserina with the biochemical characterization of two members of the P. anserina GH61 family that have revealed striking differences in regiospecificities. We have demonstrated unambiguously that PaGH61A and PaGH61B are both able to act in concert with the P. cinnabarinus CDH to degrade cellulose with the concomitant release of novel types of oxidized products, including oxidation at the C-6 position for one of the enzymes. Further characterization of the other members of the P. anserina GH61 family is needed to get insights into the role of these enzymes in vivo.
ACKNOWLEDGMENTS
This study was carried out in the framework of the Futurol project with financial support from OSEO.
We thank C. B. Faulds for his comments on the manuscript and the reviewers for their suggestions that improved the manuscript.
Footnotes
Published ahead of print 2 November 2012
REFERENCES
- 1. Chundawat SP, Beckham GT, Himmel ME, Dale BE. 2011. Deconstruction of lignocellulosic biomass to fuels and chemicals. Annu. Rev. Chem. Biomol. Eng. 2:121–145 [DOI] [PubMed] [Google Scholar]
- 2. Foust TD, Ibsen KN, Dayton DC, Hess JR, Kenney KE. 2008. The biorefinery, p 7–37 In Himmel ME. (ed), Biomass recalcitrance: deconstructing the plant cell wall for bioenergy, 1st ed Blackwell Publishing Ltd., Oxford, United Kingdom [Google Scholar]
- 3. Margeot A, Hahn-Hagerdal B, Edlund M, Slade R, Monot F. 2009. New improvements for lignocellulosic ethanol. Curr. Opin. Biotechnol. 20:372–380 [DOI] [PubMed] [Google Scholar]
- 4. Wilson DB. 2012. Processive and non processive cellulases for biofuel production—lessons from bacterial genomes and structural analysis. Appl. Microbiol. Biotechnol. 93:497–502 [DOI] [PubMed] [Google Scholar]
- 5. Lundell TK, Mäkelä MR, Hildén K. 2010. Lignin-modifying enzymes in filamentous basidiomycetes—ecological, functional and phylogenetic review. J. Basic Microbiol. 50:5–20 [DOI] [PubMed] [Google Scholar]
- 6. Sigoillot JC, Berrin JG, Bey M, Lesage-Meessen L, Levasseur A, Lomascolo A, Record E, Uzan-Boukhris E. 2012. Fungal strategies for lignin degradation, p 263–308 In Lapierre C, Jouanin L. (ed), Lignins. Elsevier, Philadelphia, PA [Google Scholar]
- 7. Eriksson KE, Pettersson B, Westermark U. 1974. Oxidation: an important enzyme reaction in fungal degradation of cellulose. FEBS Lett. 49:282–285 [DOI] [PubMed] [Google Scholar]
- 8. Beeson WT, Phillips CM, Cate JHD, Marletta MA. 2012. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134:890–892 [DOI] [PubMed] [Google Scholar]
- 9. Harris PV, Welner D, McFarland KC, Re E, Poulsen JCN, Brown K, Salbo R, Ding H, Vlasenko E, Merino S, Xu F, Cherry J, Larsen S, Leggio LL. 2010. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49:3305–3316 [DOI] [PubMed] [Google Scholar]
- 10. Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VG. 2012. Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5:45 doi:10.1186/1754-6834-5-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Langston JA, Shaghasi T, Abbate E, Xu F, Vlasenko E, Sweeney MD. 2011. Oxidoreductive cellulose depolymerisation by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl. Environ. Microbiol. 77:7007–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Phillips CM, Beeson WT, Cate JHD, Marletta MA. 2011. Cellobiose dehydrogenase and a copper dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6:1399–1406 [DOI] [PubMed] [Google Scholar]
- 13. Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen JC, Johansen KS, Krogh KB, Jørgensen CI, Tovborg M, Anthonsen A, Tryfona T, Walter CP, Dupree P, Xu F, Davies JG, Walton PH. 2011. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U. S. A. 108:15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westereng B, Ishida T, Vaaje-Kolstad G, Wu M, Eijsink VGH, Igarashi K, Samejima M, Ståhlberg J, Horn SJ, Sandgren M. 2011. The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS One 6:e27807 doi:10.1371/journal.pone.0027807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238 doi:10.1093/nar/gkn663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karlsson J, Saloheimo M, Siika-Aho M, Tenkanen M, Penttila M, Tjerneld F. 2001. Homologous expression and characterization of Cel61A (EG IV) of Trichoderma reesei. Eur. J. Biochem. 268:6498–6507 [DOI] [PubMed] [Google Scholar]
- 17. Dimarogona M, Topakas E, Olsson L, Christakopoulos P. 2012. Lignin boosts the cellulase performance of a GH-61 enzyme from Sporotrichum thermophile. Bioresour. Technol. 110:480–487 [DOI] [PubMed] [Google Scholar]
- 18. Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, Eijsink VGH. 2010. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330:219–222 [DOI] [PubMed] [Google Scholar]
- 19. Lin X, Beeson WT, Phillips CM, Marletta MA, Cate JHD. 2012. Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20:1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hallberg BM, Bergfors T, Backbro K, Pettersson G, Henriksson G, Divne C. 2000. A new scaffold for binding haem in the cytochrome domain of the extracellular flavocytochrome cellobiose dehydrogenase. Structure 8:79–88 [DOI] [PubMed] [Google Scholar]
- 21. Henriksson G, Sild V, Szabo IJ, Pettersson G, Johansson G. 1998. Substrate specificity of cellobiose dehydrogenase from Phanerochaete chrysosporium. Biochim. Biophys. Acta 1383:48–54 [DOI] [PubMed] [Google Scholar]
- 22. Mason MG, Wilson MT, Ball A, Nicholls P. 2002. Oxygen reduction by cellobiose oxidoreductase: the role of the haem group. FEBS Lett. 518:29–32 [DOI] [PubMed] [Google Scholar]
- 23. Samejima M, Eriksson KEL. 1992. A comparison of the catalytic properties of cellobiose: quinone oxidoreductase and cellobiose oxidase from Phanerochaete chrysosporium. Eur. J. Biochem. 207:103–107 [DOI] [PubMed] [Google Scholar]
- 24. Henriksson G, Johansson G, Pettersson G. 1993. Is cellobiose oxidase from Phanerochaete chrysosporium a one-electron reductase? Biochim. Biophys. Acta 1144:184–190 [DOI] [PubMed] [Google Scholar]
- 25. Igarashi K, Momohara I, Nishino T, Samejima M. 2002. Kinetics of inter-domain electron transfer in flavocytochrome cellobiose dehydrogenase from the white-rot fungus Phanerochaete chrysosporium. Biochem. J. 365:521–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ander P, Mishra C, Farrell RL, Eriksson KEL. 1990. Redox reactions in lignin degradation: interactions between laccase, different peroxidases and cellobiose: quinone oxidoreductase. J. Biotechnol. 13:189–198 [Google Scholar]
- 27. Bao WJ, Renganathan V. 1992. Cellobiose oxidase of Phanerochaete chrysosporium enhances crystalline cellulose degradation by cellulases. FEBS Lett. 302:77–80 [DOI] [PubMed] [Google Scholar]
- 28. Bey M, Berrin JG, Poidevin L, Sigoillot JC. 2011. Heterologous expression of Pycnoporus cinnabarinus cellobiose dehydrogenase in Pichia pastoris and involvement in saccharification processes. Microb. Cell Fact. 10:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Henriksson G, Ander P, Pettersson B, Pettersson G. 1995. Cellobiose dehydrogenase (cellobiose oxidase) from Phanerochaete chrysosporium as a wood degrading enzyme. Studies on cellulose, xylan and synthetic lignin. Appl. Microbiol. Biotechnol. 42:790–796 [Google Scholar]
- 30. Henriksson G, Johansson G, Pettersson G. 2000. A critical review of cellobiose dehydrogenases. J. Biotechnol. 78:93–113 [DOI] [PubMed] [Google Scholar]
- 31. Sygmund C, Kracher D, Scheiblbrandner S, Zahma K, Felice AK, Harreither W, Kittl R, Ludwig R. 2012. Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl. Environ. Microbiol. 78:6161–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Couturier M, Haon M, Coutinho PM, Henrissat B, Lesage-Meesen L, Berrin JG. 2011. Podospora anserina hemicellulases potentiate Trichoderma reesei secretome for the saccharification of lignocellulosic biomass. Appl. Environ. Microbiol. 77:237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 34. Bradford MM. 1976. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 35. Wood TM. 1988. Preparation of crystalline, amorphous, and dyed cellulase substrates. Methods Enzymol. 160:19–25 [Google Scholar]
- 36. Forsberg Z, Vaaje-Kolstad G, Westereng G, Bunæs AC, Stenstrøm Y, MacKenzie A, Sørlie M, Horn SJ, Eijsink VGH. 2011. Cleavage of cellulose by a CBM33 protein. Protein Sci. 20:1479–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Espagne E, Lespinet O, Malagnac F, Da Silva C, Jaillon O, Porcel BM, Couloux A, Aury JM, Segurens B, Poulain J, Anthouard V, Grossetete S, Khalili H, Coppin E, Dequard-Chablat M, Picard M, Contamine V, Arnaise S, Bourdais A, Berteaux-Lecellier V, Gautheret D, de Vries RP, Battaglia E, Coutinho PM, Danchin EG, Henrissat B, Khoury RE, Sainsard-Chanet A, Boivin A, Pinan-Lucarre B, Sellem CH, Debuchy R, Wincker P, Weissenbach J, Silar P. 2008. The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol. 9:R77 doi:10.1186/gb-2008-9-5-r77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parikka K, Tenkanen M. 2009. Oxidation of methyl alpha-d-galactopyranoside by galactose oxidase: products formed and optimization of reaction conditions for production of aldehyde. Carbohydr. Res. 344:14–20 [DOI] [PubMed] [Google Scholar]
- 39. Lafond M, Navarro D, Haon M, Couturier M, Berrin J-G. 2012. Characterization of a broad-specificity β-glucanase acting on β-(1,3), β-(1,4), and β-(1,6) glucans that defines a new glycoside hydrolase family. Appl. Environ. Microbiol. 78:8540–8546 [DOI] [PMC free article] [PubMed] [Google Scholar]