

Abstract
Pseudoxanthomonas spadix BD-a59, isolated from gasoline-contaminated soil, has the ability to degrade all six BTEX (benzene, toluene, ethylbenzene, and o-, m-, and p-xylene) compounds. The genomic features of strain BD-a59 were analyzed bioinformatically and compared with those of another fully sequenced Pseudoxanthomonas strain, P. suwonensis 11-1, which was isolated from cotton waste compost. The genome of strain BD-a59 differed from that of strain 11-1 in many characteristics, including the number of rRNA operons, dioxygenases, monooxygenases, genomic islands (GIs), and heavy metal resistance genes. A high abundance of phage integrases and GIs and the patterns in several other genetic measures (e.g., GC content, GC skew, Karlin signature, and clustered regularly interspaced short palindromic repeat [CRISPR] gene homology) indicated that strain BD-a59's genomic architecture may have been altered through horizontal gene transfers (HGT), phage attack, and genetic reshuffling during its evolutionary history. The genes for benzene/toluene, ethylbenzene, and xylene degradations were encoded on GI-9, -13, and -21, respectively, which suggests that they may have been acquired by HGT. We used bioinformatics to predict the biodegradation pathways of the six BTEX compounds, and these pathways were proved experimentally through the analysis of the intermediates of each BTEX compound using a gas chromatograph and mass spectrometry (GC-MS). The elevated abundances of dioxygenases, monooxygenases, and rRNA operons in strain BD-a59 (relative to strain 11-1), as well as other genomic characteristics, likely confer traits that enhance ecological fitness by enabling strain BD-a59 to degrade hydrocarbons in the soil environment.
INTRODUCTION
Monoaromatic hydrocarbons, including benzene, toluene, ethylbenzene, and o-, m-, and p-xylene, collectively called BTEX, are major components of gasoline and are thought to be the most prevalent contaminants of soil and groundwater due to frequent leakages from underground storage tanks and accidental spills (1). Moreover, due to their high aqueous solubility relative to other petroleum hydrocarbons, BTEX compounds can spread in groundwater far from their contamination sources. These compounds are toxic and/or carcinogenic to humans (2) and are listed as priority pollutants by the U.S. Environmental Protection Agency (http://www.epa.gov/waterscience/criteria/wqcriteria.html). Microbial degradation of BTEX compounds is known to be one of the most efficient ways to remove BTEX compounds from soil and groundwater (1, 3). Many BTEX-degrading bacteria, including members of the genera Pseudomonas, Ralstonia, Burkholderia, Sphingomonas, Thauera, Dechloromonas, Rhodococcus, Acinetobacter, and Marinobacter, have been isolated from diverse aerobic or anaerobic environments (4–9), which has led to extensive studies on the metabolism and genetics of these BTEX-degrading microorganisms (10–14).
Pseudoxanthomonas spadix BD-a59 was isolated from gasoline-contaminated soil in the Republic of Korea and was shown to be responsible for the in situ biodegradation of BTEX compounds in gasoline-contaminated soil (15). Strain BD-a59 is one of only a few bacterial strains, including Ralstonia pickettii PKO1 and Dechloromonas sp. strain RCB, able to mineralize all six BTEX compounds (10, 11), and until now, it has been the sole member of the genus Pseudoxanthomonas known to be a BTEX degrader. The hydrocarbon degradation properties of Pseudoxanthomonas species have rarely been studied, as they do not grow well on minimal medium without additional nutrients such as yeast extract (15). The aromatic hydrocarbon degradation properties of Pseudoxanthomonas members have been recently studied (16, 17), and the results of these studies have suggested that members of the genus Pseudoxanthomonas may be ecologically important in terms of pollutant biodegradation. However, the genetics and physiology of Pseudoxanthomonas strains with respect to hydrocarbon degradation have not been reported.
With the development of next-generation DNA sequencing technologies and bioinformatics tools, key insights into the ecological fitness traits and metabolic properties of microbes can be obtained through complete genomic analysis and comparative genomics (18, 19). Recently, the genome of strain BD-a59 was completely sequenced (20), but the genetic traits that confer the capability to metabolize BTEX compounds and facilitate ecological adaptation to gasoline-contaminated soil have yet to be thoroughly explored. Here, we analyzed the genome of strain BD-a59 bioinformatically and compared it with that of another genome-sequenced Pseudoxanthomonas strain, P. suwonensis 11-1 (GenBank accession no. CP002446), which was isolated from cotton waste compost. In addition, we also examined the special features of the genome of strain BD-a59, including genomic islands (GIs), phage integrases, dioxygenases, monooxygenases, and the components involved in hydrocarbon biodegradation. This analysis revealed a variety of genetic traits (especially hydrocarbon metabolism) likely contributing to strain BD-a59's ability to function in gasoline-contaminated soil.
MATERIALS AND METHODS
Bioinformatic analyses.
The complete genome sequences of P. spadix BD-a59 (CP003093) and P. suwonensis 11-1 (CP002446), isolated from gasoline-contaminated soil and cotton waste compost, respectively, were compared throughout this study. The NCBI (http://www.ncbi.nlm.nih.gov/) and Integrated Microbial Genomes (IMG; https://img.jgi.doe.gov/cgi-bin/er/main.cgi) servers and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway databases were the primary sources used for genome predictions and comparisons. A circular map representing the genome of strain BD-a59 (Fig. 1) was generated using the Web-based CGview program (21). The average nucleotide identity (ANI) was calculated using the Web-based JSpecies program (22). The clustered regularly interspaced short palindromic repeat (CRISPR) gene sequences were found using an online web service (http://crispr.u-psud.fr/Server/CRISPRfinder.php). The Clusters of Orthologous Groups (COG) of protein sequences from strains BD-a59 and 11-1 were analyzed using the function category comparison tool at IMG. Karlin signature skew, cumulative GC skew, and GC content were depicted using Artemis tools (sact_v9.0.5) (23). A dotplot showing chromosomal synteny between strains BD-a59 and 11-1 was constructed using the program Mummer on the IMG server, and genomic comparison was performed using the Mauve program (24). The Island Viewer was used to identify chromosomal deviations in GC content, also known as genomic islands (GIs) (http://www.pathogenomics.sfu.ca/islandviewer [25]). Genes encoding phage integrases, transposases, insertion sequence (IS) elements, dioxygenases, and monooxygenases and other functional genes were analyzed using the IMG and NCBI servers (http://www.ncbi.nlm.nih.gov/).
Fig 1.
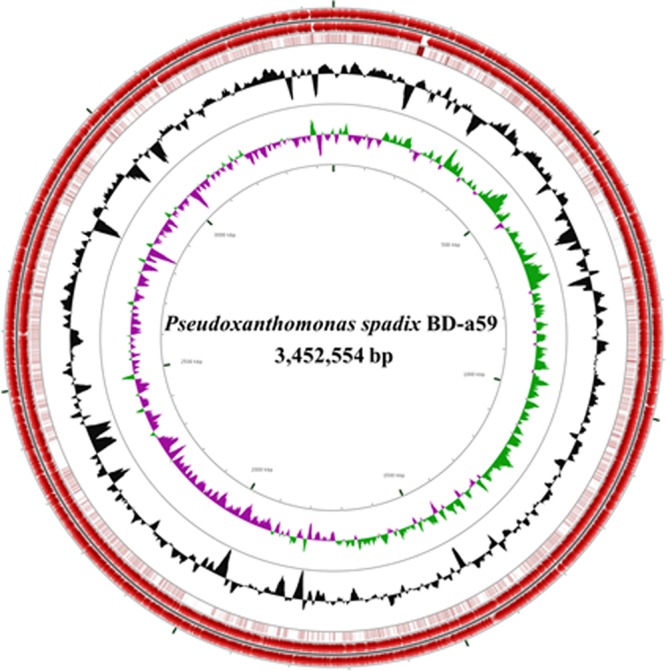
A circular map of the genome of P. spadix BD-a59. Forward-strand and reverse-strand CDSs (red) are depicted on the outermost two circles of the map, respectively. The third circle represents the BLASTN comparison of strain BD-a59's genome to strain 11-1's genome (dark brown indicates highly homologous CDSs). G+C content (black) and GC skews (positive GC skew, green; negative GC skew, violet) are shown on the fourth and fifth circles, respectively.
Hydrocarbon biodegradation.
In addition to the ability of strain BD-a59 to degrade six BTEX compounds (benzene, toluene, ethylbenzene, and o-, m-, and p-xylene), the biodegradation capabilities of strain BD-a59 for a variety of hydrocarbons (catechol, biphenyl, 4-hydroxybenzoate, phenol, phenylpropionate, 3-hydroxybenzoate, salicylate, gentisate, and hexane) were evaluated using a previously described procedure (15, 26) with some modifications. Briefly, 160-ml serum bottles containing 10 ml of MSB medium (27), 150 mg/liter of yeast extract (YE), and 150 ppm of individual hydrocarbons were prepared separately in triplicate under aerobic conditions. Strain BD-a59 cells were cultured in R2A broth at 25°C for 2 days, harvested by centrifugation (8,000 × g, 5 min), and resuspended in fresh MSB broth to a density of approximately 108 cells/ml. The resuspended cells (100 μl) were inoculated into seed serum bottles. Uninoculated serum bottles (no cells; MSB medium, YE, and each hydrocarbon only) were also prepared in triplicate as negative controls. The serum bottles were incubated at 25°C and with 150 rpm shaking for 7 days. Bottles containing hexane and phenol were extracted with 5 ml of methylene chloride, while bottles containing biphenyl and catechol were extracted with 5 ml of ethyl acetate. Bottles containing 4-hydroxybenzoate, phenylpropionate, 3-hydroxybenzoate, salicylate, and gentisate were acidified with HCl to approximately pH 3.0 and extracted with 5.0 ml ethyl acetate. All extracts were then dried over anhydrous Na2SO4 and then analyzed using a model 6890N gas chromatograph (GC; Agilent) with an HP-5 column (30-m length, 0.32-mm inner diameter, 0.25-μm film thickness [J & W Scientific]) coupled to a flame ionization detector (FID). The temperature of the GC oven was programmed to increase from 60°C (held for 1 min) to 120°C at 8°C/min and then increase to 280°C at 10°C/min, after which it was held at 280°C for 3 min.
GC-MS detection of BTEX metabolites.
To confirm the biodegradation pathways of the six BTEX compounds that were predicted based on bioinformatic analysis, metabolic intermediates were determined using a previously described procedure (28) with some modifications. Ten serum bottles containing 250 ppm of individual BTEX compounds were prepared, respectively, as described above and incubated at 25°C and with shaking at 150 rpm for 2 to 6 days. The suspensions of 10 serum bottles from the same BTEX compounds were combined and acidified with HCl to approximately pH 3.0. Metabolites were extracted from the acidified suspensions twice with 20 ml ethyl acetate and were then dried over anhydrous Na2SO4 and concentrated under an atmosphere of N2 to a volume of 300 μl. Extracts were derivatized with 25 μl BSTFA [bis(trimethylsilyl)trifluoroacetimide] for 20 min and analyzed using a HP 5890 GC (Hewlett-Packard)-Autospec mass spectrometer (MS) (Micromass, United Kingdom) with an HP-5 column. The temperature of the GC oven was programmed to increase from 70°C to 260°C at 8°C/min, after which it was held at 260°C for 5 min. The peaks were identified by matching mass spectra in a mass spectral reference library (Masslynx v.4.1; Micromass). Authentic standards of metabolites (4-hydroxybenzylaldehyde, 4-hydroxytoluene, 4-hydroxybenzoate, phenol, catechol, o-, m-, and p-methyl benzylaldehyde, and o-, m-, and p-methyl benzylalcohol) (>97% purity) were obtained from Sigma, and the metabolites were identified based on matching retention times and mass spectra to the standards.
RESULTS AND DISCUSSION
General features of the genome of strain BD-a59.
Strain BD-a59 has a single circular chromosome of 3,452,554 bp with a G+C content of 67.65%. Key structural features of strain BD-a59, including protein-coding sequences (CDSs), G+C content, and GC skew, are graphically depicted in Fig. 1. Comparative average nucleotide identity (ANI) analysis with all bacterial genomes in the IMG database showed that the genome of strain BD-a59 was most closely related to that of another sequenced Pseudoxanthomonas strain, P. suwonensis 11-1. The 16S rRNA gene sequence similarity and ANI values determined by comparisons between strains BD-a59 and 11-1 were 96% and 76.59%, respectively, which are clearly below their respective thresholds, 97% and 94%, generally accepted for species delineation (29), and these indicate that the two strains represent members of different species within the genus Pseudoxanthomonas.
The genomes of strains BD-a59 and 11-1 were compared, and their general features are summarized in Table 1. Most of the genomic features of both strains, such as genome sizes, total gene numbers, coding sequences (CDSs), and total tRNA gene numbers, were similar, although the G+C content of strain BD-a59 (67.65%) was slightly lower than that of strain 11-1 (70.21%). It has been generally accepted that multiple rRNA operons are required in prokaryotic organisms to achieve high growth rates, but their multiplicity can constitute a metabolic burden at lower growth rates (31, 32). Variations in operon numbers between different bacterial taxa have been well documented, but variations between closely related species are less often considered (33). However, although the two strains are closely related, strain BD-a59 harbored only one rRNA operon, while strain 11-1 possessed two rRNA operons (Table 1). This might explain why strain BD-a59 is capable of existing in an oligotrophic environment, i.e., soil, whereas strain 11-1 was isolated from a slightly more enriched environment, i.e., cotton waste compost. However, the multiplicity of rRNA operons is not the sole factor determining bacterial growth rate.
Table 1.
General features of the genomes of two Pseudoxanthomonas strains
| Characteristic | Value for: |
|
|---|---|---|
| BD-a59 | 11-1 | |
| Size (bp) | 3,452,554 | 3,419,049 |
| GC content (%) | 67.65 | 70.21 |
| Total no. of genes | 3,202 | 3,171 |
| No. of protein coding sequences | 3,149 | 3,110 |
| No. of proteins with function prediction | 2,254 | 2,344 |
| DNA coding density (%) | 87.48 | 89.47 |
| Protein assigned to COG (%) | 79.11 | 79.44 |
| Average gene length (bp) | 1,078.24 | 1,078.22 |
| No. of rRNA operons | 1 | 2 |
| Total no. of tRNA genes | 50 | 52 |
| ANIa (%) | 76.59 | |
| No. of confirmed CRISPRs (no. questionable)b | 1 (14) | 0 (7) |
| No. of genomic islands | 24 | 15 |
Clustered regularly interspaced short palindromic repeats (CRISPR) are distinctive features found in most prokaryotic genomes and, along with cas (CRISPR-associated) genes, are thought to be a host defense mechanism against bacteriophage predation (30). CRISPRs typically consist of several noncontiguous direct repeats separated by variable sequences called spacers. Their presence is generally accepted to be accumulated evidence of previous bacteriophage infections. The genome of strain BD-a59 contained 1 confirmed CRISPR and 14 possible CRISPRs, while the genome of strain 11-1 contained only 7 possible CRISPRs and no confirmed CRISPRs (Table 1). The confirmed CRISPR in strain BD-a59 consists of a CRISPR/cas region 3,379 bp in size that contains seven potential cas genes and 51 spacers (see Fig. S1 in the supplementary material). The presence of CRISPRs in the genome of strain BD-a59 suggests that extensive and complex genetic alterations and exchanges may have occurred as a result of bacteriophage infection during the evolutionary history of strain BD-a59.
Comparisons of gene categories in the genomes of the two Pseudoxanthomonas strains.
Comparison of the differential gene contents of the two closely related Pseudoxanthomonas strains isolated from different habitats may provide valuable clues in identifying the selective pressures and evolutionary developments that allowed strain BD-a59 to succeed in contaminated soil. The genes of strains BD-a59 and 11-1 were functionally classified based on their COG categories, and their frequencies were compared (Fig. 2). Strains BD-a59 and 11-1l had similar proportions of protein-coding genes (∼79%) that can be affiliated into respective COG categories (Table 1), and their distributions into COG functional classes were relatively similar with respect to the total numbers of protein-coding genes (Fig. 2A). However, analyzing only the genes unique to each strain, especially in the COG categories of energy production and conversion (C), carbohydrate transport and metabolism (G), lipid transport and metabolism (I), replication, recombination, and repair (L), secondary metabolite biosynthesis, transport, and catabolism (Q), general function prediction only (R), and function unknown (S), the differences were more evident (Fig. 2B).
Fig 2.
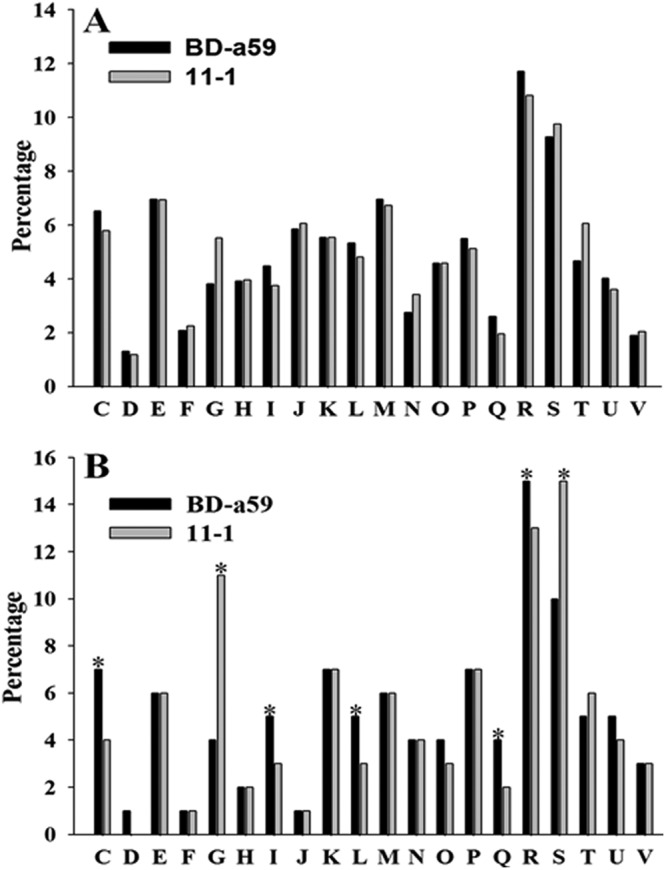
Comparison of COG functional classes of strains BD-a59 and 11-1. (A) All orthologous genes in each genome. (B) Specific orthologous genes found in each genome. The alphabetic codes represent COG functional categories as follows: C, energy production and conversion; D, cell cycle control, cell division, and chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure, and biogenesis; K, transcription; L, DNA replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, and chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolite biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; V, defense mechanisms. Asterisks appear where there is a difference of more than 20%.
Genes from the COG categories were subjected to additional scrutiny for a more refined functional analysis (Table 2). There were remarkable differences between strains BD-a59 and 11-1 in terms of numbers of mobile genetic elements such as phage integrases, transposases, and IS elements. The presence of large numbers of mobile genetic elements, in combination with CRISPRs (see above), indicates that strain BD-a59's genome may have undergone extensive genetic alterations via bacteriophage infection, horizontal gene transfer (HGT), and genetic reshuffling throughout its evolutionary history, which also may explain why more genomic islands (GIs) were identified in the genome of strain BD-a59 (24 GIs) than in that of strain 11-1 (15 GIs) (Table 1). It has been previously reported that the presence of large numbers of IS elements may be a typical characteristic associated with bacteria having slow growth and low productivity, although the reason for this is not known (34, 35). Along with the multiplicity of rRNA operons, the presence of more IS elements in strain BD-a59 than in strain 11-1 can be another reason why strain BD-a59 is capable of surviving in oligotrophic soil.
Table 2.
Abundances of genes in 11 gene classes found in the genomes of Pseudoxanthomonas strains BD-a59 and 11-1
| Gene class | No. of genes |
|
|---|---|---|
| BD-a59 | 11-1 | |
| Phage integrase | 4 | 0 |
| Transposases and IS elements | 16 | 6 |
| Sigma factors | 7 | 3 |
| Dioxygenases | 29 | 15 |
| Monooxygenases | 13 | 3 |
| TonB receptor | 23 | 47 |
| ABC transporter | 63 | 42 |
| Heavy metal-related genes | 12 | 5 |
| Histidine kinases | 20 | 36 |
| Oxidative stress-related genes | 23 | 20 |
| Chaperones | 17 | 11 |
A chromosomal synteny dot plot from a comparison between strains BD-a59 and 11-1 revealed numerous genomic translocations, inversions, and insertions that occurred over the evolutionary histories of these bacteria (Fig. 3); such rearrangements may have been mediated by the mobile genetic elements of strain BD-a59 (transposases, IS elements, and phage integrases; see above and Table 2). A global genomic comparison using the Mauve program also revealed extensive genome-wide rearrangements in strain BD-a59 relative to strain 11-1, especially in the form of reciprocal inversions (see Fig. S2 in the supplementary material). Twenty-four genomic islands were detected in the genome of strain BD-a59, and the Karlin signature skew and GC skew matched well with the locations of the genomic islands (Fig. 3), which provided additional evidence for genomic rearrangements in strain BD-a59.
Fig 3.

Karlin signature skew, GC skew, and dot-plot analysis of strain BD-a59. In the dot-plot analysis showing chromosomal synteny between strains BD-a59 and 11-1, the blue- and orange-colored circles indicate reciprocal best hits in the forward and reverse strands, respectively, for amino acid regions with >74% identity. The dot plot shows extensive rearrangement in strain BD-a59 in the form of reciprocal inversions. Plots of cumulative GC skew for strains BD-a59 and 11-1 are shown next to the axes. The Karlin signature difference and GC skew are shown above the dot plot. Twenty-four genomic islands (shaded regions) were found in the genome of strain BD-a59.
Genomic islands in strain BD-a59.
The 24 genomic islands were identified in strain BD-a59's genome using the integrated mode of IslandViewer. Table 3 shows general information regarding the GIs such as size (in kilobase pairs), number of genes, G+C content, and the number of mobile genetic elements and their predicted function. These GIs likely represent functional gene clusters acquired by strain BD-a59 via relatively recent lateral gene transfer, since the GC contents of these predicted GIs (approximately 53.4% to 64.5%) were quite different from that of the genome of strain BD-a59 (67.65%) (36, 37). Many of the GIs of strain BD-a59 were related to hydrocarbon degradation and heavy metal metabolism (e.g., GI-9, -20, and -21 contain genes encoding enzymes related to the degradation of BTEX compounds); 15 GIs were detected in the genome of strain 11-1. Interestingly, no genes related to hydrocarbon degradation or heavy metal metabolism were found in the 15 GIs of strain 11-1. Analysis of the genomic islands provided additional clues about the genomic plasticity of strain BD-a59, which is likely conferred by mobile elements such as integrases or transposases. GI-4, 6, -7, -15, -16, -17, -20, and -24 carry genes encoding integrases, transposases, or phage infections (restriction modification systems); these mobile elements contribute to the overall pool of transposase and IS elements that likely have contributed to genetic rearrangements and horizontal gene transfers in the genome of strain BD-a59. As mentioned above, the large number of genomic islands, in combination with mobile elements, provides evidence for the prominent role of horizontal gene transfer, phage attacks, and genetic rearrangements in the adaptive evolutionary history of this bacterium. This suggests that strain BD-a59 may have adapted to the oligotrophic soil habitat, as influenced by both hydrocarbons and heavy metals.
Table 3.
Characteristics of the genomic islands found in the genome of P. spadix BD-a59
| GI | Size (kbp) | No. of genes | GC content (%) | No. of hypothetical proteins | No. of transposases and integrases | Predicted function(s) |
|---|---|---|---|---|---|---|
| 1 | 12.64 | 18 | 58.6 | 16 | 0 | Unknown |
| 2 | 5.93 | 4 | 59.5 | 1 | 0 | Transport |
| 3 | 4.97 | 3 | 61.9 | 0 | 0 | Amino acid metabolism |
| 4 | 8.42 | 11 | 62.1 | 6 | 1 | Copper resistance |
| 5 | 4.80 | 5 | 61.6 | 1 | 0 | Heavy metal response |
| 6 | 16.81 | 14 | 61.1 | 3 | 2 | Cadmium transport |
| 7 | 4.86 | 9 | 64.2 | 2 | 0 | Arsenate reduction |
| 8 | 5.48 | 7 | 64.5 | 4 | 0 | Metal binding |
| 9 | 8.08 | 11 | 61.9 | 4 | 0 | Toluene degradation |
| 10 | 10.19 | 13 | 60.0 | 8 | 0 | Unknown |
| 11 | 4.56 | 5 | 62.0 | 0 | 0 | Alkane degradation |
| 12 | 8.33 | 8 | 56.5 | 0 | 0 | Phenylpropionate degradation |
| 13 | 5.34 | 7 | 62.6 | 0 | 0 | Biphenyl degradation |
| 14 | 4.04 | 3 | 57.8 | 0 | 0 | Biphenyl degradation |
| 15 | 17.16 | 14 | 60.5 | 8 | 0 | Unknown |
| 16 | 5.46 | 8 | 64.4 | 1 | 2 | Integrase/transposase |
| 17 | 55.15 | 47 | 61.2 | 33 | 1 | Restriction/modification |
| 18 | 4.12 | 7 | 63.5 | 2 | 0 | Motility |
| 19 | 17.38 | 16 | 58.8 | 4 | 0 | Hydrocarbon metabolism |
| 20 | 14.17 | 15 | 60.6 | 2 | 2 | Aromatic hydrocarbon metabolism |
| 21 | 4.55 | 4 | 53.4 | 1 | 0 | Xylene degradation |
| 22 | 5.64 | 7 | 59.9 | 0 | 0 | ABC transporter |
| 23 | 4.24 | 6 | 62.9 | 3 | 0 | Unknown |
| 24 | 5.40 | 8 | 58.8 | 5 | 0 | Unknown |
Oxidative stress tolerance, heavy metal resistance, and chaperones.
Potent mechanisms to reduce oxidative stress are required in all aerobic microorganisms that produce oxygenase-type enzymes for metabolizing pollutants (38, 39). The high abundance of oxygenase genes in strain BD-a59 (Table 1) indicates that this strain has high potential for biodegradation of hydrocarbons such as BTEX. Strain BD-a59 contains slightly more oxidative stress-related genes than strain 11-1 (Table 2). Generally, slow-growing organisms generate a smaller amount of reactive oxygen species (27). Strain BD-a59 has five peroxidase genes (DSC_01130, DSC_05235, DSC_06465, DSC_06645, and DSC_07775) and three superoxide dismutase genes (DSC_02075, DSC_02080, and DSC_12340). The same numbers of peroxidase and superoxide dismutase genes are found in strain 11-1. However, more genes coding for catalase (5 genes) and thioredoxin (7 genes) were found in the genome of strain BD-a59 than in that of strain 11-1 (2 and 5 genes, respectively). On the other hand, fewer glutaredoxin genes were identified in the genome of strain BD-a59 (3 genes) than in that of strain 11-1 (5 genes). Strain BD-a59 has more heavy metal-related genes than strain 11-1 does (Table 2). Strain BD-a59 harbors five genes related to copper, three to arsenic, three to cobalt/zinc/cadmium, and one to tellurium, while strain 11-1 contains only two copper-related and three arsenic-related genes. Most oil-contaminated soils are also polluted with heavy metals, as petroleum contains heavy metals (40). Therefore, in order to survive successfully in oil-contaminated soil, strain BD-a59 may require both hydrocarbon degradation capabilities and heavy metal resistances.
Chaperones are ubiquitous and abundant proteins that assist in the proper three-dimensional folding of proteins, especially under stress conditions such as high temperatures and conditions where pollutants and heavy metals are present (38, 41). As expected, strain BD-a59 (17 chaperones) harbors more genes coding for chaperone proteins than does strain 11-1 (11 chaperones), including heat shock proteins, DnaK, DnaJ, chaperonin (GroEL, Cpn10), an ATP-dependent chaperone (ClpB), an RNA chaperone (Hfq), and a lipoprotein chaperone (Table 2). This suggests that strain BD-a59 is more highly adapted to contaminated soil than strain 11-1. In particular, strain BD-a59 harbors 10 heat shock proteins (Hsps), while strain 11-1 contains only five Hsps. Hsps are ubiquitous proteins that prevent the stress-induced denaturation of other proteins (42). The high abundances of genes related to oxidative stress tolerances, heavy metal resistance, and chaperone function may be an adaptation strategy conferring more efficient metabolism of hydrocarbons and resistance to heavy metals.
Central metabolism.
The genes coding for the complete Embden-Meyerhof-Parnas (EMP), Entner-Doudoroff (ED), and citric acid cycle pathways are present in the two Pseudoxanthomonas strains. All genes (glucose-6-phosphate-1-dehydrogenase, DSC_01325; 6-phosphogluconolactonase, DSC_01330; transketolase, DSC_05025; 2-keto-3-deoxy-6-phosphogluconate aldolase, DSC_07850) encoding the pentose phosphate (PP) pathway, an essential process that produces reducing equivalents, ribose-5-phosphate, and erythrose-4-phosphate for the synthesis of fatty acids, nucleic acids, and amino acids, were also found in the genome of strain BD-a59. However, among the PP pathway proteins, the gene encoding 2-keto-3-deoxy-6-phosphogluconate aldolase (EC 4.1.2.14) was not identified in the genome of strain 11-1. There are two possible explanations for this: one is that strain 11-1 harbors a gene encoding an enzyme annotated differently by IMG and ER but with the same function, and the other is that strain 11-1 may have an alternative pathway that uses a different enzyme. The presence of all genes related to the central carbon metabolism pathways (EMP, ED, PP, and citric acid cycle pathways) in strain BD-a59's genome suggests that strain BD-a59 can survive efficiently in soil environments.
Catabolism of BTEX compounds and experimental verification of their degradation pathways.
Previous studies have shown that BTEX-degrading bacteria are widely distributed in diverse environments (43). There have been extensive studies demonstrating that BTEX compounds are metabolized through a variety of BTEX degradation pathways (43, 44). Based on the genome sequence of strain BD-a59 and its annotation, we bioinformatically predicted the degradation pathways of the six BTEX compounds in strain BD-a59 and confirmed them experimentally through analysis of the potential intermediates of BTEX compounds using GC-MS. The gene-associated functions of the BTEX catabolic clusters were predicted on the basis of the NCBI genomic database, the Integrated Microbial Genomes (IMG) database, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database.
Benzene/toluene degradation in strain BD-a59.
Previously, five toluene biodegradation pathways, initiated by oxidation of the methyl group (45), ring monooxidation at position 2, 3, or 4 (11, 46, 47), or ring 2,3-dioxidation (48) of toluene, have been reported as being typical aerobic toluene degradation pathways. A benzene/toluene catabolic gene cluster was identified from GI-9 of the genome of strain BD-a59 (Table 3). Figure 4A shows the physical map of the benzene/toluene catabolic genes and their associated functions. The gene cluster contains genes coding for a complete toluene 4-monooxygenase (T4MO) (orf2 to orf7, DSC_04380 to DSC_04405) and a Zn-dependent dehydrogenase (orf1, DSC_04375) as putative toluene degradation-associated proteins (Fig. 4A). Based on gene annotation, we propose that toluene degradation in strain BD-a59 is initiated by ring monooxidation at position 4, as shown in Fig. 4B. This pathway for the toluene biodegradation has been described for Pseudomonas mendocina KR1 (47). This degradation pathway converts toluene to 4-hydroxytoluene (p-cresol) by T4MO, followed by the conversion of 4-hydroxytoluene to 4-hydroxybenzaldehyde and 4-hydroxybenzoate by subsequent dehydrogenation (Fig. 4B). To confirm the potential biodegradation pathway, we searched for the presence of these intermediates using GC-MS after growing strain BD-a59 on toluene as a sole carbon and energy source. We detected 4-hydroxytoluene, 4-hydroxybenzaldehyde, and 4-hydroxybenzoate as intermediates in toluene-grown cells (see Fig. S3 in the supplementary material). These three compounds are not metabolic intermediates in the other four toluene biodegradation pathways; therefore, we conclude that strain BD-a59 metabolizes toluene through the pathway of ring monooxidation at position 4.
Fig 4.

Physical map of the toluene/benzene degradation gene cluster (A) and the proposed degradation pathways of toluene (B) and benzene (C) in strain BD-a59. The putative functions of the genes in the toluene/benzene degradation gene cluster were predicted as follows: orf1 (DSC_04375), Zn-dependent dehydrogenases; orf2 (DSC_04380), toluene monooxygenase oxidoreductase; orf3 (DSC_04385), toluene-4-monooxygenase protein E; orf4 (DSC_04390), toluene-4-monooxygenase protein D; orf5 (DSC_04395), toluene-4-monooxygenase protein C; orf6 (DSC_04400), toluene-4-monooxygenase protein B; orf7 (DSC_04405), toluene-4-monooxygenase protein A; orf8 (DSC_04410), catechol 2,3-dioxygenase; orf9 (DSC_04415), phenol 2-monooxygenase P5 subunit; orf10 (DSC_04420), phenol 2-monooxygenase P4 subunit; orf11 (DSC_04425), phenol 2-monooxygenase P3 subunit; orf12 (DSC_04430), phenol 2-monooxygenase P2 subunit; orf13 (DSC_04435), phenol 2-monooxygenase P1 subunit; orf14 (DSC_04440), phenol hydroxylase subunit; orf15 (DSC_04445), transcriptional regulator.
The benzene/toluene catabolic gene cluster in strain BD-a59 also harbors genes coding for phenol 2-hydroxylase and catechol 2,3-dioxygenase in the upstream region (Fig. 4A). Studies of benzene degradation have shown that there are two primary pathways for the metabolism of benzene, which are clearly distinguished by the conversion of benzene to phenol or, alternatively, to cis-dihydrobenzenediol in the first oxidation step (12, 49). However, no genes annotated as encoding benzene monooxygenase or benzene 1,2-dioxygenase (for the conversion of benzene to phenol or cis-dihydrobenzenediol, respectively) were found in the genome of strain BD-a59. Interestingly, it has been reported that the toluene monooxygenases of P. mendocina KR1 and R. pickettii PKO1 have the ability to oxidize both toluene and benzene (12). Based on this, we propose a potential benzene degradation pathway, which has phenol and catechol as intermediates, for strain BD-a59 as shown in Fig. 4C. To confirm the presence of this benzene degradation pathway, we searched for benzene intermediates using GC-MS after benzene was provided as the sole carbon and energy source. Phenol and catechol were detected as intermediates in benzene-grown cells (see Fig. S3 in the supplemental material). Phenol was detected as a major intermediate, while catechol was detected as a minor intermediate. Hydroxylation of phenol to catechol by phenol 2-hydroxylase is thought to be the rate-limiting step in the aerobic phenol-degrading pathway (50), which might be why phenol was accumulated as a key intermediate. The detection of phenol and catechol as intermediates and the metabolic gene cluster analysis suggest that benzene is likely metabolized in strain BD-a59 through the phenol degradation pathway, using T4MO for the first benzene oxidation.
Xylene degradation in strain BD-a59.
The degradation of o-xylene in Burkholderia cepacia MB2 proceeds through the oxidation of a methyl substituent of o-xylene to 2-methylbenzyl alcohol by xylene monooxygenase, which is followed by the subsequent conversion of 2-methylbenzyl alcohol to 2-methylbenzaldehyde and 2-methylbenzoate by benzylalcohol dehydrogenase and benzaldehyde dehydrogenase, respectively (51). Gibson et al. (52) reported that Pseudomonas putida also uses xylene monooxygenase to oxidize m- and p-xylene to m- and p-methylbenzyl alcohol, respectively. While methyl group oxidation of xylenes can follow the pathways described above for toluene, Jang et al. (53) reported that o- and p-xylene may also be metabolized through direct oxidation of the aromatic ring by xylene dioxygenase in Rhodococcus sp. strain YU6.
A xylene catabolic gene cluster was identified in GI-21 of the genome of strain BD-a59 (Table 3), and its physical map and associated gene functions are shown in Fig. 5A. The xylene catabolic gene cluster harbors xylene metabolic genes coding for xylene monooxygenase (orf2 and orf3, DSC_04805 and DSC_04810), benzylalcohol dehydrogenase (orf6, DSC_14825), benzaldehyde dehydrogenase (NAD positive [NAD+]) (orf7, DSC_14830), and methyl benzoate dioxygenase (orf4, DSC_14815). The predicted functions of the associated genes suggest that o-, m-, and p-xylene compounds are potentially metabolized by the oxidation of a methyl substituent of xylene to an alcohol group, as shown in Fig. 5B. We searched for potential metabolic intermediates of xylene compounds using GC-MS to confirm the presence of the proposed xylene degradation pathway after the addition of o-, m-, and p-xylene to strain BD-a59 as the sole carbon source. As expected, methylbenzaldehyde, 1,2-dihydroxy-methyl-cyclohexa-3,5-diene-carboxylate, and methyl benzylalcohol were detected as key metabolic intermediates of all xylene compounds (see Fig. S3 in the supplemental material). The detection of this suite of intermediates indicates that all three xylene isomers were metabolized by the pathways shown in Fig. 5B but that the detected abundances of particular metabolites were likely governed by differing affinities of the various xylene metabolic enzymes for these xylene compounds and their metabolites.
Fig 5.
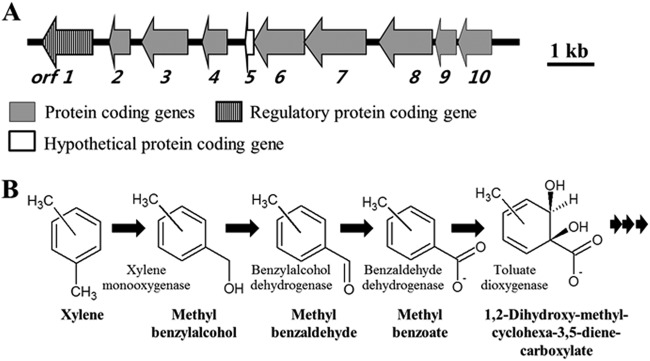
Physical maps of the xylene degradation gene cluster (A) and the proposed degradation pathway of xylene (B) in strain BD-a59. The putative functions of the genes in the xylene degradation cluster were predicted as follows: orf1 (DSC_14800), transcriptional regulator (Fis family); orf2 (DSC_14805), xylene monooxygenase electron transfer subunit (oxydoreductase); orf3 (DSC_14810), xylene monooxygenase subunit 1; orf4 (DSC_14815), benzoate 1,2-dioxygenase alpha subunit; orf5 (DSC_14820), hypothetical protein; orf6 (DSC_14825), benzylalcohol dehydrogenase; orf7 (DSC_14830), benzaldehyde dehydrogenase; orf8 (DSC_14835), phenylpropionate dioxygenase large subunit; orf9 (DSC_14840), aromatic oxygenase small subunit; orf10 (DSC_14845), ABC transporter.
Ethylbenzene degradation in strain BD-a59.
Initial aerobic ethylbenzene degradation proceeds via two established pathways. One pathway involves dioxygenation of the aromatic ring by ethylbenzene dioxygenase, causing extradiol ring cleavage; this pathway is found in Pseudomonas sp. strain NCIB 10643 (54). The other pathway involves the oxidation of ethylbenzene by naphthalene dioxygenase to styrene or 1-phenethyl alcohol (55). We searched for intermediates of ethylbenzene using GC-MS after the addition of ethylbenzene as the sole carbon source. From the results of this analysis, phenethyl alcohol (but not styrene) was detected as a metabolic intermediate (see Fig. S3 in the supplemental material). Therefore, we surmise that strain BD-a59 metabolizes ethylbenzene through the conversion of ethylbenzene to 1-phenethyl alcohol. 1-Phenethyl alcohol is typically next converted either to 2-hydroxyacetophenone (via naphthalene dioxygenase) or to benzoylacetate (via acetophenone carboxylase; 44, 55); neither of the latter metabolites was detected. Regarding genes catalyzing the initial attack of ethylbenzene, inspection of the genome annotation for strain BD-a59 failed to uncover an ethylbenzene dioxygenase; however, this bacterium carries 29 dioxygenases (Table 2). In light of the established roles of naphthalene dioxygenase in catalyzing multiple steps (44, 55) in ethylbenzene biodegradation, we propose that strain BD-a59 utilizes the pathway shown in Fig. 6.
Fig 6.
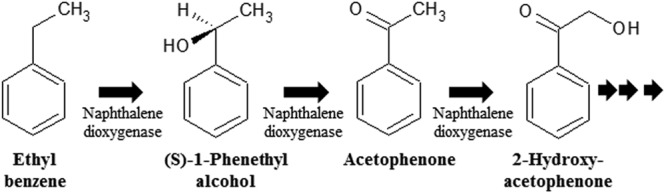
Proposed degradation pathway of ethyl benzene in strain BD-a59. The first step in the pathway can also be mediated by an alternative enzyme, ethylbenzene dehydrogenase. The second step in the pathway can also be mediated by an alternative enzyme, 1-phenylethanol dehydrogenase.
We tested the degradation ability of strain BD-a59 for other organic compounds: salicylate, catechol, biphenyl, phenylpropionate, gentisate, naphthalene, 3-hydroxybenzoate, hexane, and phenol. We observed that strain BD-a59 has the ability to degrade catechol, biphenyl, phenylpropionate, and phenol, which further supported the proposed metabolic pathways for BTEX compounds.
In conclusion, comparative genomics based on the analysis of complete genomes of closely related strains can provide valuable insights into the acquisition, loss, and evolution of genes (56). We compared the genomic features of strain BD-a59 with those of strain 11-1 and found bioinformatic evidence that the genome of strain BD-a59 may have been altered via horizontal gene transfer (HGT), phage attack, and/or genetic reshufflings during its evolutionary history. Three genomic features of strain BD-a59 contrast strikingly with those of strain 11-1: genomic islands (GIs), rRNA operons, and oxygenase genes. These genetic traits have likely contributed to the successful adaptation of strain BD-a59 both to oligotrophic soil conditions and to exploitation of hydrocarbons as carbon sources. Additionally, we predicted the degradation pathways of BTEX compounds in strain BD-a59 by analyzing BTEX degradation gene clusters, and the identified pathways were partially experimentally confirmed. The availability of the complete genome of strain BD-a59 will allow us to continue to advance the understanding of the physiology, evolution, and ecological fitness of this BTEX-biodegrading bacterium in contaminated soil environments.
Supplementary Material
ACKNOWLEDGMENTS
These efforts were supported by the “National Research Foundation of Korea (no. 2010-0026359)” grant funded by the Korean government (MEST), Republic of Korea. Funding to E.L.M. was provided by NSF grant DEB-0841999.
Footnotes
Published ahead of print 16 November 2012
Supplemental material for this article may be found at 10.1128/AEM.02809-12.
REFERENCES
- 1. Bowlen GF, Kosson DS. 1995. In situ processes for bioremediation of BTEX and petroleum fuel products, p 515–542 In Young LY, Cerniglia CE. (ed), Microbial transformations and degradation of toxic organic chemicals. Wiley-Liss, Inc., New York, NY [Google Scholar]
- 2. Dean BJ. 1985. Recent findings on the genetic toxicology of benzene, toluene, xylenes and phenols. Mutat. Res. 154:153–181 [DOI] [PubMed] [Google Scholar]
- 3. Atlas RM, Cerniglia CE. 1995. Bioremediation of petroleum pollutants: diversity and environmental aspects of hydrocarbon biodegradation. Bioscience 45:332–338 [Google Scholar]
- 4. Berlendis S, Cayol JL, Verhé F, Laveau S, Tholozan JL, Ollivier B, Auria R. 2010. First evidence of aerobic biodegradation of BTEX compounds by pure cultures of Marinobacter. Appl. Biochem. Biotechnol. 160:1992–1999 [DOI] [PubMed] [Google Scholar]
- 5. Chakraborty R, O'Connor SM, Chan E, Coates JD. 2005. Anaerobic degradation of benzene, toluene, ethylbenzene, and xylene compounds by Dechloromonas strain RCB. Appl. Environ. Microbiol. 71:8649–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim D, Kim YS, Kim SK, Kim SW, Zylstra GJ, Kim YM, Kim E. 2002. Monocyclic aromatic hydrocarbon degradation by Rhodococcus sp. strain DK17. Appl. Environ. Microbiol. 68:3270–3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim JM, Jeon CO. 2009. Isolation and characterization of a new benzene, toluene, and ethyl benzene degrading bacterium, Acinetobacter sp. B113. Curr. Microbiol. 58:70–75 [DOI] [PubMed] [Google Scholar]
- 8. Shinoda Y, Sakai Y, Uenishi H, Uchihashi Y, Hiraishi A, Yukawa H, Yurimoto H, Kato N. 2004. Aerobic and anaerobic toluene degradation by a newly isolated denitrifying bacterium, Thauera sp. strain DNT-1. Appl. Environ. Microbiol. 70:1385–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zylstra GJ, Kim E. 1997. Aromatic hydrocarbon degradation by Sphingomonas yanoikuyae B1. J. Ind. Microbiol. Biotechnol. 19:408–414 [DOI] [PubMed] [Google Scholar]
- 10. Jahn MK, Haderlein SB, Meckenstock RU. 2005. Anaerobic degradation of benzene, toluene, ethylbenzene, and o-xylene in sediment-free iron-reducing enrichment cultures. Appl. Environ. Microbiol. 71:3355–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olsen RH, Kukor JJ, Kaphammer B. 1994. A novel toluene-3-monooxygenase pathway cloned from Pseudomonas pickettii PKO1. J. Bacteriol. 176:3749–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tao Y, Fishman A, Bentley WE, Wood TK. 2004. Oxidation of benzene to phenol, catechol, and 1, 2, 3-trihydroxybenzene by toluene 4-monooxygenase of Pseudomonas mendocina KR1 and toluene 3-monooxygenase of Ralstonia pickettii PKO1. Appl. Environ. Microbiol. 70:3814–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Williams PA, Jones RM, Shaw LE. 2002. A third transposable element, ISPpu12, from the toluene-xylene catabolic plasmid pWW0 of Pseudomonas putida mt-2. J. Bacteriol. 184:6572–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zylstra GJ, McCombie WR, Gibson DT, Finette BA. 1988. Toluene degradation by Pseudomonas putida F1: genetic organization of the tod operon. Appl. Environ. Microbiol. 54:1498–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim JM, Le NT, Chung BS, Park JH, Bae JW, Madsen EL, Jeon CO. 2008. Influence of soil components on the biodegradation of benzene, toluene, ethylbenzene, and o-, m-, and p-xylenes by the newly isolated bacterium Pseudoxanthomonas spadix BD-a59. Appl. Environ. Microbiol. 74:7313–7320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nayak AS, Sanjeev Kumar S, Santosh Kumar M, Anjaneya O, Karegoudar TB. 2011. A catabolic pathway for the degradation of chrysene by Pseudoxanthomonas sp. PNK-04. FEMS Microbiol. Lett. 320:128–134 [DOI] [PubMed] [Google Scholar]
- 17. Patel V, Cheturvedula S, Madamwar D. 2012. Phenanthrene degradation by Pseudoxanthomonas sp. DMVP2 isolated from hydrocarbon contaminated sediment of Amlakhadi canal, Gujarat, India. J. Hazard. Mater. 201-202:43–51 [DOI] [PubMed] [Google Scholar]
- 18. Math RK, Jin HM, Kim JM, Hahn Y, Park W, Madsen EL, Jeon CO. 2012. Comparative genomics reveals adaptation by Alteromonas sp. SN2 to marine tidal-flat conditions: cold tolerance and aromatic hydrocarbon metabolism. PLoS One 7:e35784 doi:10.1371/journal.pone.0035784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shendure J, Ji H. 2008. Next-generation DNA sequencing. Nat. Biotechnol. 26:1135–1145 [DOI] [PubMed] [Google Scholar]
- 20. Lee SH, Jin HM, Lee HJ, Kim JM, Jeon CO. 2012. Complete genome sequence of the BTEX-degrading bacterium Pseudoxanthomonas spadix BD-a59. J. Bacteriol. 194:544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stothard P, Wishart DS. 2005. Circular genome visualization and exploration using CGView. Bioinformatics 21:537–539 [DOI] [PubMed] [Google Scholar]
- 22. Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106:19126–19131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945 [DOI] [PubMed] [Google Scholar]
- 24. Darling AE, Mau B, Perna NT. 2010. prograssiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147 doi:10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hsiao W, Wan I, Jones SJ, Brinkman FSL. 2003. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19:418–420 [DOI] [PubMed] [Google Scholar]
- 26. Jin HM, Kim JM, Lee SH, Madsen EL, Jeon CO. 2012. Alteromonas as a key agent of polycyclic aromatic hydrocarbon biodegradation in crude oil contaminated coastal sediment. Environ. Sci. Technol. 46:7731–7740 [DOI] [PubMed] [Google Scholar]
- 27. Stanier RY, Palleroni NJ, Doudorhoff M. 1966. The aerobic pseudomonads: a taxonomic study. J. Gen. Microbiol. 43:159–271 [DOI] [PubMed] [Google Scholar]
- 28. Pumphrey GM, Madsen EL. 2007. Naphthalene metabolism and growth inhibition bynaphthalene in Polaromonas naphthalenivorans strain CJ2. Microbiology 153:3730–3738 [DOI] [PubMed] [Google Scholar]
- 29. Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U. S. A. 102:2567–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712 [DOI] [PubMed] [Google Scholar]
- 31. Klappenbach JA, Dunbar JM, Schmidt TM. 2000. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Mirobiol. 66:1328–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vieira-Silva S, Rocha EP. 2010. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 6:e1000808 doi:10.1371/journal.pgen.1000808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. 2004. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186:2629–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ivars-Martinez E, Martin-Cuadrado A, D'Auria G, Mira A, Ferriera S, Johnson J, Friedman R, Rodriguez-Valera F. 2008. Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2:1194–1212 [DOI] [PubMed] [Google Scholar]
- 35. Martin-Cuadrado AB, Rodriguez-Valera F, Moreira D, Alba JC, Ivars-Martínez E, Henn MR, Talla E, López-García P. 2008. Hindsight in the relative abundance, metabolic potential and genome dynamics of uncultivated marine archaea from comparative metagenomic analyses of bathypelagic plankton of different oceanic regions. ISME J. 2:865–886 [DOI] [PubMed] [Google Scholar]
- 36. Gogarten JP, Townsend JP. 2005. Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 3:679–687 [DOI] [PubMed] [Google Scholar]
- 37. Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304 [DOI] [PubMed] [Google Scholar]
- 38. Storz G, Imlayt JA. 1999. Oxidative stress. Curr. Opin. Microbiol. 2:188–194 [DOI] [PubMed] [Google Scholar]
- 39. Yeom S, Yeom J, Park W. 2010. Molecular characterization of FinR, a novel redox-sensing transcriptional regulator in Pseudomonas putida KT2440. Microbiology 156:1487–1496 [DOI] [PubMed] [Google Scholar]
- 40. Bada BS, Olarinre TA. 2012. Characteristics of soils and heavy metal content of vegetation in oil spill impacted land in Nigeria. Proceedings of the Annual International Conference on Soils, Sediments, Water and Energy, vol 17, article 2 University of Massachusetts Amherst, Amherst, MA: http://scholarworks.umass.edu/soilsproceedings/vol17/iss1/2 [Google Scholar]
- 41. Almeida JA, Diniz YS, Marques SFG, Faine LA, Ribas BO, Burneiko RC, Novelli ELB. 2002. The use of the oxidative stress responses as biomarkers in Nile tilapia (Oreochromis niloticus) exposed to in vivo cadmium contamination. Environ. Int. 27:673–679 [DOI] [PubMed] [Google Scholar]
- 42. Feder EM, Hofmann GE. 1999. Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu. Rev. Physiol. 61:243–282 [DOI] [PubMed] [Google Scholar]
- 43. Farhadian M, Vachelard C, Duchez D, Larroche C. 2008. In situ bioremediation of monoaromatic pollutants in groundwater: a review. Bioresour. Technol. 99:5296–5308 [DOI] [PubMed] [Google Scholar]
- 44. Gao J, Ellis LBM, Wackett LP. 2010. The University of Minnesota Biocatalysis/Biodegradation Database: improving public access. Nucleic Acids Res. 38:D488–D491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shaw JP, Harayama S. 1992. Purification and characterisation of the NADH:acceptor reductase component of xylene monooxygenase encoded by the TOL plasmid pWW0 of Pseudomonas putida mt-2. Eur. J. Biochem. 209:51–61 [DOI] [PubMed] [Google Scholar]
- 46. Shields MS, Reagin MJ, Gerger RR, Campbell R, Somerville C. 1995. TOM, a new aromatic degradative plasmid from Burkholderia (Pseudomonas) cepacia G4. Appl. Environ. Microbiol. 61:1352–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yen KM, Karl MR, Blatt LM, Simon MJ, Winter RB, Fausset PR, Lu HS, Harcourt AA, Chen KK. 1991. Cloning and characterization of a Pseudomonas mendocina KR1 gene cluster encoding toluene-4-monooxygenase. J. Bacteriol. 173:5315–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wackett LP, Kwart LD, Gibson DT. 1988. Benzylic monooxygenation catalyzed by toluene dioxygenase from Pseudomonas putida. Biochemistry 27:1360–1367 [DOI] [PubMed] [Google Scholar]
- 49. Zamanian M, Mason JR. 1987. Benzene dioxygenase in Pseudomonas putida. Subunit composition and immuno-cross-reactivity with other aromatic dioxygenases. Biochem. J. 244:611–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu C, Zhang L, Zhao L. 2008. Molecular cloning, genetic organization of gene cluster encoding phenol hydroxylase and catechol 2,3-dioxygenase in Alcaligenes faecalis IS-46. World J. Microbiol. Biotechnol. 24:1687–1695 [Google Scholar]
- 51. Jørgensen C, Nielsen B, Jensen BK, Mortensen E. 1995. Transformation of o-xylene to o-methyl benzoic acid by a denitrifying enrichment culture using toluene as the primary substrate. Biodegradation 6:141–146 [DOI] [PubMed] [Google Scholar]
- 52. Gibson DT, Mahadevan V, Davey JF. 1974. Bacterial metabolism of para- and meta-xylene: oxidation of the aromatic ring. J. Bacteriol. 119:930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jang JY, Kim D, Bae HW, Choi KY, Chae JC, Zylstra GJ, Kim YM, Kim E. 2005. Isolation and characterization of a rhodococcus species strain able to grow on ortho- and para-xylene. J. Microbiol. 43:325–330 [PubMed] [Google Scholar]
- 54. Gibson DT, Gschwendt B, Yeh WK, Kobal VM. 1973. Initial reactions in the oxidation of ethylbenzene by Pseudomonas putida. Biochemistry 12:1520–1528 [DOI] [PubMed] [Google Scholar]
- 55. Lee K, Gibson DT. 1996. Toluene and ethylbenzene oxidation by purified naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816-4. Appl. Environ. Microbiol. 62:3101–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, Yang Q, Miyada CG, Lory S. 2003. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:8484–8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.