

Abstract
Bifidobacteria are a major microbial component of infant gut microbiota, which is believed to promote health benefits for the host and stimulate maturation of the immune system. Despite their perceived importance, very little is known about the natural development of and possible correlations between bifidobacteria in human populations. To address this knowledge gap, we analyzed stool samples from a randomly selected healthy cohort of 87 infants and their mothers with >90% of vaginal delivery and nearly 100% breast-feeding at 4 months. Fecal material was sampled during pregnancy, at 3 and 10 days, at 4 months, and at 1 and 2 years after birth. Stool samples were predicted to be rich in the species Bifidobacterium adolescentis, B. bifidum, B. dentium, B. breve, and B. longum. Due to high variation, we did not identify a clear age-related structure at the individual level. Within the population as a whole, however, there were clear age-related successions. Negative correlations between the B. longum group and B. adolescentis were detected in adults and in 1- and 2-year-old children, whereas negative correlations between B. longum and B. breve were characteristic for newborns and 4-month-old infants. The highly structured age-related development of and correlation networks between bifidobacterial species during the first 2 years of life mirrors their different or competing nutritional requirements, which in turn may be associated with specific biological functions in the development of healthy gut.
INTRODUCTION
Mother-to-child transmission and temporal development of the human gut microbiota are population-based processes. Understanding these processes is essential to the identification of gut microbiota-associated functionalities. Certain members of the genus Bifidobacterium represent very abundant early colonizers of the infant gut (1), making them a prime target for investigation.
The high abundance of Bifidobacterium species in infants is considered to promote development and maturation of the immune system to sustain health (2–4). Furthermore, our recent studies suggest that the succession of bifidobacteria is important for the proper immunological development (5, 6).
Eight Bifidobacterium species have been associated with the human gastrointestinal tract (GIT): Bifidobacterium adolescentis, B. breve, B. longum subsp. longum, B. longum subsp. infantis, B. pseudolongum, B. bifidum, B. pseudocatenulanum, and B. dentium. Some bifidobacterial strains, e.g., B. pseudolongum, appear to be exclusively associated with adult gut microbiota, and some, especially B. longum subsp. infantis, are typically isolated from infants (7). The population-wise, age-related development of bifidobacteria in infants remains to be investigated.
Due to the fact that there is evidence for previously uncharacterized diversity of bifidobacteria in the human gut (8, 9), we believe that to obtain a comprehensive description of bifidobacterial composition, the study should include a large number of individuals, and various techniques should be applied. It is also very important to avoid targeting specific bifidobacteria, since this may lead to the exclusion of as-yet-undiscovered, but potentially important bifidobacterial groups.
The aim of the present study, therefore, was to describe bifidobacterial composition, temporal development, and possible correlations in a large, randomly selected cohort of mothers and their children using a combination of both culture-dependent and -independent techniques. This was done by analyzing the series of stool samples from the IMPACT cohort (10). The samples were collected during early and late pregnancy stages and at 3 days, 10 days, 4 months, and 1 and 2 years after birth. We present results showing a highly structured, age-related succession of bifidobacterial species within the study population, as well as correlations between the abundances of these species during the first 2 years of life.
MATERIALS AND METHODS
The simplified workflow of the data analysis is represented in Fig. 1.
Fig 1.
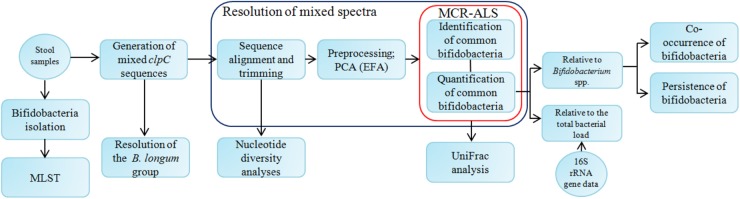
Flow diagram of the study. Boxes and circles represent processes and the data or materials obtained from them, respectively. At first, generated mixed spectra were trimmed to ensure that each spectrum brings an equal amount of information into the system. Then these spectra were analyzed using principal component analysis (PCA), evolving factor analysis (EFA), and multivariate curve resolution analysis with alternating least squares (MCR-ALS) to identify and quantify common bifidobacterial species in the data set. Quantification was performed relative to both the Bifidobacterium group and the total bacterial load. The B. longum group was resolved separately. Diversity was assessed using both taxon-based (UniFrac analysis based on MCR-ALS predictions) and taxon-independent (based on mixed spectra) techniques. The co-occurrence of various bifidobacteria, as well as their persistence over time, was also evaluated. Also, bifidobacteria were isolated from stool samples, and MLST analysis of several isolates was performed.
Study cohort.
The PACT study (Prevention of Allergy among Children in Trondheim), a controlled nonrandomized prospective intervention study of pregnant women and their children up to 2 years of age, was started in 2000 (11). At the same time, a prospective observational substudy (IMPACT [Immunology and Microbiology in PACT]) of 720 pregnant women and their offspring, recruited from the PACT control cohort, was also initiated (10). All pregnant women in the control cohort were eligible for participation if they were willing and able to answer a questionnaire in Norwegian and supply biological material. We analyzed a randomly selected subset of the IMPACT study comprising stool samples of 87 infants and their mothers. At the point of inclusion participants were normally healthy children not screened for allergic disease hereditary diseases, allergy or atopy in the family, exposure factors, lifestyle, parental health, siblings health, smoking exposure, or any possible confounder. The samples were collected during the first or second trimester (8 to 20 gestation weeks) and the third trimester (30 to 40 gestation weeks) from mothers, and at 3 days, 10 days, 4 months, and 1 year and 2 years after birth from children. Most children (∼90%) were delivered vaginally. The information on infants' diet (Table 1) was received from a questionnaire filled out by parents at 2 years. More details about the IMPACT study cohort can be found in Storrø et al. (5).
Table 1.
Number of infants who were breast- and formula-fed and who received solid food at 6 weeks, 4 months, and 1 year of age
| Diet (no. of infants) | % infantsa receiving different diets |
||
|---|---|---|---|
| 6 wk | 4 mo | 1 yr | |
| Breast-feeding (84) | 97.6 | 95.2 | 29.8 |
| Formula feeding (84) | 2.3 | 4.8 | 29.8 |
| Combined (85) | 10.6 | 21.2 | 55.3 |
| Solids (87) | NA | 13.8 | 98.9 |
Data are based on the questionnaire filled out by parents at 2 years. The total number of infants in the study was 87. NA, not applicable.
In some cases, the stool sample from one or several collection time points was missing, whereas for some other individuals, more than one sample was present for the same age point (see Fig. S1A in the supplemental material). Moreover, several samples had sequence spectra of poor quality (many abnormally shaped fluorescence peaks resembling dye blobs or a low fluorescence signal) that would compromise the performance of multivariate curve resolution (MCR) analysis (see Fig. S2 in the supplemental material) and therefore had to be removed. In all, there were 330 samples in the final data set. For 52 children, bifidobacteria in the sample from the mother were also detected. The numbers of samples included in the study at each time point, as well as the numbers of mother-child pairs that contained data for all or just a few time points, are summarized in Fig. S1B and C in the supplemental material.
Isolation of DNA from stool samples.
Fecal specimens were stored in sterile Cary Blair transport and holding medium (BD Diagnostics, Sparks, MD). Each specimen was frozen at −20°C no later than 2 h after sampling and transported to the laboratory for further storage at −80°C. Stool samples were first diluted 1:1 in solution 1 (50 mM glucose, 25 mM Tris-HCl [pH 8.0], 10 mM EDTA [pH 8.0]). The resulting suspension was then diluted 1:4 in 4 M guanidinium thiocyanate. The sample was then transferred to a sterile FastPrep-tube (Qbiogene, Inc., USA) with 250 mg of acid-washed glass beads (≤106 μm; Sigma-Aldrich, Germany), homogenized in FastPrep Instrument (Qbiogene) for 40 s and then centrifuged at 13,500 rpm for 5 min. Then, 170 μl of supernatant, together with 10 μl of Silica particles (Merck, Germany), was transferred to a 96-well Greiner U-plate (Greiner Bio-One, Germany), which was then placed in a Biomek 2000 Workstation (Beckman Coulter, USA). After the addition of Sarkosyl (1%), the plate was incubated at 65°C for 10 min, followed by 10 min at room temperature. The supernatant was discarded, and a bead pellet was washed twice with 50% ethanol. The beads were finally suspended in 100 μl of buffer C (1 mM EDTA [pH 8.0], 10 mM Tris-HCl [pH 8.0]), followed by incubation at 65°C for 30 min, and the solution with the eluted DNA was collected.
Generation of mixed clpC sequences.
Based on the evaluation of six housekeeping genes (12), we chose a mixed Bifidobacterium clpC (caseinolytic protease C) gene sequencing approach to obtain a comprehensive description of the bifidobacterial composition. Bifidobacterium clpC gene was amplified using clpC-specific primers developed by Ventura et al. (12). Sequencing of the forward strand was performed using BigDye Terminator v1.1 chemistry (Applied Biosystems, USA). The B. longum group-specific primer (5′-AGAAGCTGGAAGCCGAT-3′) was designed based on 34 Bifidobacterium clpC alleles downloaded from the Bifidobacterium multilocus sequence typing (MLST) database (13).
Mixed sequence analysis.
The simplified scheme of mixed sequence analysis steps is represented in Fig. 2.
Fig 2.

Simplified scheme of mixed sequence analysis. (A) Alignment of mixed sequences. All sequences have the same start and end, as well as a variable region in the middle. (B) MCR-ALS. Pure spectra common for all (the majority of) samples and their relative amounts in each sample are identified. (C) Base calling of pure MCR-ALS-resolved spectra. The nucleotide sequence of each of the pure resolved spectra is identified.
At first, mixed sequence spectra were aligned to small (∼20 bp) start and end fragments flanking the variable region of the clpC gene and trimmed (Fig. 2A). To correct for retention shift differences and to increase the linearity of the data, all spectra were preprocessed after the alignment by using correlation-optimized warping (14, 15). Preprocessed aligned mixed sequences were then analyzed using a multivariate curve resolution (MCR-ALS) approach (Fig. 2B). This method allows simultaneous qualitative and quantitative identification of the components (in this case, groups of Bifidobacterium species), which are common to all of the samples in the data set of interest (16). The specific feature of the MCR-ALS method is that it particularly searches for components that are common for all of the samples in the data set, whereas other information is regarded as noise. Mathematically, the MCR-ALS method presents the initial experimental matrix of spectral data D (m × n) as the combination of the concentration matrix C (m × i) and the matrix of pure components S (i × n), and a residual term E (m × n):
| (1) |
To define the initial number of components (initial estimates i), we used both principal component analysis (PCA) and evolving factor analysis (EFA) with nucleotide fluorescence intensities at each position being used as variables. PCA transforms the initial data into a new coordinate system, and the number of components is determined by the amount of variance explained (L. Smith, unpublished data). EFA on the other hand, repeatedly applies PCA to sections of the data set starting from the first two samples and subsequently adding the next one every run of the PCA (17). In both cases, the number of suggested components, which is defined based on the drop in eigenvalue, is used as the initial number of components i for MCR-ALS.
Resolved spectra were then base called using an in-house-developed program that works in MATLAB, the same programming environment in which MCR-ALS is performed (Fig. 2C [the source code can be made available upon request]). Identification of resolved components was then performed using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) searches against the National Center for Biotechnology Information (NCBI) database.
Due to a high noise ratio in the T-channel, which could compromise results of MCR-ALS analysis, the information on T nucleotides was excluded from all spectra prior to resolution and then reintroduced at the base-calling stage. The validity of T-channel information removal was tested using predefined mixtures of bifidobacterial strains. MCR-ALS resolution of spectra without information on T nucleotides was comparable to that of spectra with information on all four nucleotides available (see Table S1 in the supplemental material).
All of the analyses of sequence spectra were performed using MATLAB R2010a software (The MathWorks, Inc., Natick, MA), Statistical and Bioinformatics toolboxes for MATLAB. For EFA, PCA, and MCR-ALS analyses, PLS Toolbox v5.8 for MATLAB (Eigenvector Research, Inc., USA) was used.
Diversity measures.
Nucleotide α-diversity was measured using a modified Simpson's index, calculated based on the fraction of each nucleotide's fluorescence intensity at every position, normalized for the length of the spectrum:
| (2) |
The rationale is that in case of a pure sequence, there is only one nucleotide at each position (indexed by i), while in a very unified mixed sample, each position would contain all four nucleotides, and their intensity fractions would approach 0.25. To prevent misinterpretation based on the variation in length of the sequence (n), the value is then normalized. Furthermore, we consider the inverse relationship 1/cmixed to support the more intuitive interpretation: the higher the cmixed value, the more mixed the sample is (1 corresponds to pure, not mixed, samples, whereas a cmixed value of 4 is the maximum and indicates a uniform mixture). In the case of bifidobacteria, however, the maximum of 4 will never be achieved due to high GC content. Although the information from the T-channel was noisy, we included these data into diversity measures analyses, since the fluorescence intensity at each position was normalized to 1, and abnormally high T-peaks would diminish the fraction of other nucleotides only in few positions.
To calculate individual diversity (i.e., the diversity within one individual), the raw mixed spectrum of each sample was used. The diversity within the population was calculated based on the average spectrum for each age group.
The β-diversity at various ages within individuals, as well as within the whole population (using the average spectra for each age group), was assessed using the modified Bray-Curtis dissimilarity index as follows:
| (3) |
where Nki and Nkj denote the fluorescence intensity fractions of a given nucleotide N at position k of the spectra belonging to ages i and j, respectively. In the extreme case when there is no single position with a shared nucleotide between the two sequences, the sum of the differences between the fluorescence intensity fractions divided by the sum of the fractions equals 1. When both sequences are the same, the similarity index equals 0.
For taxon-based assessment of diversity, we used UniFrac analysis (18) of species composition predicted by MCR-ALS. We also calculated Simpson's index of diversity and the Bray-Curtis dissimilarity index of MCR-ALS-predicted bifidobacterial composition.
Temporal development.
The percentage and development in time were calculated relative to the Bifidobacterium group (MCR-predicted percentages). However, these percentages only reflect within-group composition, not taking into account the abundance of bifidobacteria as a whole compared to the other bacterial groups. In the previous study of 16S rRNA amplicons of IMPACT samples (see Text S1 in the supplemental material), one of MCR-ALS-resolved components was classified as Bifidobacterium genus (see Table S2 in the supplemental material). Therefore, using the information on relative abundance of bifidobacteria component in the samples (see Table S3 in the supplemental material), we also recalculated the percentages of Bifidobacterium species relative to total bacterial load. The significance of the change in the abundance between two subsequent time points was calculated with the Friedman test, which is a nonparametric version of two-way analysis of variance for repeated measurements (MATLAB documentation, 2010). The null hypothesis was rejected at the level of 5%.
Correlations in the amount of bifidobacteria.
Pairwise comparisons between relative amounts of bifidobacteria species at each age group were performed using the Pearson correlation coefficient. To minimize false significant correlations, results were adjusted for multiple testing within each age category using the Bonferroni correction.
To test the correlations between the levels of every bacteria at two subsequent time points, all values were binarized. Values higher than mean abundance of a given bifidobacterial species at a particular point, were marked as “high” values, and smaller values were marked as “low” values. The co-occurrence of high and low values was tested using the Fisher exact test.
Cloning.
To ensure the correct identification of Bifidobacterium spp. in the study cohort, we selected stool samples for the cloning of clpC amplicons using a TOPO TA cloning kit (Invitrogen, USA) according to the manufacturer's instructions. The plasmid inserts from all positively transformed Escherichia coli TOP10 colonies were then PCR amplified using primer pairs specific for the vector and then sequenced with the clpC primer following ExoI treatment.
Quantitative RT-PCR.
The fractions of B. adolescentis, B. longum, B. infantis, B. breve, B. bifidum, and B. dentium in 11 selected stool samples relative to all Bifidobacterium spp. and to the total microbiota were quantified by real-time PCR (RT-PCR) with the double-stranded DNA-specific EvaGreen fluorescent dye (Solis BioDyne, Estonia) using 16S-23S ITS primer pairs specific for the named species and designed by Haarman and Knol (19). Each quantitative PCR (qPCR; 20 μl) contained 5× HOT FIREPol EvaGreen qPCR Mix (Solis BioDyne), 200 nM forward primer, 200 nM reverse primer, 1 μl of template DNA, and H2O. Initial denaturation was performed at 95°C for 15 min, followed by 40 cycles of denaturation at 95°C for 15 s, primer annealing at 60°C for 20 s, and elongation at 72°C for 20 s. PCR efficiency for each reaction was calculated using a linear regression (20).
Isolation of bifidobacteria.
Stool samples were diluted 10-, 100-, and 1,000-fold in 0.1% peptone water with the addition of l-cysteine (0.025%) and sodium thioglycolate (0.025%), followed by incubation at 37°C for 4 h to aid the recovery of injured cells (21). Although higher dilution rates are recommended, experiments are normally performed on fresh stool samples; however, in the present study, stool samples were kept at −80°C for up to 8 years. To create anaerobic conditions, Oxyrase for Broth (Oxyrase, United Kingdom) was added to each tube. After the initial incubation, diluted samples were streaked on Bifidobacterium agar plates (BD Diagnostics, USA) and further incubated anaerobically at 37°C for 72 h. After incubation, the plates were examined, and randomly selected colonies were inoculated into liquid MRS medium (Merck, Germany) and grown anaerobically at 37°C for 1 day.
Isolates were classified by sequencing of the clpC gene. We then selected 13 isolates representing all identified species for MLST analysis performed as described by Deletoile et al. (22).
Pyrosequencing data analysis.
16S rRNA amplicon pyrosequencing data for stool samples from seven randomly selected mother-children pairs were taken from a parallel study of IMPACT samples addressing the total microbial composition (see Text S1 in the supplemental material). Briefly, pyrosequencing data were processed using the QIIME pipeline (23). Sequences were filtered, and chimeras were removed using UCHIME algorithm (24) in addition to the ChimeraSlayer reference-based method. Next, sequences were clustered at the 97% similarity level, and the RDP classifier (25) was used to assign taxonomic identity to the resulting operational taxonomic units (OTUs). We only present results here based on clusters assigned to the Bifidobacteriaceae family. To obtain relative estimates of various bifidobacterial species detected by pyrosequencing, OTUs were searched against the NCBI database using BLAST. All of those assigned to a certain bifidobacterial species were grouped together, and their relative abundances were recalculated.
Ethical Committee approval.
The Regional Committee for Medical Research Ethics for Central Norway approved the study (reference number 120-2000). The study was approved by the Norwegian Data Inspectorate, granting license to process personal health data, and one of the parents of each child signed a written informed consent form (reference number 2003/953-3 KBE/-). The Current Controlled Trials registration number is ISRCTN28090297.
RESULTS
Diversity determined directly from the mixed sequence spectra.
Nucleotide diversity analysis of the raw mixed spectra (average length of 221 bp) by determination of modified Simpson's α-diversity index showed that the diversities both within individuals and within the population ranged from mean of 1.6 (standard deviation of 0.19) to 1.7 (0.14) and from 1.7 to 1.9, respectively, throughout the whole duration of the study, with 1 corresponding to pure samples and 4 corresponding to the most mixed spectra. The maximum diversity was observed in stool samples from adults (Fig. 3). Immediately after birth, the nucleotide diversity significantly decreased (P = 0.0016) and remained unchanged up to the age of 4 months. Interestingly, from 4 months to 1 year, the diversities both within individuals and within the study population increased. However, the diversity did not change from 1 to 2 years of age.
Fig 3.
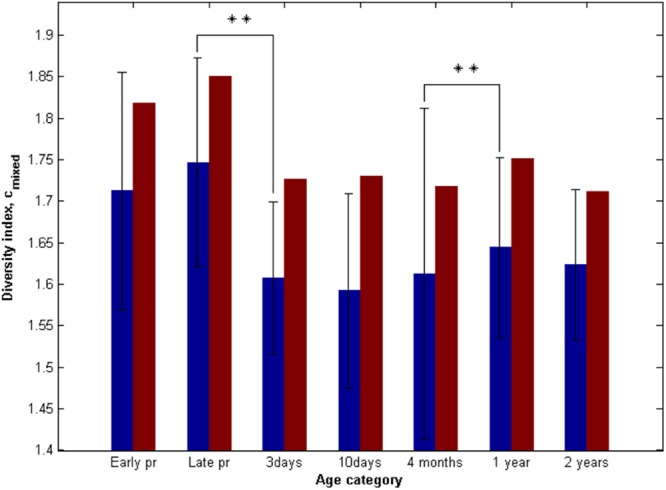
Modified Simpson's index of nucleotide spectra diversity cmixed at various ages. Early pr and Late pr, early (8 to 20 weeks) and late (30 to 40 weeks) pregnancy periods, respectively. Blue bars represent the average nucleotide diversity for individuals with the standard deviation as shown, whereas red bars represent the diversity of the average nucleotide intensity spectra for each age category. The significance in difference between diversity indexes at two subsequent time points was calculated with the Friedman's test. **, P < 0.01.
The Bray-Curtis β-diversity index revealed high variability of sequence spectra between subsequent time points within individuals (Fig. 4A). The highest similarity, in this case, was detected between the two pregnancy stages (0.08 [0.03]), whereas the highest dissimilarity was detected between mother-newborn pairs (0.14 [0.05]) and in the period from 4 months to 1 year (0.13 [0.05]). No significant difference between matching mother-newborn pairs and nonmatching mother-newborn pairs was detected. The similarity of the population average, however, was much higher, with the Bray-Curtis index ranging from 0.02 for the period between the two pregnancy stages up to 0.09 when comparing the average mixed spectra from the age of 3 days to that of the mothers in their late pregnancy stage. Clustering of the average mixed sequence spectra, however, suggests a clear age-dependent pattern of the bifidobacterial composition (Fig. 4B).
Fig 4.

Comparison of mixed sequence spectra. (A) Modified Bray-Curtis index of nucleotide similarity (BC) between the subsequent time points. E-L pr, period between early (8 to 20 weeks) and late (30 to 40 weeks) pregnancy periods; L pr-3 d, comparison between 3-day-old newborns and their mothers during the late pregnancy stage; 3 d-10 d, comparison between 3 and 10 days of age; 10 d-4 m, comparison between 10 days and 4 months of age; 4 m-1 y, comparison between 4 months and 1 year of age; 1 y-2 y, comparison between 1 and 2 years of age. Blue bars represent the average similarity indices of nucleotide intensity spectra within every individual (with the standard deviation as shown), whereas red bars represent the similarity indices between the average nucleotide intensity spectra for each age category. (B) Clustering of the average nucleotide intensity spectra.
Resolution of mixed sequence spectra into species components.
To define the initial number of components to be resolved by MCR-ALS from stool samples, we used both PCA and EFA. Although iterative methods, such as EFA, are commonly recommended for MCR-ALS analysis (16), the use of PCA also allows identifying outliers, which are best excluded from data sets prior to analysis. Both PCA and EFA analyses proposed six components (see Fig. S3 in the supplemental material). However, when we performed MCR-ALS analysis with six components, one of them was poorly resolved, resulting in a longer nucleotide sequence spectrum with many mixed peaks which were hard to interpret (see Fig. S2 in the supplemental material). Therefore, we repeated the MCR-ALS analysis with five components. All five components were well resolved. The base-called components' spectra were identified by using the resulting sequence as a query in BLAST searches against the NCBI nucleotide database (Table 2). These components are further denoted by their closest matches in the NCBI database, which are, for components 1 to 5, B. bifidum, B. dentium, B. adolescentis, B. breve, and B. longum, respectively.
Table 2.
Lowest E-value BLAST search hits of MCR-ALS-resolved spectra sequences against the NCBI nucleotide databasea
| MCR-ALS-resolved spectrum | Sequence length (bp) | Closest BLAST search hit | GenBank accession no. | E value | % identity |
|---|---|---|---|---|---|
| Component 1 | 221 | B. bifidum | DQ206821.1 | 1 × 10−102 | 98 |
| Component 2 | 215 | B. dentium | AY722387.1 | 3 × 10−109 | 98 |
| Component 3 | 219 | B. adolescentis | DQ238016.1 | 3 × 10−107 | 99 |
| Component 4 | 216 | B. breve | AB437352.1 | 4 × 10−101 | 98 |
| Component 5 | 221 | B. longum | AP010889.1 | 4 × 10−111 | 100 |
Query coverage, 100%.
The nucleotide intensity of the residual, not extracted, spectra was on average three times lower than that of the resolved components. We then compared the residual spectral information against the NCBI database using BLAST. The analysis revealed the presence of such species as B. pseudolongum, B. longum suis, B. magnum, B. mongoliense, B. scardovii, and B. asteroides. However, in most cases, the hit length was low (∼40 bp), indicating that these sequences might rather come from uncharacterized bifidobacteria. There were 33 samples, mostly belonging to 3- to 10-day-old newborns (see Fig. S1C in the supplemental material), which had comparatively high residual MCR component (the score was more than two times higher than the mean score of the residual component). In 25 of these samples, the residual spectra did not show any significant homology to known bifidobacterial species, whereas in the others, B. adolescentis, B. breve, B. longum, B. cuniculi, and B. magnum were detected. Empirical evaluation of raw spectra suggested that these residuals may reflect technical noise. Despite the noise, the MCR prediction of the most prevalent component in these samples corresponded to the lowest E-value BLAST search hit for raw unresolved sequences.
These results suggest that there were five most commonly identified bifidobacteria species in the study cohort. However, other species were also present and might have been present in high relative amounts in some individuals, but they were not shared among the majority of the samples.
Diversity analyses based on the resolved species components.
The species composition based on MCR predictions was used as input in the taxon-based diversity analyses. In addition, we included the sequence information from the residual component representing a taxon. The Simpson's diversity index showed high correlation to that of the modified Simpson's for the direct analyses of the mixed sequence spectra (Pearson correlation coefficient = 0.49, P = 1.35 × 10−21). We also found high correlation between the Bray-Curtis dissimilarity index based on the resolved sequence components, and its modified version used for the analyses of the mixed sequence spectra (Pearson correlation coefficient = 0.47, P = 1.34 × 10−10). Furthermore, the UniFrac clustering supported the direct mixed sequence analyses, with a large diversity at the individual level (see Fig. S4 in the supplemental material).
Temporal changes in Bifidobacterium species composition.
There was a large variation between individuals at every age, and the confidence levels for the mean varied from 0.6% up to 10 to 13% depending on the species and the age (see Table S4 in the supplemental material). Based on the analysis of total 16S rRNA gene content, bifidobacteria comprised 2% of total bacterial load in adults. In newborns, it constituted nearly one-fourth of the bacterial load, reaching the level of 60% by the age of 4 months (Fig. 5B). Relative to total bifidobacteria, stool samples from pregnant women were predicted to be rich in the B. adolescentis, B. longum, and B. bifidum group, whereas B. dentium was present in smaller amounts, and B. breve was nearly absent (Fig. 5A). The Friedman test revealed a significant decrease in the relative amount of B. adolescentis (P = 0.0005) and increase of B. breve (P = 0.0348) in stool samples of newborns compared to their mothers. The B. longum group represented the majority of bifidobacterial load at 3 and 10 days after the birth, whereas B. breve was the second most abundant species. Interestingly, B. breve separated all 10-day-old infants in two distinct groups. In one group, it accounted for <15%, whereas in the other it accounted for >75% of the bifidobacterial load. At 4 months of age, B. breve became the most predominant species in stool samples. However, by the age of 1 year, the percentage of B. breve decreased drastically (see Fig. S5A in the supplemental material), and B. longum regained its position as the most abundant group. Like adults, in children at 2 years of age, the majority of bifidobacterial load consisted of B. adolescentis and B. longum, although, unlike adults, B. longum was the most prevalent. B. bifidum comprised around one fourth of the bifidobacterial load in adults and 1- to 2-year-old infants, whereas in newborns and 4-month-old infants, its levels comprised to 9 to 15%.
Fig 5.

Bifidobacterium species composition in stool samples of infants (from 3 days to 2 years of age) and their mothers during pregnancy (pr) based on the results of MCR-ALS analysis relative to the bifidobacteria group (A) and relative to the total bacterial load (B).
Relative to the total microbial load, however, the amount of B. adolescentis comprised ca. 1 to 2.5% during the whole duration of the study (Fig. 5B), with the only significant difference in abundance occurring between newborns and mothers (see Fig. S5B in the supplemental material). There also was a significant fluctuation in the relative amount of B. bifidum from 10 days to 1 year of age, where it first increased up to 8% at 4 months (P = 0.0093) and then decreased to ca. 3% by the age of 1 year (P = 0.0090) and further down to 1.5%, still comprising twice as much of the bacterial load compared to adults (see Fig. S5B in the supplemental material). The most pronounced changes, however, were detected with regard to B. breve and B. longum. The relative amounts of B. breve were found to vary significantly between subsequent time points starting from 10 days on (see Fig. S5B in the supplemental material). B. longum was also found at significantly higher levels in 10-day-old infants compared to those at 3 days of age (P = 0.0067). From 4 months to 1 year of age, however, it decreased significantly (P = 0.0112; see Fig. S5B in the supplemental material).
Co-occurrence of bifidobacterial species.
Pairwise comparisons of the bifidobacterial co-occurrence revealed that in adulthood, as well as in 2-year-old children, there was a negative correlation between the amount of B. adolescentis and B. longum (P = 2.27 × 10−7, P = 4.6 × 10−5, and P = 1.77 × 10−10 for early pregnancy, late pregnancy, and 2 years, respectively; Fig. 6A). In newborns and 4-month-old infants, on the other hand, a negative correlation between B. breve and B. longum (P = 8.37 × 10−5, P = 7.12 × 10−7, and P = 1.8 × 10−17 for 3 days, 10 days, and 4 months, respectively) was detected (Fig. 6B). Interestingly, in 1-year-olds, negative correlations both between B. adolescentis and B. longum (P = 0.007) and between B. longum and B. breve (P = 0.048) was detected.
Fig 6.

Co-occurrence of five dominant bifidobacterial species. Ellipses represent correlations, detected more than in two various ages. Pink ellipses, correlations detected between B. adolescentis and B. longum group; green ellipses, correlations detected between B. longum group and B. breve. (A) Mothers during early (8 to 20 weeks) and late (30 to 40 weeks) pregnancy stages, as well as in infants at 1 and 2 years of age. (B) Infants at 3 and 10 days and 4 months after the birth.
Correlations between the changes of bifidobacterial species loads from one to another time point were also detected. As such, the change in the relative amount of B. longum from mother to newborn was negatively correlated to that of B. breve. Fluctuations between these two species loads were also negatively correlated from 3 to 10 days of age and further to 4 months, 1 year of age, and 2 years of age (see Fig. S6 in the supplemental material). The change in the relative amount of B. longum from 1 to 2 years was also negatively correlated to that of B. adolescentis (see Fig. S6 in the supplemental material).
Mode of delivery and diet effects.
Due to high frequency of vaginal delivery, we were unable to test differences in bifidobacterial composition which might have been caused by mode of delivery. For the same reasons, the effect of breast-feeding versus formula feeding could not be investigated. The only factor we could test was solid food (rice, corn, and wheat) consumption at 4 months. Using two-sided permutation test with 106 permutations, we did not find any significant difference in relative amounts of bifidobacterial species (P = 0.11, P = 0.74, P = 0.14, P = 0.84, and P = 0.85 for B. bifidum, B. dentium, B. adolescentis, B. breve, and B. longum, respectively).
Resolution of B. longum group.
Most of the nucleotide variation in clpC gene amplicons between B. longum subsp. longum and B. longum subsp. infantis is located downstream from the part of the clpC gene that was used for the MCR-ALS resolution. Therefore, to distinguish between B. longum subspecies, we designed a specific sequencing primer that binds closer to a variable site between B. longum subsp. longum and B. longum subsp. infantis (see Table S5 in the supplemental material). By resequencing clpC gene amplicons with this primer, B. longum was detected in 80% of all samples. We then binarized MCR-ALS predictions on the abundance of B. longum group in stool samples as present or absent and compared the data to resequencing results obtained with B. longum specific primer. In total, there was a high correlation between MCR-ALS predictions and resequencing data (according chi-squared analysis, χ2 = 28.58 and P = 8.99 × 10−8). Interestingly, all of the obtained sequences were pure. Sequences with a similar nucleotide variation pattern were aligned together, and the consensus sequence of the alignment was queried against the NCBI nucleotide database using BLAST. Four separate clusters were identified. The largest cluster belonged to B. longum subsp. longum, comprising 251 sequences. The second largest cluster comprised 12 sequences, belonging to B. longum subsp. infantis, whereas the third cluster, containing 5 sequences, comprised sequences that strongly resembled B. longum group but could not be assigned to either B. longum subsp. longum or B. longum subsp. infantis. Two sequences belonging to this cluster showed the closest homology to B. longum subsp. suis. One of the clusters, comprising 8 sequences, could not be assigned to any of the B. longum group species. In samples obtained from individuals both during pregnancy and right after birth, only B. longum subsp. longum was identified. Most of the B. longum subsp. infantis sequences were characteristic for children aged 4 months; there was only one baby, in whom it was detected earlier, and only two children where it persisted up to 1 year of age. By the age of 2 years, B. longum subsp. longum was found in 59 of 61 children in whom B. longum group was detected (see Fig. S7 in the supplemental material).
Validation of bifidobacterial species composition.
To assess the ability of MCR-ALS analysis to resolve five-species mixtures, we verified the method using predefined mixtures of five bifidobacterial species (B. bifidum DSM20456, B. dentium DSM20436, B. adolescentis DSM20083, B. breve DSM20213, and B. longum subsp. longum DSM20219). In general, there was 83 to 97% correlation between actual mixture composition and that predicted by MCR-ALS (see Table S1 in the supplemental material).
To evaluate the MCR resolution for the stool sample, we selected 13 samples for the cloning-based analysis of bifidobacterial diversity. Sequencing of cloned inserts from these samples confirmed the presence of species predicted by MCR (see Table S6 in the supplemental material). All cloned inserts shared 98 to 99% identity to the corresponding reference sequences deposited in GenBank. In one sample, B. animalis was detected, and the analysis of the sample's residual spectral information, not resolved by MCR, confirmed the findings. Representative sequences of cloned inserts from all identified bifidobacterial species were deposited in GenBank (accession numbers JQ288967 to JQ288972).
We have previously estimated the relative amounts of B. breve and B. longum in the same stool samples by RT-PCR (5). When we compared MCR predictions for these species to those from obtained RT-PCR, there was a high correlation between the estimates (χ2 = 149.32 [P = 2.44 × 10−34] and χ2 = 16.75 [P = 4.26 × 10−5] for B. breve and B. longum, respectively). For the comparison, we binarized all of the data as high or low values based on the mean value and performed the chi-squared test. To verify MCR predictions with regard to all detected bifidobacterial species, we then analyzed 11 samples using RT-PCR with 16S-23S-ITS-region-targeting primers, specific for B. bifidum, B. dentium, B. longum subsp. longum, B. longum subsp. infantis, B. breve, and B. adolescentis. The relative composition revealed by RT-PCR amplification significantly correlated to that predicted by MCR-ALS (Pearson correlation coefficient = 0.76, P = 8.0·10−12; see Fig. S8 in the supplemental material). However, in four samples, the composition predicted by MCR was more diverse. In particular, B. dentium and B. bifidum were nearly absent according to RT-PCR. We then performed RT-PCR analysis on DNA isolated from pure bacterial cultures of B. bifidum (DSM20456) and B. dentium (DSM20436). Both of these species were detected only after 30 cycles, whereas with a universal bacterial primer pair they were detected on the 15th cycle. In silico PCR analysis (http://insilico.ehu.es/PCR/) of B. bifidum PRL2010, B. bifidum S17, and B. dentium Bd1 genome sequences with ITS-targeting primer pairs specific for the given species failed to produce any PCR product in silico. This may indicate that B. bifidum and B. dentium primer pairs target regions not universally conserved among all of the species strains.
We have previously analyzed stool samples from seven mother-child pairs using deep 454 sequencing of 16S rRNA amplicons (see Text S1 in the supplemental material). To compare MCR predictions to pyrosequencing findings, we extracted the information on bifidobacterial OTUs, grouped them according to the assigned bifidobacterial species, and calculated their relative abundances. In general, there was a significant correlation between MCR predictions and pyrosequencing data (Pearson correlation coefficient 0.60, P = 2.11 × 10−15; see Fig. S9 in the supplemental material). There was not a single OTU identified as B. adolescentis. We therefore reanalyzed the raw unfiltered data, and we found that sequences with high homology to B. adolescentis were removed from the final data set during the chimera filtering procedure. However, we were unable to introduce the information about these sequences to the final pyrosequencing data set.
We also isolated Bifidobacterium species from nine stool samples belonging to 4-month-old infants. Sequencing of clpC gene PCR products revealed that isolates belong to B. longum subsp. infantis, B. longum subsp. longum, B. adolescentis, B. breve, and B. animalis corresponding well with qualitative predictions of MCR analysis on expected bifidobacterial species. We then selected 13 isolates for MLST analysis (22). However, in addition to clpC gene, we managed to amplify only four genes (fusA, GTP-binding protein chain elongation factor EF-G; ileS, isoleucyl-tRNA synthetase; rplB, 50S ribosomal subunit protein L2; and gyrB, DNA gyrase, subunit B). Apart from the gyrB and fusA genes, which had very low resolution (see Fig. S13 and S14 in the supplemental material), clustering of sequences with all known profile sequences of a given gene corresponded to the expected clpC-gene based species delineations (see Fig. S10 to S12 in the supplemental material). Representative sequences of bifidobacterial isolates were deposited in GenBank (accession numbers JQ288937 to JQ288965).
DISCUSSION
Despite a major interest in bifidobacteria, there is still some inconsistency in the literature with regard to bifidobacterial loads in the human intestine. Estimates of bifidobacterial load in adults range from ca. 4% (26) up to 15% of the total gut microbiota (3). For infants, the range is higher. Some researchers suggest that this genus reaches up to 90% of the total microbiota (27, 28), whereas others claim much lower abundances (ca. 1 to 2%) of these species among infants (29). The inconsistencies in the literature may be due to both methodological differences in identifying the species (28, 30, 31) and the lack of precision among small study populations.
In our study, bifidobacteria comprised ca. 2% of the total bacterial load in adults, whereas the peak of its abundance was observed in 4-month-old infants, when this bacterial group constituted ca. 60% of intestinal microbiota. To our knowledge, our study with 87 mothers and their infants represents the largest comprehensive (including all bifidobacterial species) temporal study of natural development of bifidobacteria within a cohort thus far. Due to the large variation between individuals, and also taking into account the high frequency of missing data in longitudinal human study cohorts, we believe it is essential to analyze large cohorts.
Clustering of the population average mixed sequence spectra indicates that the system seems to be very structured, with significant differences between various ages. Analyses of mixed sequence spectra information at the individual level (UniFrac clustering), however, revealed only minor age-related patterns, apart from the cluster comprising the majority of stool samples from pregnant women and 2-year-old children. As illustrated both by Simpson's diversity index, the Bray-Curtis similarity index, and the information extracted from the residual spectra, there are large individual variations in the composition. This individuality probably brings the “noise” into the system and makes it difficult to deduce structured information when comparing every single individual with one another.
B. bifidum is predicted to possess lacto-N-biosidase and galacto-N-biosidase activity, enabling it to ferment human milk oligosaccharides (32). However, its relative abundance compared to other Bifidobacterium groups was higher in adults and 2-year-old children than in newborns and 4-month-old infants. On the other hand, relative to the total bacterial load, this group of bacteria exhibited peak abundance at 4 months, when most infants were breast-fed, and became nearly absent in 2-year-olds and adults. These observations suggest that B. bifidum is less affected by the total reduction of bifidobacteria with age. It has been demonstrated that B. bifidum is capable of utilizing host-derived glycans (33, 34), which we believe may partially explain the observed stability of this species. The levels of B. dentium remained nearly the same regardless of host age, also pointing to stability of this species. If B. dentium is commonly present in the mouth, then it would most likely be transferred constantly to the intestines with food and then excreted. The levels of B. dentium detected, however, suggest growth of this species in the gut lumen.
B. breve was nearly absent in adults, whereas in newborns and 4-month-old infants it was, on average, the second most abundant bifidobacterial group both relative to the bifidobacterial and total bacterial loads. Interestingly, with regard to the amount of B. breve, all 10-day-old infants were separated into two groups: (i) infants where B. breve constituted up to 15% of the bifidobacterial load, and (ii) infants where it accounted for >75% of the bifidobacterial load. B. breve was shown to be an efficient inducer of serum IgA (35), a main immunoglobulin found in mucous secretion of intestinal and respiratory tract (36), which averts penetration of pathogenic microorganisms by preventing their adsorption to mucosal epithelium. Therefore, in the future it would be very interesting to investigate whether such a huge gap in B. breve abundance has a host-related cause or can be explained by bacterial competition. At 4 months, the difference in B. breve abundance was smaller, since all of the infants had quite high numbers of this species. B. breve is regarded to be a very important species during the weaning period, since all of the tested strains possess the gene encoding amylopullulanase, the enzyme responsible for starch degradation (37). Because nearly half of infants at this age in the study cohort were given starch containing solid food, we could address the question of whether or not starch consumption would increase B. breve fraction. However, we did not detect a significant difference in the relative amount of this species between infants who were given solid food and those who were not. This may indicate that, apart from starch, there are probably other factors that promote B. breve at 4 months.
The human gut is a very densely populated site with interactions between numerous microorganisms (38), potentially leading to intricate correlation patterns. We believe that it is crucially important to identify correlation patterns between various bacteria, since this will very likely lead to a better understanding of disease development and health maintenance in individuals. In a recent study by Turroni et al. (28), the researchers noticed underlying interactions between bifidobacteria, although the number of individuals was too low to obtain more precise information. In our work, we could find two distinct patterns of correlation separating all seven ages into two major groups (Fig. 6). Adult-profile correlations comprised stool samples from adults, as well as from 1- and 2-year-old children. Infant-profile correlations comprised stool samples from newborns and 4-month-old infants. Negative correlations between the numbers of the B. adolescentis and B. longum groups were characteristic of the adult profile. At the same time, in pregnant women, there was also a negative correlation between B. adolescentis and B. breve, which only reappeared in children at the age of 2 years, also indicating that by the age of 1 year the development of Bifidobacterium species is not completely finished. For the infant profile, on the other hand, a negative correlation between the B. longum group and B. breve was detected. Starting from 3 days of age and up to 4 months, in infants who had a low amount of B. breve, B. longum accounted for ca. 70% of the load, whereas in those who had high B. breve levels, B. longum was nearly absent.
Within both adult and infant profiles, B. longum comprised one of the central groups for which correlations were detected. This stability of the group regardless of the changes in the gut indicates the high competitiveness of B. longum and its vast ability to adjust to various conditions. All sequences obtained from stool samples with the specific B. longum group primer were pure. Both during pregnancy and right after the birth, only B. longum subsp. longum was identified in stool samples (see Fig. S7). This is in accordance with a recent model suggesting the transmittance of B. longum subsp. longum from mother to child (39). B. longum subsp. infantis was detected only in 12 samples, mostly belonging to 4-month-old infants, which might indicate horizontal transmittance of these bacteria. In the children who had B. longum subsp. infantis at 4 months of age, B. longum subsp. longum was detected earlier and later in life, suggesting a competition between the two subspecies. Genomic analysis of these two subspecies has revealed that B. longum subsp. longum, although able to ferment HMOs, is more suited for the fermentation of plant-derived oligosaccharides, whereas B. longum subsp. infantis harbors breakdown machinery for milk-derived oligosaccharides (40, 41).
In conclusion, our analyses of a large unselected cohort revealed structured age-related successions of bifidobacteria and correlations between the species of this genus within the population. We also showed that the B. longum group is probably one of the most central bifidobacteria in the human gut with respect to the correlation networks. In the future, it will be very interesting to determine whether the development patterns and correlations revealed here are also valid for populations in other geographical regions. At the same time, it will also be important to address the mechanisms of Bifidobacterium transmittance and the biological role of Bifidobacterium spp. in the human gut.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Norwegian Department of Health and Social Affairs and Hedmark Sparebank.
We thank Eric de Muinck (University of Oslo, Oslo, Norway), Signe Marie Drømtorp (Nofima, Norway), and Monika Sekelja (Genetic Analysis AS, Norway) for their help with DNA isolation, sequencing, and optimization of the MATLAB script for sequence alignment.
Footnotes
Published ahead of print 2 November 2012
Supplemental material for this article may be found at 10.1128/AEM.02359-12.
REFERENCES
- 1. Tannock GW. 2010. Analysis of bifidobacterial populations in bowel ecology studies, p 1–15 In Mayo B, van Sinderen D. (ed), Bifidobacteria: genomics and molecular aspects. Caister Academic Press, Norfolk, England [Google Scholar]
- 2. Gronlund MM, Gueimonde M, Laitinen K, Kociubinski G, Gronroos T, Salminen S, Isolauri E. 2007. Maternal breast-milk and intestinal bifidobacteria guide the compositional development of the Bifidobacterium microbiota in infants at risk of allergic disease. Clin. Exp. Allergy 37:1764–1772 [DOI] [PubMed] [Google Scholar]
- 3. Menard O, Butel MJ, Gaboriau-Routhiau V, Waligora-Dupriet AJ. 2008. Gnotobiotic mouse immune response induced by Bifidobacterium sp. strains isolated from infants. Appl. Environ. Microbiol. 74:660–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Young SL, Simon MA, Baird MA, Tannock GW, Bibiloni R, Spencely K, Lane JM, Fitzharris P, Crane J, Town I, Addo-Yobo E, Murray CS, Woodcock A. 2004. Bifidobacterial species differentially affect expression of cell surface markers and cytokines of dendritic cells harvested from cord blood. Clin. Diagn. Lab. Immunol. 11:686–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Storrø O, Øien T, Langsrud RK, Dotterud CK, Johnsen R. 2011. Temporal variations in early gut microbial colonization are associated with allergen-specific immunoglobulin E but not atopic eczema at 2 years of age. Clin. Exp. Allergy 41:1545–1554 [DOI] [PubMed] [Google Scholar]
- 6. Vebo HC, Sekelja M, Nestestog R, Storro O, Johnsen R, Oien T, Rudi K. 2011. Temporal development of the infant gut microbiota in immunoglobulin E-sensitized and nonsensitized children determined by the GA-map infant array. Clin. Vaccine Immunol. 18:1326–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ventura M, Turroni F, Bottacini F, Giubellini V, van Sinderen D. 2010. Bifidobacterial ecology and comparative genomics: perspectives and prospects, p 31–44 In Mayo B, van Sinderen D. (ed), Bifidobacteria: genomics and molecular aspects. Caister Academic Press, Norfolk, England [Google Scholar]
- 8. Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P, Cagnasso P, Bizzarri B, de'Angelis GL, Shanahan F, van Sinderen D, Ventura M. 2009. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl. Environ. Microbiol. 75:1534–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Turroni F, Marchesi JR, Foroni E, Gueimonde M, Shanahan F, Margolles A, van Sinderen D, Ventura M. 2009. Microbiomic analysis of the bifidobacterial population in the human distal gut. ISME J. 3:745–751 [DOI] [PubMed] [Google Scholar]
- 10. Øien T, Storrø O, Johnsen R. 2006. Intestinal microbiota and its effect on the immune system—a nested case-cohort study on prevention of atopy among small children in Trondheim: the IMPACT study. Contemp. Clin. Trials 27:389–395 [DOI] [PubMed] [Google Scholar]
- 11. Storrø O, Øien T, Dotterud CK, Jenssen JA, Johnsen R. 2010. A primary health-care intervention on pre- and postnatal risk factor behavior to prevent childhood allergy. BMC Public Health 10:443 doi:10.1186/1471-2458-10-4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ventura M, Canchaya C, Del Casale A, Dellaglio F, Neviani E, Fitzgerald GF, van Sinderen D. 2006. Analysis of bifidobacterial evolution using a multilocus approach. Int. J. Syst. Evol. Microbiol. 56:2783–2792 [DOI] [PubMed] [Google Scholar]
- 13. Brisse S. 2009. Bifidobacterium MLST database: Bifidobacterium clpC alleles. Institut Pasteur, Paris, France: http://www.pasteur.fr/cgi-bin/genopole/PF8/mlstdbnet.pl?page=alleles&format=FASTA&locus=clpC&file=bifido_profiles.xml [Google Scholar]
- 14. Zimonja M, Rudi K, Trosvik P, Næs T. 2008. Multivariate curve resolution of mixed bacterial DNA sequence spectra: identification and quantification of bacteria in undefined mixture samples. J. Chemometrics 22:309–322 [Google Scholar]
- 15. Tomasi G, van den Berg F, Andersson C. 2004. Correlation optimized warping and dynamic time warping as preprocessing methods for chromatographic data. J. Chemometrics 18:231–241 [Google Scholar]
- 16. Tauler R, Smilde A, Kowalski B. 1995. Selectivity, local rank, 3-way data-analysis and ambiguity in multivariate curve resolution. J. Chemometrics 9:31–58 [Google Scholar]
- 17. Garrido M, Rius FX, Larrechi MS. 2008. Multivariate curve resolution-alternating least squares (MCR-ALS) applied to spectroscopic data from monitoring chemical reactions processes. Anal. Bioanal. Chem. 390:2059–2066 [DOI] [PubMed] [Google Scholar]
- 18. Lozupone C, Hamady M, Knight R. 2006. UniFrac: an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371 doi:10.1186/1471-2105-7-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haarman M, Knol J. 2005. Quantitative real-time PCR assays to identify and quantify fecal Bifidobacterium species in infants receiving a prebiotic infant formula. Appl. Environ. Microbiol. 71:2318–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37:e45 doi:10.1093/nar/gkp045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nebra Y, Jofre J, Blanch AR. 2002. The effect of reducing agents on the recovery of injured Bifidobacterium cells. J. Microbiol. Methods 49:247–254 [DOI] [PubMed] [Google Scholar]
- 22. Deletoile A, Passet V, Aires J, Chambaud I, Butel MJ, Smokvina T, Brisse S. 2010. Species delineation and clonal diversity in four Bifidobacterium species as revealed by multilocus sequencing. Res. Microbiol. 161:82–90 [DOI] [PubMed] [Google Scholar]
- 23. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lay C, Rigottier-Gois L, Holmstrom K, Rajilic M, Vaughan EE, de Vos WM, Collins MD, Thiel R, Namsolleck P, Blaut M, Dore J. 2005. Colonic microbiota signatures across five northern European countries. Appl. Environ. Microbiol. 71:4153–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, Welling GW. 2000. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J. Pediatr. Gastroenterol. Nutr. 30:61–67 [DOI] [PubMed] [Google Scholar]
- 28. Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, Kerr C, Hourihane J, Murray D, Fuligni F, Gueimonde M, Margolles A, De Bellis G, O'Toole PW, van Sinderen D, Marchesi JR, Ventura M. 2012. Diversity of bifidobacteria within the infant gut microbiota. PLoS One 7:e36957 doi:10.1371/journal.pone.0036957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. 2007. Development of the human infant intestinal microbiota. PLoS Biol. 5:e177 doi:10.1371/journal.pbio.0050177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sergeant MJ, Constantinidou C, Cogan T, Penn CW, Pallen MJ. 2012. High-throughput sequencing of 16S rRNA gene amplicons: effects of extraction procedure, primer length, and annealing temperature. PLoS One 7:e38094 doi:10.1371/journal.pone.0038094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sim K, Cox MJ, Wopereis H, Martin R, Knol J, Li MS, Cookson WO, Moffatt MF, Kroll JS. 2012. Improved detection of bifidobacteria with optimised 16S rRNA-gene based pyrosequencing. PLoS One 7:e32543 doi:10.1371/journal.pone.0032543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wada J, Ando T, Kiyohara M, Ashida H, Kitaoka M, Yamaguchi M, Kumagai H, Katayama T, Yamamoto K. 2008. Bifidobacterium bifidum lacto-N-biosidase, a critical enzyme for the degradation of human milk oligosaccharides with a type 1 structure. Appl. Environ. Microbiol. 74:3996–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turroni F, Bottacini F, Foroni E, Mulder I, Kim JH, Zomer A, Sanchez B, Bidossi A, Ferrarini A, Giubellini V, Delledonne M, Henrissat B, Coutinho P, Oggioni M, Fitzgerald GF, Mills D, Margolles A, Kelly D, van Sinderen D, Ventura M. 2010. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc. Natl. Acad. Sci. U. S. A. 107:19514–19519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turroni F, Milani C, van Sinderen D, Ventura M. 2011. Genetic strategies for mucin metabolism in Bifidobacterium bifidum PRL2010: an example of possible human-microbe co-evolution. Gut Microbes 2:183–189 [DOI] [PubMed] [Google Scholar]
- 35. Yasui H, Mike NNA, Hayakawa K, Ohwaki M. 1992. Detection of Bifidobacterium strains that induce large quantities of IgA. Microb. Ecol. Health Dis. 5:155–162 [Google Scholar]
- 36. Wood P. 2006. Understanding immunology, 2nd ed Pearson Prentice Hall, Harlow, England [Google Scholar]
- 37. Ventura M, Canchaya C, Fitzgerald GF, Gupta RS, van Sinderen D. 2007. Genomics as a means to understand bacterial phylogeny and ecological adaptation: the case of bifidobacteria. Antonie Van Leeuwenhoek 91:351–372 [DOI] [PubMed] [Google Scholar]
- 38. Ley RE, Peterson DA, Gordon JI. 2006. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124:837–848 [DOI] [PubMed] [Google Scholar]
- 39. Makino H, Kushiro A, Ishikawa E, Muylaert D, Kubota H, Sakai T, Oishi K, Martin R, Ben Amor K, Oozeer R, Knol J, Tanaka R. 2011. Transmission of intestinal Bifidobacterium longum subsp. longum strains from mother to infant, determined by multilocus sequencing typing and amplified fragment length polymorphism. Appl. Environ. Microbiol. 77:6788–6793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. LoCascio RG, Desai P, Sela DA, Weimer B, Mills DA. 2010. Broad conservation of milk utilization genes in Bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Appl. Environ. Microbiol. 76:7373–7381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, Lapidus A, Rokhsar DS, Lebrilla CB, German JB, Price NP, Richardson PM, Mills DA. 2008. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. U. S. A. 105:18964–18969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36:W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum-parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.