

Abstract
TRIF is an adaptor molecule important in transducing signals from intracellularly signaling Toll-like receptor 3 (TLR3) and TLR4. Recently, TLR2 was found to signal from intracellular compartments. Using a synthetic ligand for TLR2/1 heterodimers, as well as Borrelia burgdorferi, which is a strong activator of TLR2/1, we found that TLR2 signaling can utilize TRIF. Unlike TRIF signaling by other TLRs, TLR2-mediated TRIF signaling is dependent on the presence of another adaptor molecule, MyD88. However, unlike MyD88 deficiency, TRIF deficiency does not result in diminished control of infection with B. burgdorferi in a murine model of disease. This appears to be due to the effects of MyD88 on phagocytosis via scavenger receptors, such as MARCO, which are not affected by the loss of TRIF. In mice, TRIF deficiency did have an effect on the production of inflammatory cytokines, suggesting that regulation of inflammatory cytokines and control of bacterial growth may be uncoupled, in part through transduction of TLR2 signaling through TRIF.
INTRODUCTION
Innate immunity plays an important role in early responses to microbial invaders through recognition of conserved molecular patterns by receptors, such as those of the Toll-like receptor (TLR) family. Innate immune responses to Borrelia burgdorferi, the causative agent of Lyme disease, have been shown to be mediated by multiple TLR receptors, including TLR2/1 heterodimers, TLR5, TLR7, TLR8, and TLR9 (1–7). All of these TLRs are thought to utilize the adaptor molecule myeloid differentiation primary response 88 (MyD88). TLR2 responses to B. burgdorferi have been the most extensively studied as B. burgdorferi express a large number of triacylated lipoproteins that activate TLR2/1 heterodimers (1, 2, 5, 7–10). In murine models, loss of TLR2 or MyD88 results in a greatly increased replication of the organism and up to several hundredfold increases in tissue numbers of bacteria (6, 11, 12).
TIR-domain-containing adaptor-inducing beta interferon (TRIF) is another adaptor molecule used by some intracellularly signaling TLRs—specifically TLR3 and TLR4 (13). While TLR3 utilizes TRIF exclusively, TLR4 utilizes MyD88 to signal from its plasma membrane location and TRIF to signal from endosomal compartments. TRIF is thought to mediate induction of type I interferons, whereas activation of MyD88 is more closely related to release of inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-12 (IL-12), and IL-6 (13, 14).
Although a very early study reported a relationship between TLR2 and TRIF, subsequent studies did not confirm this, and the general consensus has been that TLR2 does not utilize TRIF as an adaptor molecule (15–17). This viewpoint has been bolstered by the fact that signaling through TLR2 has not been closely associated with induction of type I interferons and that TLR2 has long been felt to signal exclusively from the plasma membrane (14, 18, 19). In addition, the complete abrogation of activation of inflammatory signaling in the absence of MyD88 implied that TLR2 did not activate a MyD88-independent pathway for signaling (19, 20). However, more recently, multiple studies have shown that TLR2 is present in intracellular compartments and is involved in signaling from these compartments (3, 21–26). Stimulation with TLR2 specific lipopeptides was shown to lead to the activation of type I interferons and proinflammatory cytokines, such as IL-6, whose activation and release was dependent on endosomal acidification (23, 26). Localization of TLR2 signaling thus appears to shape downstream induction of cytokines, with TLR2-induced activation of type I interferons taking place solely from intracellular compartments.
Studies from our group and others have shown that internalization of B. burgdorferi is important for activating inflammatory cellular responses and, in particular, those mediated by TLR2 (1, 2, 23, 27–29). The recent findings that TLR2 can signal from intracellular compartments and can induce type I interferon production, coupled with the fact that TRIF is a major adaptor for signaling from intracellular compartments for other TLRs, led us to reexamine the role of TRIF in mediating TLR2 signals. Here, we show that TLR2 can utilize the TRIF adaptor from intracellular compartments. TLR2/TRIF signaling was fully dependent on the presence of MyD88, with no residual signaling in MyD88-deficient cells, suggesting an as-yet-unrecognized component of the TRIF pathway which depends either directly or indirectly on MyD88 activation. We show that TRIF is not involved in the control of bacterial loads in vivo, but it does play a role in controlling both inflammatory and anti-inflammatory cytokines.
MATERIALS AND METHODS
Mouse and bacterial strains.
MyD88- and TRIF-deficient mice (developed by S. Akira) were backcrossed to the C57BL/6 background for six generations (16, 30) and were subsequently crossbred to generate the double knockout line and reestablish the wild-type and the single knockout lines by breeding with a pure C57BL/6 background mouse (Jackson Laboratories). TLR2 deficient mice were obtained from Jackson Laboratories. MARCO-deficient mice on a BALB/c background were derived as previously described (31). All mice were housed in specific-pathogen-free rooms according to institutional guidelines for humane care and use of laboratory animals.
An infectious clone of B. burgdorferi strain B31 (5A11) (32) was used for all infection studies in mice and were cultured in Barbour-Stoenner-Kelley (BSK)-H (Sigma, St. Louis, MO) complete media at 33°C to early stationary phase, and the cell density was determined by using a Petroff-Hauser counting chamber. Clonal isolates of infectious, low-passage B. burgdorferi sensu stricto (strain N40, clone D10E9) were cultured as described previously (23), and used at multiplicity of infection (MOI) of 10:1 in in vitro experiments.
Ligands and inhibitors.
The TLR2 ligand Pam3CSK4 was purchased from Invivogen and resuspended to a concentration of 1 mg/ml in endotoxin-free water. The purity of the ligand from contaminants that may activate other receptors was subsequently tested by stimulating TLR2-deficient cells and measuring secretion of cytokines and activation of type I interferons as described below. There was no increase in cytokine or type I interferon expression of TLR2 knockout cells when stimulated with Pam3CSK4.
Generation of BMDM.
Bone marrow-derived macrophages (BMDM) were generated as previously described (33). Bone marrow cells were flushed from mouse femurs and tibiae with sterile Dulbecco modified Eagle medium (DMEM) and cultured on plastic petri dishes (100 mm by 15 mm) for 5 to 7 days in DMEM supplemented with 30% L929 cell conditioned medium, 20% fetal bovine serum (FBS), and 1% penicillin-streptomycin.
Infection of BMDM cultures.
B. burgdorferi were washed three times with DMEM with 10% FBS, counted, and resuspended in the same medium. Medium from BMDM was removed and replaced with the same medium containing B. burgdorferi at MOIs of 10 in the presence of polymyxin B (Sigma) at 50 μg/ml to ensure no endotoxin contamination. Cells were harvested at various time points by collecting the cell culture supernatant and then adding TRIzol (Invitrogen) to the remaining cells.
Mouse infections.
Mouse infection protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the Harvard School of Public Health, Tufts University, and Ohio State University. Sex- and age-matched wild-type and knockout mice were infected at 3 to 12 weeks of age with a dose of 105 spirochetes by subcutaneous inoculation. B. burgdorferi infection was confirmed by serology and by culturing (in BSK-H complete medium containing 20 μg of phosphomycin/ml, 50 μg of rifampin/ml, and 2.5 μg of amphotericin B/ml) of live spirochetes from skin, heart, and joint. At the time of necropsy, the tissue samples of skin, heart, joint, and bladder were immediately frozen in liquid nitrogen and stored at −80°C for RNA and/or DNA extraction at a later time.
ELISA measurements.
Supernatants were collected from BMDM cultures at 6 and 16 h poststimulation. Cytokines were measured by enzyme-linked immunosorbent assay (ELISA) using the IL-10 (R&D Systems) and IL-6 (e-Bioscience) kits according to the manufacturer's instructions.
Microscopy.
B. burgdorferi phagocytosis experiments were performed and stained as previously described (23). Coverslips were examined using a Zeiss Axiolan 2 microscope. Images were captured with a digital charge-coupled device camera (Hamamatsu). Images were merged using Volocity software (Improvision, Inc.). The percent internalized bacteria was determined by dividing the number of macrophages with at least one internalized (red) bacterium by the total number of macrophages in the view field determined by DAPI (4′,6′-diamidino-2-phenylindole) staining. Lysosomal marker antibody against Lamp-1 was obtained from the hybridoma facility at the Iowa State University. Anti-B. burgdorferi antibody was kindly provided by Allen Steere.
RNA purification and cDNA synthesis.
Mouse joint tissues were pulverized in TRIzol using the Precellys lysing kit containing metal beads (Precellys). RNA was extracted from BMDM cells and mouse tissues using TRIzol (Invitrogen) according to the manufacturer's instructions. Contaminating DNA was removed using the Turbo DNA-free kit (Roche) or by RNase-free DNase I (Invitrogen) digestion, followed by further purification on RNeasy columns (Qiagen). Quality of the RNA was checked by the examination of RNA integrity on formamide gels and by 5′-3′ analysis. cDNA was synthesized from 1 μg of RNA measured by spectrophotometer using a ImPromII kit (Promega) according to the manufacturer's instructions. The absence of DNA contamination in the RNA samples was verified either by the use of no-RT controls or by using primer-spanning introns.
DNA purification.
Concomitant DNA purification from the TRIzol extraction of mouse joint samples was carried out according to the manufacturer's instructions and further purified on DNeasy columns (Qiagen).
Real-time quantitative (qPCR) analysis.
Quantification of target genes from cDNA or total DNA was performed either using the iQ SYBR green Supermix 2× (Bio-Rad) or the TaqMan Universal PCR Master Mix (Applied Biosystems). Each primer was added to a final concentration to 0.4 μM. Cycling parameters for SYBR green-based reactions were 95°C for 15 min, 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s, followed by 95°C for 1 min and 55°C for 1 min. Melting curve analysis for purity was performed on each sample by performing 80 cycles of increasing temperature for 10 s, each beginning at 55°C. Primers used in qPCR analysis of murine irf7, il-6, il-10, and ifn-α universal primers; cxcl10, ifit1, and β-actin were as previously described (33, 34). All samples for each biological replicate were run in duplicates and checked for intra-run variation and, if needed, analyzed again. Different genes/transcripts were analyzed in the same run to avoid inter-run variations. β-Actin was analyzed for every run for all samples tested, as a positive control and inter-run calibrator, by inclusion of a standard sample in each run. Cytokine and β-actin gene expression was normalized from cDNA using the ΔΔCT method, where the amount of target, normalized to an endogenous reference and relative to a calibrator, is given by 2−ΔΔCT, where CT is the cycle number of the detection threshold. All analyses and calculations were performed using the iCycler software.
TaqMan probe-based qPCR analyses of B. burgdorferi flaB gene and mouse beta-actin gene were carried out as described previously (35). Serial dilutions of a standard were included in every plate to establish a standard curve, which was used calculate the gene copy number of an unknown sample from the threshold cycle number.
Statistics.
For ELISA and qPCR, the mean percentage of cytokine expression relative to the control is reported, with statistical significance determined by Mann-Whitney U analysis.
RESULTS
TLR2-mediated induction of inflammatory responses utilizes MyD88 and TRIF.
Although it has been shown that TLR2 signals from the endosome (3, 21, 23–26), it is unclear how the molecular TLR2 signaling complex at the endosome differs from the one at the cell surface. TRIF had long been thought not to be involved in transduction of signals from TLR2. However, since the recent discovery of TRIF's role in intracellular TLR4 signaling (14) and the reports of TLR2 signaling from the endosome, some groups have begun to reexamine the role of TRIF in TLR2 signaling (36).
In our studies, we used a synthetic lipopeptide, Pam3CSK4, a TLR2/1 heterodimer ligand to specifically address the role of TRIF in TLR2 signaling. To investigate the role of TRIF and TLR2 in a bacterial model, we chose Borrelia burgdorferi, a pathogen that has a large number of TLR2 lipoprotein ligands and signals for proinflammatory cytokines primarily via TLR2. Maximal release of proinflammatory cytokines in response to B. burgdorferi has been shown to require phagocytosis, processing of the organism, and signaling from acidified endosomal compartments (1, 12, 23, 27–29). B. burgdorferi is not known to express any ligands for the known TRIF utilizing TLRs (TLR3 and TLR4), and TLR2 signaling has been shown to be the major contributor to induction of proinflammatory cytokines in response to the organism (1, 23). Indeed, when we stimulated TLR2-deficient cells with B. burgdorferi at an MOI of 10, we observed a significant decrease in the expression of inflammatory cytokines, including type I interferons (Fig. 1A).
Fig 1.
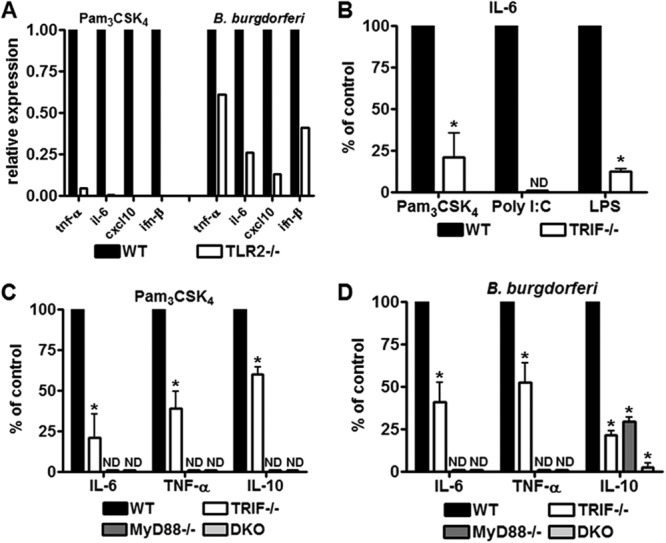
TRIF mediates TLR2 proinflammatory signaling. (A) BMDM isolated from wild-type (WT) and TLR2-deficient mice (TLR2−/−) were stimulated with 1 μg of Pam3CSK4/ml or B. burgdorferi at MOI of 10, in the presence of polymyxin B for 6 h. Expression of ifn-β, tnf-α, cxcl10, and il6 was measured by RT-qPCR. Values represent mean induction of expression relative to control cells. (B) BMDM isolated from wild-type (WT) and TRIF-deficient mice (TRIF−/−) were stimulated with 1 μg of Pam3CSK4/ml, 10 μg of poly(I·C)/ml and 5 ng of LPS/ml for 6 h. Except for LPS, stimulations were performed in the presence of polymyxin B. Values represent mean secretion of IL-6 in deficient BMDM relative to wild-type cells measured by ELISA. For IL-6, wild-type cells stimulated with Pam3CSK4 secreted a mean of 2.52 ng/ml, TRIF-deficient cells secreted a mean of 0.47 ng/ml. Wild-type cells stimulated with poly(I·C) secreted a mean of 1.3 ng of IL-6/ml, whereas TRIF-deficient cells secreted no detectable (ND) levels. Wild-type cells stimulated with LPS secreted a mean of 7.08 ng of IL-6/ml, while TRIF-deficient cells secreted a mean of 0.56 ng/ml. *, P < 0.05 by Mann-Whitney U. (C) BMDM isolated from wild-type (WT), MyD88-deficient mice (MyD88), TRIF-deficient mice (TRIF), and MyD88 and TRIF doubly deficient mice (DKO) were stimulated with Pam3CSK4 at 1 μg/ml. For IL-6, measured by ELISA at 6 h postinfection, wild-type cells secreted a mean of 2.52 ng/ml, MyD88-deficient cells had undetectable levels (ND), TRIF-deficient cells secreted a mean of 0.47 ng/ml and DKO cells had undetectable levels. *, P < 0.05 by Mann-Whitney U. For TNF-α, measured by ELISA at 6 h postinfection, wild-type cells secreted a mean of 1.87 ng/ml, MyD88-deficient cells and DKO-deficient cells had no detectable levels, and TRIF-deficient cells secreted a mean of 0.8 ng/ml. *, P < 0.05 by Mann-Whitney U. For IL-10, measured by ELISA at 16 h postinfection, wild-type cells secreted a mean of 0.99 ng/ml, MyD88-deficient cells, and DKO-deficient cells had no detectable levels and TRIF-deficient cells secreted a mean of 0.61 ng/ml. *, P < 0.05 by Mann-Whitney U. (D) BMDM isolated from wild-type (WT), MyD88-deficient mice (MyD88), TRIF-deficient mice (TRIF) and MyD88 and TRIF doubly deficient mice (DKO) were stimulated with B. burgdorferi at an MOI of 10. For IL-6, measured by ELISA at 6 h postinfection, wild-type cells secreted a mean of 2.64 ng/ml, MyD88- and DKO-deficient cells had no detectable (ND) levels, TRIF-deficient cells secreted a mean of 0.99 ng/ml. *, P < 0.05 by Mann-Whitney U. For TNF-α, measured by ELISA at 6 h postinfection, wild-type cells secreted a mean of 1.81 ng/ml, MyD88-deficient cells and DKO-deficient cells had no detectable levels and TRIF-deficient cells secreted a mean of 0.98 ng/ml. *, P < 0.05 by Mann-Whitney U. For IL-10, measured by ELISA at 16 h postinfection, wild-type cells secreted a mean of 1.28 ng/ml, MyD88-deficient cells a mean of 0.39 ng/ml, DKO-deficient cells a mean of 0.19 ng/ml, and TRIF-deficient cells secreted a mean of 0.3 ng/ml. *, P < 0.05 by Mann-Whitney U.
To address the role of TRIF in Pam3CSK4 and B. burgdorferi signaling, we stimulated BMDM from wild-type, TRIF-, MyD88-, or TRIF/MyD88-deficient mice with B. burgdorferi at an MOI of 10 and Pam3CSK4 at 1 μg/ml. We measured secretion of IL-6, TNF-α at 6 h, and IL-10 at 16 h by ELISA (Fig. 1B, C, and D). The purity of the TLR2 synthetic ligand against lipopolysaccharide (LPS) or other contamination was confirmed by stimulating TLR2-deficient BMDM with Pam3CSK4 and showing a lack of proinflammatory and type I interferon induction (Fig. 1A), and all stimulations with ligands were performed in the presence of polymyxin B. To ensure our TRIF-deficient cells behaved as expected, we stimulated cells with LPS at 5 ng/ml and poly(I·C) at 10 μg/ml, both of which utilize the TRIF pathway (Fig. 1B). Our results show that there was a significant decrease in all inflammatory mediators tested in the TRIF-deficient cells in response to Pam3CSK4 and B. burgdorferi (Fig. 1C and D). Interestingly, MyD88-deficient cells showed no secretion or activation of inflammatory mediators in response to Pam3CSK4, suggesting that all Pam3CSK4/TLR2 signaling relied on MyD88 and thus TRIF signaling was also dependent on MyD88 (Fig. 1C).
B. burgdorferi induces TLR2-dependent type I interferons via both MyD88 and TRIF.
Recently, TLR2 signaling from the endosome has been implicated in the activation of type I interferon responses (3, 21, 23, 26). B. burgdorferi has also been shown by several groups to elicit type I interferon activation (2–4, 23, 37, 38). To determine whether TRIF plays a role in this process, we stimulated wild-type, TRIF-deficient, MyD88-deficient, and TRIF/MyD88 doubly deficient cells with Pam3CSK4 at 1 μg/ml (Fig. 2A) and B. burgdorferi at an MOI of 10 (Fig. 2B).
Fig 2.
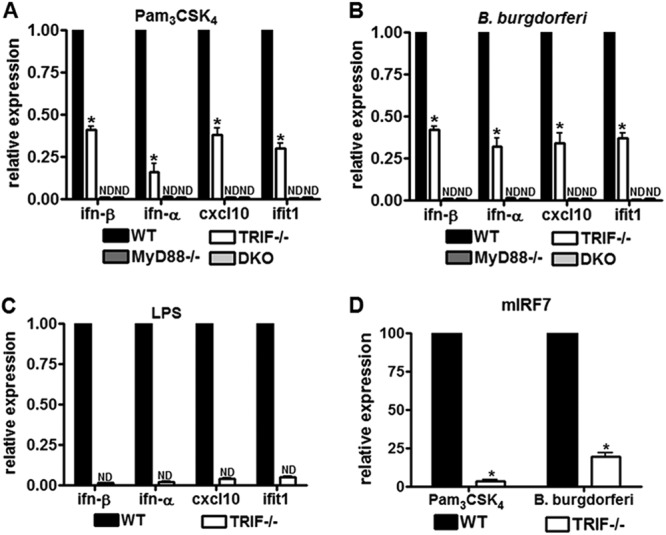
TRIF mediates activation of type I interferons in response to TLR2 ligands. (A) BMDM isolated from wild type (WT), MyD88-deficient mice (MyD88), TRIF-deficient mice (TRIF), and MyD88 and TRIF doubly deficient mice (DKO) were stimulated with 1 μg of Pam3CSK4/ml. Expression of ifn-β, cxcl10 and ifit1, measured at 6 h postinfection, and ifn-α, measured at 16 h postinfection, was determined by RT-qPCR. Values represent mean induction of expression relative to control cells and the standard errors of the mean (SEM) of three independent experiments. *, P < 0.05 by Mann-Whitney U. (B) BMDM isolated from wild type (WT), MyD88-deficient mice (MyD88), TRIF-deficient mice (TRIF), and MyD88 and TRIF doubly deficient mice (DKO) were stimulated with B. burgdorferi at an MOI of 10. Expression of ifn-β, ifn-α, cxcl10, and ifit1 was measured by RT-qPCR. Expression of ifn-β, cxcl10, and ifit1, measured at 6 h postinfection, and ifn-α, measured at 16 h postinfection, was determined by RT-qPCR. Values represent mean induction of expression relative to control cells and SEM of three independent experiments. *, P < 0.05 by Mann-Whitney U. (C) BMDM isolated from wild-type (WT) and TRIF-deficient (TRIF−/−) mice were stimulated with 5 ng of LPS/ml. Expression of ifn-β, cxcl10, and ifit1, measured at 6 h postinfection, and ifn-α, measured at 16 h postinfection, was determined by RT-qPCR. Values represent mean induction of expression relative to control cells and the SEM of three independent experiments. *, P < 0.05 by Mann-Whitney U. (D) BMDM isolated from wild-type (WT) and TRIF-deficient (TRIF) mice were stimulated with 1 μg of Pam3CSK4/ml and B. burgdorferi at an MOI of 10 for 6 h. Expression of irf7 was measured by RT-qPCR. Values represent the mean induction of expression relative to control cell and the SEM of three independent experiments. *, P < 0.05 by Mann-Whitney U.
The induction levels for the type I interferons ifn-β and ifn-α were assessed by RT-qPCR at 6 and 16 h postinfection, respectively, and TRIF pathway-associated transcripts cxcl10 and ifit-1 were also assessed by RT-qPCR at 6 h. We observed a reduction in the secretion and activation of these inflammatory mediators in TRIF-deficient BMDM in response to both stimuli, showing that TRIF is involved in type I interferon activation downstream of Pam3CSK4/TLR2 and B. burgdorferi. Similar to what we observed for the secretion of other inflammatory mediators (Fig. 1), the reduction in activation in TRIF deficient cells was partial, in contrast to type I interferon activation downstream of LPS signaling in TRIF-deficient cells (Fig. 2C).
To determine whether TRIF utilizes interferon regulatory factors (IRFs) to activate type I interferons downstream of TLR2 signaling, we evaluated expression levels of interferon regulatory factor 7 (IRF7), shown to be important in the activation of type I interferons upon TLR2 ligand stimulation (26) (Fig. 2D). Induction of IRF7 was dramatically decreased in TRIF-deficient cells upon Pam3CSK4 and B. burgdorferi stimulation, suggesting that type I interferon induction downstream of IRF7 was regulated by TRIF.
TRIF deficiency does not have a significant effect on control of B. burgdorferi infection.
To address the role of TRIF in B. burgdorferi infection in vivo, wild-type C57BL/6 and TRIF-deficient mice were subcutaneously infected with the spirochetes. Mice were sacrificed at 3 weeks postinfection, and we measured the spirochete burden in joint tissue by qPCR, which is shown as copies of the B. burgdorferi flaB gene per 1,000 copies of mouse β-actin (Fig. 3A). Our analysis showed no significant difference in the bacterial load of TRIF-deficient mice in comparison to the wild type. This suggests that TRIF, unlike MyD88, does not play a major role in control of B. burgdorferi infection.
Fig 3.

TRIF deficiency has no effect on the control of B. burgdorferi infection in vivo or B. burgdorferi phagocytosis in vitro. (A) WT (n = 10), MyD88-deficient (n = 10), TRIF-deficient (n = 10), and MyD88/TRIF doubly deficient (DKO) mice (n = 8) were infected with 105 bacteria and sacrificed at 3 weeks postinfection. Bacterial loads in joints were quantified by qPCR for the bacterial flaB gene and normalized to copies of mouse β-actin. For MyD88 and DKO mice, P < 0.0001 by Mann-Whitney U. (B) BMDM from wild-type and TRIF-deficient mice were stimulated with B. burgdorferi at an MOI of 10 for 60 min and stained by immunofluorescence as described in panel C. Percent internalized B. burgdorferi was determined by counting numbers of cells with internalized bacteria versus cells not containing internalized bacteria. The data represent the mean of internalized bacteria ± the SEM of three independent experiments. *, P < 0.05 by Mann-Whitney U. (C) Internalization of spirochetes in wild-type (WT) or TRIF-deficient cells was determined by immunofluorescent staining before (A488/green-labeled) and after (A549/red-labeled) permeabilization of the cells. Nuclei are stained with DAPI.
Role of TRIF in phagocytosis.
One possible explanation for the difference observed between TRIF- and MyD88-deficient mice in the control of infection is through the effect each molecule has on phagocytosis. MyD88 has previously been reported to play a role in phagocytosis of multiple bacteria, including B. burgdorferi (1, 11, 12, 39). We found that TRIF-deficient BMDM do not show a significant decrease in B. burgdorferi phagocytosis compared to the wild type (Fig. 3B and C). This suggests that one possible explanation for the difference in bacterial load observed in MyD88-deficient mice, but not in TRIF-deficient mice, is due to their differential effect on B. burgdorferi phagocytosis.
Effect of scavenger receptor MARCO on phagocytosis of B. burgdorferi.
To further explore the differential effect in phagocytosis of B. burgdorferi in TRIF- and MyD88-deficient cells, we examined specific receptors involved in phagocytosis. We first examined the effects of B. burgdorferi infection on the expression of different scavenger receptors from BMDM, type A (SR-A), type B (SR-B), and macrophage receptor with collagenous structure (MARCO). Of the tested scavenger receptors, MARCO showed the greatest induction after incubation with B. burgdorferi (Fig. 4A). To determine whether MARCO might have a role in phagocytosis of B. burgdorferi, we examined phagocytosis by wild-type and MARCO-deficient BMDM. MARCO-deficient BMDM were significantly impaired in their ability to internalize B. burgdorferi at 60 min (Fig. 4B and C). In vitro, MARCO expression from TRIF-deficient cells measured by real-time RT-qPCR was decreased approximately 50% in comparison to the wild type after the addition of B. burgdorferi (Fig. 4D). In contrast, MARCO expression in MyD88-deficient cells was nearly undetectable in both B. burgdorferi stimulated and unstimulated cells. This was paralleled with expression of MARCO in vivo (Fig. 4E). In the joints of TRIF-deficient mice there was no observable difference with MARCO expression in wild-type mouse joints, as opposed to MyD88-deficient mice where MARCO expression was significantly reduced. Given the role of MARCO in phagocytosis of B. burgdorferi, the effects of TRIF and MyD88 deficiency in vivo are consistent with the bacterial numbers seen in these knockout mice compared to the wild type.
Fig 4.

MyD88, but not TRIF deficiency, has an effect on expression of MARCO in vitro and in vivo and B. burgdorferi phagocytosis is impaired in MARCO-deficient mice. (A) Wild-type and MyD88-deficient BMDM were incubated with B. burgdorferi at an MOI of 10 for 5, 20, and 60 min. Cells were harvested and processed for RNA using TRIzol. RT-qPCR was performed using primers for Lox-1, SR-A, and MARCO and values represent mean induction of expression relative to wild-type unstimulated cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by Mann-Whitney U. (B) BMDM from wild-type and MARCO-deficient mice were stimulated with B. burgdorferi at an MOI of 10 for 60 min. Internalization of spirochetes was determined by immunofluorescent staining before (blue) and after (red) permeabilization of the cells. Lysosomes were stained with anti-Lamp1 antibodies (green). (C) Percent internalized B. burgdorferi was determined by counting numbers of cells with internalized bacteria versus cells not containing internalized bacteria. The data represent the means of internalized bacteria ± the SEM of three independent experiments. *, P < 0.05 by Mann-Whitney U. (D) Expression of MARCO in wild-type, TRIF-deficient, and MyD88-deficient BMDM stimulated with B. burgdorferi at an MOI of 10 was measured by RT-qPCR. Values represent mean induction of expression relative to control cells and the SEM of three independent experiments. *, P < 0.05 by Mann-Whitney U. (E) Levels of marco transcript were measured by RT-qPCR from joint tissue in wild type (n = 10), TRIF-deficient (n = 10), MyD88-deficient (n = 10), and doubly deficient (n = 9) mice. One mouse in the wild-type group was arbitrarily set to a value of 1, and all other values are shown in reference to that value using the 2−ΔΔCT method after normalization for the value of β-actin. Each circle in the data represents one mouse, and the lines indicate median values. P < 0.0001 or not significant (NS) by Mann-Whitney U.
TRIF deficiency leads to an altered cytokine profile in response to B. burgdorferi infection in vivo.
Because our TRIF knockout mice were on a C57BL/6 background, which is resistant to the development of arthritis and carditis, we could not measure the histological or phenotypic changes at these sites. However, previous studies have shown that C57BL/6 mice still have increased levels of proinflammatory cytokines in their joints after B. burgdorferi infection (33, 37, 40, 41). We determined the effects of TRIF deficiency on B. burgdorferi-induced induction of proinflammatory cytokines by measuring the levels of IL-6 and IFN-β mRNA transcripts in infected mouse joints. Mouse il-6 and ifn-β transcripts were increased in TRIF, MyD88, and doubly deficient animals compared to wild-type transcripts (P = 0.0029/P = 0.0177, P = 0.0002/P = 0.0159, and P = 0.0033) (Fig. 5A and B). This appears to be contrary to its in vitro role in inducing these two cytokines. Therefore, we wanted to examine levels of anti-inflammatory cytokines to evaluate whether they are inversed to the levels of proinflammatory mediators in the mouse joints at 3 weeks postinfection. We measured levels of il-10 transcripts in joint tissue from TRIF-, MyD88-, and TRIF/MyD88-deficient mice infected with B. burgdorferi.
Fig 5.
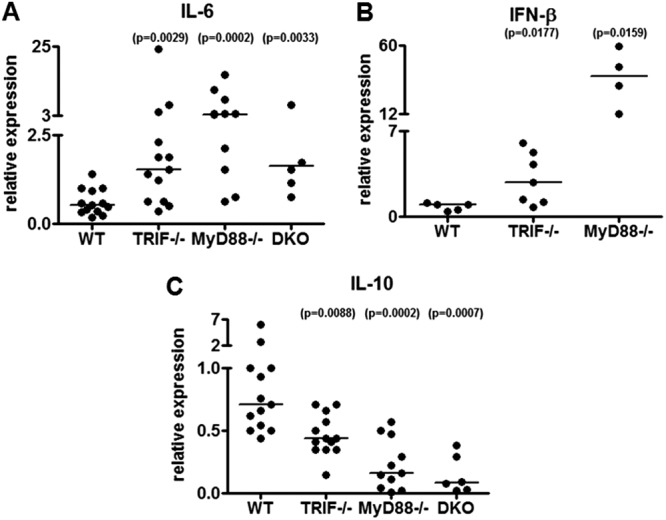
TRIF deficiency results in altered cytokine responses in vivo. (A) Levels of il-6 transcript were measured by RT-qPCR from joint tissue in wild-type (n = 13), TRIF-deficient (n = 13), MyD88-deficient (n = 12), and doubly deficient (DKO) (n = 6) mice from two independent experiments. The lowest value in the wild-type group was arbitrarily set to a value of 1, and all other values are shown in reference to that value using the 2−ΔΔCT method after normalization for the value of β-actin. Each circle in the data represents one mouse and the lines indicate median values P = 0.0029, P = 0.0002, and P = 0.0033 by Mann-Whitney U. (B) Levels of ifn-β transcript were measured by RT-qPCR from joint tissue in wild-type (WT) (n = 5), TRIF-deficient (n = 7), and MyD88-deficient (n = 4) mice. One mouse in the wild-type group was arbitrarily set to a value of 1, and all other values are shown in reference to that value using the 2−ΔΔCT method after normalization for the value of β-actin. Each circle in the data represents one mouse and the lines indicate median values. P = 0.0177 and P = 0.0159 by Mann-Whitney U. (C) The levels of il-10 transcript were measured by RT-qPCR from joint tissue in wild-type (WT) (n = 13), TRIF-deficient (n = 14), MyD88-deficient (n = 11), and doubly deficient (DKO) (n = 6) mice. One mouse in the wild-type group was arbitrarily set to a value of 1, and all other values are shown in reference to that value using the 2−ΔΔCT method after normalization for the value of β-actin. Each circle in the data represents one mouse and the lines indicate median values. P = 0.0088, P = 0.0002, and P = 0.0007 by Mann-Whitney U.
All three knockout mice showed a significant decrease in il-10 expression in comparison to wild-type mice (Fig. 5B). These data suggest that a decrease in expression and/or secretion of the anti-inflammatory IL-10 cytokine contributes to the increase in inflammatory cytokines observed in TRIF-deficient, MyD88-deficient, and doubly deficient mice.
DISCUSSION
TRIF and MyD88 are the two major adaptor molecules utilized by TLRs. All known TLRs utilize either TRIF or MyD88 or, in the case of TLR4, both. Although there is overlap in the downstream pathways and in the specific inflammatory mediators activated by stimulation through TRIF and MyD88, the two adaptors have been thought to function independently. TRIF, which transduces signals from TLR3 and TLR4, is thought to be recruited to intracellularly located TLRs and to be important in the induction of interferons and interferon-related genes. Early studies evaluating the role of TRIF in TLR2 signaling suggested a role for TRIF in the TLR2/6 lipopeptide MALP-2-dependent activation of NF-κB (17). Also, an interaction of TLR2 and TRIF in immunoprecipitation experiments with tagged proteins was observed, although another group was not able to confirm the interaction (15, 17). This was followed by studies examining stimulation of TRIF-deficient cells with peptidoglycan, another TLR2/6 ligand, which did not find a reduction in IL-12p40 proinflammatory cytokine secretion (16). When combined with studies that showed that MyD88-deficient cells did not have any residual TLR2-associated activation of most cytokines, the accepted paradigm has been that TLR2 utilizes only MyD88 as an adaptor (13, 19, 20). This view was consistent with data suggesting that TLR2 ligands, such as MALP-2, could not induce IFN-β (15). However, more recently, multiple studies, using a number of different TLR2 ligands and cell types, have found that TLR2 is indeed able to induce a type I interferon response, and it does so from endosomal compartments (3, 21, 23, 26). Given the relationship of TRIF with type I interferon responses, research has again focused at the possible role of TRIF in TLR2 signaling. A recent study by Aubry et al. showed that a TLR2/TRIF cascade was responsible for IFN-β activation downstream of Listeria monocytogenes in vitro (36). In our studies, we found that TRIF mediates signals from TLR2/1 for synthetic ligands, such as Pam3CSK4, as well as from a live pathogenic organism, B. burgdorferi. Loss of TRIF results in decreased induction of proinflammatory and anti-inflammatory molecules in response to Pam3CSK4 and B. burgdorferi. Among the reasons why our results with Pam3CSK4 may differ from early results with peptidoglycan include the possibility that TRIF is differentially utilized by TLR2/1 heterodimers compared to TLR2/6 heterodimers. Also, peptidoglycan is a complex ligand containing the TLR2/6 ligand lipoteichoic acid, as well as components shown to activate intracellular cytoplasmic receptors such as Nod1 and Nod2, and the activation of additional receptors may have masked the role of TRIF for TLR2/6 signaling (42).
For TLR4, intracellular responses to LPS, which utilize TRIF, were differentiated from plasma membrane signaling utilizing MyD88 by differences in induction of cytokines. For example, induction of TNF-α was shown to occur from the plasma membrane in response to LPS and could not be blocked by acidification inhibitors. Similarly, TRIF deficiency completely abolishes LPS induced type I interferon activation. In our studies, TRIF deficiency decreased, but did not eliminate, the induction of IL-6 and type I interferons in response to Pam3CSK4 or B. burgdorferi, in contrast to MyD88 deficiency which completely eliminated induction. Our data suggest an inter-play between TRIF and MyD88 for TLR2 signaling where induction of proinflammatory molecules by TRIF requires the presence of MyD88. Cross talk between TRIF and MyD88 signaling in the activation of inflammatory mediators has been well established; however, the mechanisms mediating this cross talk are not well understood. Some studies have suggested that the Jun N-terminal kinase (JNK) pathway and the interferon regulatory factor 5 (IRF5) are mechanisms of cross talk (43, 44). It remains unclear how the MyD88/IRF5 platform bridges the TRIF pathway or at which point in the JNK pathway the two intercept. Alternatively, MyD88 may more directly regulate the expression or localization of TRIF to intracellular compartments.
Another possible mechanism to explain the dependence of TRIF signaling on the presence of MyD88 is that loss of MyD88 results in decreased MARCO-dependent phagocytosis, which affects the localization of ligands to intracellular compartments involved in TRIF signaling. Although the defect in phagocytosis in the absence of MyD88 is partial (1), we cannot rule out the possibility that the partial loss of phagocytosis drops the amount of ligand localized to the proper compartments below the range needed to activate TRIF. Since we are unable to eliminate cell surface signaling while leaving intracellular signaling intact, it is uncertain exactly how much internalized B. burgdorferi is necessary to activate intracellular pathways.
Our findings that TRIF is necessary for induction of type I interferons downstream of B. burgdorferi are in contrast to the findings published by Miller et al. (38). While there was an effect of TRIF deficiency on IL-6 secretion in response to B. burgdorferi stimulation, they did not observe an effect on interferon-inducible gene expression in TRIF-deficient cells. Although the cause of this discrepancy is not clear, there were differences in the conditions of the trial. Miller et al. performed their studies under serum-free conditions, while we used 10% serum. The presence of serum can alter some immune responses, although it is unclear why the effect would be limited to type I interferons. Another difference includes the manner in which the TRIF-deficient mice were generated. Miller et al. used mice generated by chemical mutagenesis leading to a single base pair deletion at codon 708 and the early termination of TRIF transcripts (45), while the knockout line used in the present study is a result of a gene deletion where the entire TRIF open reading frame was replaced with a neomycin resistance cassette (16).
Although our study does not specifically show that TRIF mediates TLR2-dependent B. burgdorferi inflammatory cytokines, the similarities in the B. burgdorferi responses and those observed to TLR2-specific ligands suggest that at least a portion of the B. burgdorferi response is mediated by TLR2 and TRIF. Although other TLRs that induce type I interferons downstream of B. burgdorferi (TLR7/8 and TLR9) are believed to exclusively utilize MyD88, we cannot exclude the possibility that B. burgdorferi TRIF-mediated signaling takes place via these or other TLRs as well. Besides TLR4, TLR3 is the only other TLR that has been shown to utilize TRIF. B. burgdorferi does not activate inflammatory cytokine release through TLR4 (46). Although B. burgdorferi does not express known TLR3 ligands, the lack of activation has not been shown directly, and thus we cannot exclude the possibility of an unrecognized TLR3 ligand of B. burgdorferi.
Surprisingly, despite the role of TRIF in induction of proinflammatory mediators by TLR2 ligands in vitro, our animal model of B. burgdorferi infection revealed the opposite effect in TRIF-deficient mice with increased rather than decreased levels of many of the proinflammatory cytokines. A similar reverse phenotype has previously been observed in TLR2-, MyD88-, and CD14-deficient mice infected with B. burgdorferi (5, 6, 11, 12, 39, 47–49). Part of this response is perhaps due to higher levels of bacteria in the tissues of TLR2-, MyD88-, and CD14-deficient animals due to their important role in control of infection. While TRIF-deficient animals showed similar increases in inflammatory cytokines as TLR2- and MyD88-deficient mice, we found that TRIF was not important in control of infection and numbers of spirochetes were similar to the wild type. The reason for the difference in control of spirochete numbers between TRIF and MyD88 may be due to the role of MyD88 in phagocytosis of the organism. This may be partially mediated by the effect of MyD88 on scavenger receptors such as MARCO. MARCO, a member of a subclass of scavenger receptors type A, was shown to be important in TLR ligand internalization and signaling and for effective cytokine responses to Mycobacterium tuberculosis (50, 51). To our knowledge, MARCO and other scavenger receptors have not previously been shown to mediate phagocytosis of B. burgdorferi. We found that MARCO, which has very low levels of basal expression, was significantly induced upon B. burgdorferi stimulation in wild-type cells but not in MyD88-deficient cells. MARCO-deficient BMDM had a significant defect in the uptake of B. burgdorferi. In TRIF-deficient cells, as opposed to MyD88-deficient cells, we did not observe a complete decrease in MARCO expression, although we did observe a smaller change. It is possible that the levels of MARCO expression that remain in TRIF-deficient BMDM are sufficient to carry out phagocytosis as opposed to MyD88 where the expression of MARCO is abolished. Expression of scavenger receptors has been shown to be regulated by TLR signals and TNF-α (52–54); thus, the partial defect in TNF-α secretion observed in TRIF-deficient cells versus the complete abrogation observed in MyD88-deficient cells correlates with the differences observed in MARCO expression in TRIF- and MyD88-deficient cells. Interestingly, the in vivo expression of MARCO in TRIF-deficient mouse joints was not statistically different from the expression observed in joints of wild-type mice. Differences in the effect of TRIF deficiency in vivo versus in vitro may be due to the cell types tested or to differences in responses between naive BMDM in vitro and activated cells in vivo. Taken together, our data suggest a biological relevance in MARCO expression controlled by MyD88 versus TRIF. This lack of a role in phagocytosis of TLR2/TRIF signaling, as opposed to TLR2/MyD88 signaling, may explain its lack of importance in the control of B. burgdorferi infection.
In our mouse studies, TRIF deficiency results in an increase in proinflammatory cytokines in the joint, which, unlike in the case of MyD88, CD14, or TLR2 deficiency, cannot be explained by an increase in bacterial numbers in vivo. Although it is counterintuitive that deficiencies in pathways important in proinflammatory responses result in an increase in expression of inflammatory mediators, this effect has been seen with multiple innate immune receptors in response to B. burgdorferi (5, 11, 39, 55). There are many potential explanations for why this may occur. In studies of TLRs in other systems, they have been involved in induction of anti-inflammatory mediators, downregulation of inflammatory receptors and activation of suppressor pathways, all of which could result in increased release of inflammatory mediators in the absence of the receptor (56–59). Many of these effects have been seen with prolonged stimulation of TLRs which fits well with the model of B. burgdorferi causing long-term infection. Further studies to determine the nature of the changes in the inflammatory response during long-term infections could contribute to our understanding of these phenotypes.
ACKNOWLEDGMENTS
We thank Erin Troy, Mekki Bensaci, Elizabeth Tenorio, Brian Klein, Chari Cortez, and Debaditya Bhattacharya for helpful discussions and critical reading of the manuscript.
This study was supported by R01AI071107 (L.T.H.), R56AI80646 (L.T.H.), R21AI097971 (L.T.H.), and T32AI007329 (T.P.O.).
Footnotes
Published ahead of print 19 November 2012
REFERENCES
- 1. Shin OS, Isberg RR, Akira S, Uematsu S, Behera AK, Hu LT. 2008. Distinct roles for MyD88 and Toll-like receptors 2, 5, and 9 in phagocytosis of Borrelia burgdorferi and cytokine induction. Infect. Immun. 76:2341–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Salazar JC, Duhnam-Ems S, La Vake C, Cruz AR, Moore MW, Caimano MJ, Velez-Climent L, Shupe J, Krueger W, Radolf JD. 2009. Activation of human monocytes by live Borrelia burgdorferi generates TLR2-dependent and -independent responses which include induction of IFN-β. PLoS Pathog. 5:e1000444 doi:10.1371/journal.ppat.1000444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cervantes JL, Dunham-Ems SM, La Vake CJ, Petzke MM, Sahay B, Sellati TJ, Radolf JD, Salazar JC. 2011. Phagosomal signaling by Borrelia burgdorferi in human monocytes involves Toll-like receptor (TLR) 2 and TLR8 cooperativity and TLR8-mediated induction of IFN-beta. Proc. Natl. Acad. Sci. U. S. A. 108:3683–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Petzke MM, Brooks A, Krupna MA, Mordue D, Schwartz I. 2009. Recognition of Borrelia burgdorferi, the Lyme disease spirochete, by TLR7 and TLR9 induces a type I IFN response by human immune cells. J. Immunol. 183:5279–5292 [DOI] [PubMed] [Google Scholar]
- 5. Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, Weis JJ. 2002. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J. Immunol. 168:348–355 [DOI] [PubMed] [Google Scholar]
- 6. Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, Weis JJ. 2002. Toll-like receptor 2 plays a pivotal role in host defense and inflammatory response to Borrelia burgdorferi. Vector Borne Zoonotic Dis. 2:275–278 [DOI] [PubMed] [Google Scholar]
- 7. Hirschfeld M, Kirschning CJ, Schwandner R, Wesche H, Weis JH, Wooten RM, Weis JJ. 1999. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by Toll-like receptor 2. J. Immunol. 163:2382–2386 [PubMed] [Google Scholar]
- 8. Fikrig E, Narasimhan S, Neelakanta G, Pal U, Chen M, Flavell R. 2009. Toll-like receptors 1 and 2 heterodimers alter Borrelia burgdorferi gene expression in mice and ticks. J. Infect. Dis. 200:1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cassiani-Ingoni R, Cabral ES, Lunemann JD, Garza Z, Magnus T, Gelderblom H, Munson PJ, Marques A, Martin R. 2006. Borrelia burgdorferi induces TLR1 and TLR2 in human microglia and peripheral blood monocytes but differentially regulates HLA-class II expression. J. Neuropathol. Exp. Neurol. 65:540–548 [DOI] [PubMed] [Google Scholar]
- 10. Alexopoulou L, Thomas V, Schnare M, Lobet Y, Anguita J, Schoen RT, Medzhitov R, Fikrig E, Flavell RA. 2002. Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat. Med. 8:878–884 [DOI] [PubMed] [Google Scholar]
- 11. Bolz DD, Sundsbak RS, Ma Y, Akira S, Kirschning CJ, Zachary JF, Weis JH, Weis JJ. 2004. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J. Immunol. 173:2003–2010 [DOI] [PubMed] [Google Scholar]
- 12. Liu N, Montgomery RR, Barthold SW, Bockenstedt LK. 2004. Myeloid differentiation antigen 88 deficiency impairs pathogen clearance but does not alter inflammation in Borrelia burgdorferi-infected mice. Infect. Immun. 72:3195–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650 [DOI] [PubMed] [Google Scholar]
- 14. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. 2008. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 9:361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 4:161–167 [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. 2003. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301:640–643 [DOI] [PubMed] [Google Scholar]
- 17. Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 169:6668–6672 [DOI] [PubMed] [Google Scholar]
- 18. Kawai T, Akira S. 2009. The roles of TLRs, RLRs, and NLRs in pathogen recognition. Int. Immunol. 21:317–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. 2002. TLR4, but not TLR2, mediates IFN-β-induced STAT1α/β-dependent gene expression in macrophages. Nat. Immunol. 3:392–398 [DOI] [PubMed] [Google Scholar]
- 20. Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, Muhlradt PF, Akira S. 2000. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a Toll-like receptor 2- and MyD88-dependent signaling pathway. J. Immunol. 164:554–557 [DOI] [PubMed] [Google Scholar]
- 21. Barbalat R, Lau L, Locksley RM, Barton GM. 2009. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 10:1200–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dieterich R, Hammerschmidt C, Richter D, Skerka C, Wallich R, Matuschka FR, Zipfel PF, Kraiczy P. 2010. Inadequate binding of immune regulator factor H is associated with sensitivity of Borrelia lusitaniae to human complement. Infect. Immun. 78:4467–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marre ML, Petnicki-Ocwieja T, Defrancesco AS, Darcy CT, Hu LT. 2010. Human integrin α3β1 regulates TLR2 recognition of lipopeptides from endosomal compartments. PLoS One 5:e12871 doi:10.1371/journal.pone.0012871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ip WK, Sokolovska A, Charriere GM, Boyer L, Dejardin S, Cappillino MP, Yantosca LM, Takahashi K, Moore KJ, Lacy-Hulbert A, Stuart LM. 2010. Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J. Immunol. 184:7071–7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ip WKE, Takahashi K, Moore KJ, Stuart LM, Ezekowitz RAB. 2008. Mannose-binding lectin enhances Toll-like receptors 2 and 6 signaling from the phagosome. J. Exp. Med. 205:169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dietrich N, Lienenklaus S, Weiss S, Gekara NO. 2010. Murine Toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS One 5:e10250 doi:10.1371/journal.pone.0010250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shin OS, Miller LS, Modlin RL, Akira S, Uematsu S, Hu LT. 2009. Downstream signals for MyD88-mediated phagocytosis of Borrelia burgdorferi can be initiated by TRIF and are dependent on PI3K. J. Immunol. 183:491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cruz AR, Moore MW, La Vake CJ, Eggers CH, Salazar JC, Radolf JD. 2008. Phagocytosis of Borrelia burgdorferi, the Lyme disease spirochete, potentiates innate immune activation and induces apoptosis in human monocytes. Infect. Immun. 76:56–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moore MW, Cruz AR, LaVake CJ, Marzo AL, Eggers CH, Salazar JC, Radolf JD. 2007. Phagocytosis of Borrelia burgdorferi and Treponema pallidum potentiates innate immune activation and induces gamma interferon production. Infect. Immun. 75:2046–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adachi O, Kawai T, Takeda M, Matsumoto M, Tsutsui H, Sakagami K, Nakanishi K, Akira S. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143. [DOI] [PubMed] [Google Scholar]
- 31. Ghosh S, Gregory D, Smith A, Kobzik L. 2011. MARCO regulates early inflammatory responses against influenza: a useful macrophage function with adverse outcome. Am. J. Respir. Cell Mol. Biol. doi:10.1165/rcmb.2010-0349OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Purser JE, Norris SJ. 2000. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. U. S. A. 97:13865–13870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Petnicki-Ocwieja T, DeFrancesco AS, Chung E, Darcy CT, Bronson RT, Kobayashi KS, Hu LT. 2011. Nod2 suppresses Borrelia burgdorferi mediated murine Lyme arthritis and carditis through the induction of tolerance. PLoS One 6:e17414 doi:10.1371/journal.pone.0017414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marie I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li X, Liu X, Beck DS, Kantor FS, Fikrig E. 2006. Borrelia burgdorferi lacking BBK32, a fibronectin-binding protein, retains full pathogenicity. Infect. Immun. 74:3305–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aubry C, Corr SC, Wienerroither S, Goulard C, Jones R, Jamieson AM, Decker T, O'Neill LA, Dussurget O, Cossart P. 2012. Both TLR2 and TRIF contribute to interferon-beta production during Listeria infection. PLoS One 7:e33299 doi:10.1371/journal.pone.0033299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller JC, Ma Y, Bian J, Sheehan KC, Zachary JF, Weis JH, Schreiber RD, Weis JJ. 2008. A critical role for type I IFN in arthritis development following Borrelia burgdorferi infection of mice. J. Immunol. 181:8492–8503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller JC, Maylor-Hagen H, Ma Y, Weis JH, Weis JJ. 2010. The Lyme disease spirochete Borrelia burgdorferi utilizes multiple ligands, including RNA, for interferon regulatory factor 3-dependent induction of type I interferon-responsive genes. Infect. Immun. 78:3144–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Behera AK, Hildebrand E, Bronson RT, Perides G, Uematsu S, Akira S, Hu LT. 2006. MyD88 deficiency results in tissue-specific changes in cytokine induction and inflammation in interleukin-18-independent mice infected with Borrelia burgdorferi. Infect. Immun. 74:1462–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oosting M, van de Veerdonk FL, Kanneganti TD, Sturm P, Verschueren I, Berende A, van der Meer JW, Kullberg BJ, Netea MG, Joosten LA. 2011. Borrelia species induce inflammasome activation and IL-17 production through a caspase-1-dependent mechanism. Eur. J. Immunol. 41:172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu N, Belperron AA, Booth CJ, Bockenstedt LK. 2009. The caspase 1 inflammasome is not required for control of murine Lyme borreliosis. Infect. Immun. 77:3320–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS. 2009. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. U. S. A. 106:15813–15818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhu Q, Egelston C, Vivekanandhan A, Uematsu S, Akira S, Klinman DM, Belyakov IM, Berzofsky JA. 2008. Toll-like receptor ligands synergize through distinct dendritic cell pathways to induce T cell responses: implications for vaccines. Proc. Natl. Acad. Sci. U. S. A. 105:16260–16265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ouyang X, Negishi H, Takeda R, Fujita Y, Taniguchi T, Honda K. 2007. Cooperation between MyD88 and TRIF pathways in TLR synergy via IRF5 activation. Biochem. Biophys. Res. Commun. 354:1045–1051 [DOI] [PubMed] [Google Scholar]
- 45. Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. 2003. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424:743–748 [DOI] [PubMed] [Google Scholar]
- 46. Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD, Golenbock DT. 1999. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 274:33419–33425 [DOI] [PubMed] [Google Scholar]
- 47. Behera AK, Hildebrand E, Uematsu S, Akira S, Coburn J, Hu LT. 2006. Identification of a TLR-independent pathway for Borrelia burgdorferi-induced expression of matrix metalloproteinases and inflammatory mediators through binding to integrin alpha 3 beta 1. J. Immunol. 177:657–664 [DOI] [PubMed] [Google Scholar]
- 48. Sahay B, Patsey RL, Eggers CH, Salazar JC, Radolf JD, Sellati TJ. 2009. CD14 signaling restrains chronic inflammation through induction of p38-MAPK/SOCS-dependent tolerance. PLoS Pathog. 5:e1000687 doi:10.1371/journal.ppat.1000687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X, Ma Y, Yoder A, Crandall H, Zachary JF, Fujinami RS, Weis JH, Weis JJ. 2008. T cell infiltration is associated with increased Lyme arthritis in TLR2−/− mice. FEMS Immunol. Med. Microbiol. 52:124–133 [DOI] [PubMed] [Google Scholar]
- 50. Mukhopadhyay S, Varin A, Chen Y, Liu B, Tryggvason K, Gordon S. 2011. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood 117:1319–1328 [DOI] [PubMed] [Google Scholar]
- 51. Bowdish DM, Sakamoto K, Kim MJ, Kroos M, Mukhopadhyay S, Leifer CA, Tryggvason K, Gordon S, Russell DG. 2009. MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathog. 5:e1000474 doi:10.1371/journal.ppat.1000474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hsu HY, Twu YC. 2000. Tumor necrosis factor-alpha-mediated protein kinases in regulation of scavenger receptor and foam cell formation on macrophage. J. Biol. Chem. 275:41035–41048 [DOI] [PubMed] [Google Scholar]
- 53. Hsu HY, Nicholson AC, Hajjar DP. 1996. Inhibition of macrophage scavenger receptor activity by tumor necrosis factor-alpha is transcriptionally and posttranscriptionally regulated. J. Biol. Chem. 271:7767–7773 [DOI] [PubMed] [Google Scholar]
- 54. Zamora C, Canto E, Nieto JC, Angels Ortiz M, Juarez C, Vidal S. 2012. Functional consequences of CD36 downregulation by TLR signals. Cytokine doi:10.1016/j.cyto.2012.06.020 [DOI] [PubMed] [Google Scholar]
- 55. Sahay B, Singh A, Gnanamani A, Patsey RL, Blalock JE, Sellati TJ. 2011. CD14 signaling reciprocally controls collagen deposition and turnover to regulate the development of Lyme arthritis. Am. J. Pathol. 178:724–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Biswas SK, Lopez-Collazo E. 2009. Endotoxin tolerance: new mechanisms, molecules, and clinical significance. Trends Immunol. 30:475–487 [DOI] [PubMed] [Google Scholar]
- 57. Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. 1999. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J. Exp. Med. 189:615–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115–122 [DOI] [PubMed] [Google Scholar]
- 59. Kobayashi K, Hernandez LD, Galan JE, Jr, Medzhitov R, Flavell RA. 2002. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110:191–202 [DOI] [PubMed] [Google Scholar]