

Abstract
Histoplasma capsulatum is a fungal respiratory pathogen that survives and replicates within the phagolysosome of macrophages. The molecular factors it utilizes to subvert macrophage antimicrobial defenses are largely unknown. Although the ability of H. capsulatum to prevent acidification of the macrophage phagolysosome is thought to be critical for intracellular survival, this hypothesis has not been tested since H. capsulatum mutants that experience decreased phagosomal pH have not been identified. In a screen to identify H. capsulatum genes required for lysis of bone marrow-derived macrophages (BMDMs), we identified an insertion mutation disrupting the H. capsulatum homolog of 3-hydroxy-methylglutaryl coenzyme A (HMG CoA) lyase (HCL1). In addition to its inability to lyse macrophages, the hcl1 mutant had a severe growth defect in BMDMs, indicating that HMG CoA lyase gene function is critical for macrophage colonization. In other organisms, HMG CoA lyase catalyzes the last step in the leucine catabolism pathway. In addition, both fungi and humans deficient in HMG CoA lyase accumulate acidic intermediates as a consequence of their inability to catabolize leucine. Consistent with observations in other organisms, the H. capsulatum hcl1 mutant was unable to grow on leucine as the major carbon source, caused acidification of its growth medium in vitro, and resided in an acidified vacuole within macrophages. Mice infected with the hcl1 mutant took significantly longer to succumb to infection than mice infected with the wild-type strain. Taken together, these data indicate the importance of Hcl1 function in H. capsulatum replication in the harsh growth environment of the macrophage phagosome.
INTRODUCTION
Macrophages possess an arsenal of antimicrobial defenses that are designed to neutralize and exterminate invading microbes (reviewed in reference 1). During phagocytosis by macrophages, microbes are exposed to highly reactive and toxic radicals. As the phagosome matures, it progressively acidifies due to the activity of host vacuolar ATPases that pump protons into the phagosome. Ultimately, the phagosome fuses with the lysosome, allowing maximal acquisition of lysosomal hydrolases in the resultant compartment. In addition, the phagosome is thought to be a glucose-poor environment, necessitating that phagosomal pathogens utilize alternate nutrient acquisition pathways to survive intracellularly. In the present study, we examine the role of a metabolic enzyme, 3-hydroxy-methylglutaryl coenzyme A (HMG CoA) lyase, in promoting the survival and replication of the fungal pathogen Histoplasma capsulatum within the macrophage phagosome.
H. capsulatum is a dimorphic intracellular fungal pathogen that parasitizes macrophages. Endemic to the Ohio and Mississippi Valley regions of the United States, H. capsulatum is one of the most common causes of fungal respiratory infection, with up to 500,000 new cases arising each year (2). Infection is initiated when mammalian hosts inhale infectious spores and mycelial fragments that have aerosolized from the soil. Upon entering the alveolar space of the lung, these infectious particles undergo a morphological conversion to budding yeast cells, which parasitize alveolar macrophages. Remarkably, these yeast cells are able to survive and replicate to high levels intracellularly, ultimately lysing host immune cells.
Much of the previous work examining the interactions between H. capsulatum yeast cells and the host has characterized fungal survival strategies that promote colonization of its intracellular niche. For example, H. capsulatum yeast cells fail to stimulate the production of toxic reactive oxygen species during phagocytosis by resting murine macrophages (3). The phagolysosome containing H. capsulatum does not acidify and instead remains at a near-neutral pH of 6.5 (4, 5). Presumably, lysosomal hydrolyases are not maximally active at this pH, allowing survival and growth of H. capsulatum within the phagolysosome. In addition, the near-neutral pH of the H. capsulatum phagolysosome is thought to promote release of iron from host transferrin, thereby facilitating iron acquisition by the pathogen during its intracellular growth (6, 7).
To identify molecular factors that mediate H. capsulatum survival, growth, and pathogenesis, we performed an unbiased genetic screen for H. capsulatum insertion mutants that were unable to lyse host macrophages. One of the lysis-defective mutants identified in this screen contained an insertion in an H. capsulatum gene that encodes the metabolic enzyme HMG CoA lyase. This protein has been studied in Pseudomonas species, Aspergillus species, and humans and is known to catalyze the last step in leucine catabolism. Here, we show that the H. capsulatum HMG CoA lyase (HCL1) is required for full virulence of H. capsulatum in host macrophages and in mice.
MATERIALS AND METHODS
Strains and culture conditions.
A. tumefaciens strain LBA1100 was kindly provided by Thomas Sullivan and Bruce Klein with permission from Paul Hooykas (Leiden University, Leiden, Netherlands). H. capsulatum strain G217B ura5Δ (WU15) was kindly provided by William Goldman (University of North Carolina, Chapel Hill). The wild-type, hcl1 mutant, and complemented strains were generated in the present study as described below in Materials and Methods. Yeast cells were grown in either Histoplasma macrophage medium (HMM) or minimal medium (3M) (8). Liquid cultures were grown in an orbital shaker at 37°C with 5% CO2. Stock cultures were maintained by passaging every 2 to 3 days with a 1:25 dilution. HMM agarose or 3M agarose plates were incubated in a humidified chamber at 37°C with 5% CO2.
For both macrophage and mouse infections, an overnight, mid-log culture of yeast cells (optical density at 600 nm [OD600] = 5 to 7) was prepared. Approximately 18 h prior to the infection, a 2-day late log/stationary phase culture (OD600 = 10 to 12) was diluted 1:5 into HMM media. The diluted cells were then incubated at 37°C with 5% CO2 overnight to obtain mid-log cultures at the time of infection. Culture ODs were measured using an Eppendorf BioPhotometer.
Genetic screen of H. capsulatum lysis defective mutants.
Mutant FE6-C3 (the hcl1 mutant) was initially identified in a genetic screen for mutants that did not lyse macrophages (D. T. Isaac et al., unpublished data). Briefly, H. capsulatum insertion mutants were generated using Agrobacterium-mediated transformation of G217B ura5Δ (WU15) as previously described (9). We selected for hygromycin-resistant transformants on HMM plates containing 400 μg of uracil/ml, 200 μg of hygromycin B/ml, and 200 μM cefotaxime. A total of 14,000 insertion mutants were screened for their ability to lyse BMDMs and J774.1 macrophages. Forty-seven mutants were defective for macrophage lysis, including mutant FE6-C3.
Complementation of the hcl1 mutant.
pAC04, an integrating complementation plasmid, was generated. This complementation construct contained the HCL1 open reading frame (ORF), 900 bp of 5′ flanking sequence, and 886 bp of 3′ flanking sequence. pAC04 was transformed into the hcl1 mutant using Agrobacterium-mediated gene transfer as previously described (9), generating the hcl1+HCL1 strain that is designated as the “complemented strain” for the experiments described here. Briefly, the A. tumefaciens strain LBA1100 was transformed with pAC04 and induced overnight with 200 μM acetosyringone (AS; Sigma-Aldrich). H. capsulatum yeast cells were harvested from 4-day patches on HMM+uracil agarose plates and diluted to 5 × 108 cells/ml. Equal volumes of the H. capsulatum and A. tumefaciens cultures were mixed, and 400 μl of the mix was spread onto Biodyne A nylon membranes on IM agarose plates containing 200 μM AS and 200 μg of uracil/ml. Cocultivation plates were incubated at 28°C for 3 days. The membranes were then transferred onto HMM plates with no added uracil and incubated at 37°C for 2 to 3 weeks. The hcl1 mutant was also transformed with pVN47, the parental plasmid for pAC04, which contains the URA5 gene but does not carry HCL1. The resultant strain is designated as the “hcl1 mutant” for the experiments described in the present study. The G217B ura5Δ parental strain, WU15, was also transformed with pVN47, generating a Ura+ wild-type control, which is the strain designated as “wild-type” for the experiments described in this study. Several wild-type, mutant, and complemented transformants were analyzed in each experiment, but only one representative strain is shown.
HCL1 expression analysis. (i) Northern blot.
Total RNA was isolated from logarithmically growing yeast cells using a guanidinium thiocyanate lysis protocol as previously described (10). A total of 4 μg of total RNA from all strains was separated on a 1.5% formaldehyde agarose gel and transferred to a positively charged nylon membrane (Roche 11209299001). The membrane was then subjected to Northern blot analysis according to the digoxigenin (DIG) Northern blot protocol (Roche Applied Sciences). DIG-labeled probes were generated from WU15 genomic DNA using the PCR DIG probe synthesis kit (Roche, catalog no. 11636090910). The membrane was then exposed to film and developed.
(ii) Quantitative reverse transcription-PCR (qRT-PCR).
Total RNA was treated with DNase I (Promega) and cDNA was synthesized using Stratascript reverse transcriptase (Stratagene) and oligo(dT). Quantitative PCR was performed using SYBR green PCR Master Mix (Applied Biosystems) and 200 nM primer on the Mx3000P QPCR system (Stratagene) with Comparative Quantitation program, using actin (ACT1) as the normalizing transcript. Primer sequences used are as follows: HCL1 (HCL1 sense, 5′-AAGACGGGATTGACCACGATTGAG; HCL1 antisense, 5′-ATGCGATCGAGTGAGATGACTTGG) and ACT1 (ACT1 sense, 5′-GAAGGAGATTACCGCTCTCG; and ACT1 antisense, 5′-CGACAACAACGAAAACCTTAGA). All primers spanned an exon-exon boundary.
Cytotoxicity/LDH release assay.
Bone marrow-derived macrophages (BMDMs) were isolated as described previously (11). In 24-well tissue-culture treated dishes, 2 × 105 BMDMs were infected, in duplicate, with H. capsulatum strains at a multiplicity of infection (MOI) = 2 yeasts per macrophage. In preparation for the infections, logarithmically growing H. capsulatum cultures were pelleted, resuspended in Dulbecco modified Eagle medium (DMEM) without phenol red, sonicated for 3 s on setting 2 using a Fisher Scientific Sonic Dismembrator Model 100, and counted by hemacytometer. The colony-forming units (CFU) were enumerated before and after sonication to confirm that sonication did not affect yeast-cell viability (data not shown). After a 2-h incubation period, the medium was removed from the infected BMDMs and the monolayers were washed twice with DMEM without phenol red. A total of 750 μl of bone marrow macrophage medium (BMM) without phenol red (consisting of DMEM high glucose without phenol red (UCSF Cell Culture Facility), 20% fetal bovine serum (FBS), 10% (vol/vol) CMG supernatant (conditioned medium obtained from CMG cells [3T3 fibroblasts transfected with the murine CSF-1 cDNA, kindly provided by Mary Nakamura, UCSF]), 2 mM glutamine, and 110 mg of sodium pyruvate, penicillin, and streptomycin/ml) was added to each well. The infected macrophages were then incubated at 37°C with 5% CO2. Approximately 48 h postinfection (hpi), 250 μl of fresh BMM was added to each well. At various time points postinfection, lactate dehydrogenase (LDH) levels in the infected-macrophage supernatants were measured to monitor BMDM lysis. At each time point, the volume in each well was brought up to 1 ml with BMM. A total of 200 μl of the culture supernatant from the infected macrophages was transferred to two wells of a 96-well plate. To measure the total LDH, two mock-infected BMDM monolayers were lysed at 2 hpi with 1 ml of lysis solution (1% Triton X-100 in DMEM without phenol red). A total of 200 μl of this mock-infected macrophage lysate was transferred to two wells of the 96-well plate, which was then centrifuged to pellet any cells or debris that might be present in the supernatants or lysate. A total of 20 μl of the clarified culture supernatant was transferred to a fresh 96-well plate containing 30 μl of DMEM-phenol red in each well. A total of 60 μl of complete LDH solution was then added to each well. Complete LDH solution is made from equal volumes of the following solutions: (i) 2 mg of INT (IodoNitroTetrazolium chloride)/ml in phosphate-buffered saline (PBS), (ii) 36 mg of lithium l-lactate/ml in 10 mM Tris (pH 8.5), and (iii) 1× NAD+/diaphorase in PBS containing 1% bovine serum albumin (BSA), diluted from a 10× stock solution (13.5 U of diaphorase/ml, 3 mg of NAD+/ml, 0.03% BSA, and 1.2% sucrose in PBS). The plate was then incubated for 30 min in the dark. A total of 40 μl of 1 M acetic acid was added to stop the reaction. The OD490 was then measured using a Molecular Devices Spectramax Plus 384 plate reader. The percent BMDM lysis at each time point was calculated as the percentage of the total LDH from lysed uninfected cells at 2 hpi. Since wells of infected macrophages also contain uninfected BMDMs that continue to replicate over the course of the experiment, the total LDH at later time points is greater than the total LDH from the 2-h time point. This increase in total LDH results in an apparent percent lysis that is >100% when normalized to the total LDH from the 2-h time point. We confirmed that H. capsulatum G217B cells did not give rise to any LDH activity when subjected to this LDH release assay (data not shown).
Intracellular replication assay.
In 24-well tissue culture treated dishes, 2 × 105 BMDMs were infected, in duplicate, with H. capsulatum strains at an MOI of 2. After a 2-h incubation period, the culture supernatants were removed, the monolayers were washed twice with DMEM and then 1 ml of BMM was added to each well. The infected macrophages were then incubated at 37°C with 5% CO2. The medium was changed at 48 hpi and every day thereafter. At various time points postinfection, the medium was removed from each well and 500 μl of double-distilled H2O was added. After a 5-min incubation at room temperature, the infected macrophages were mechanically lysed by vigorous pipetting. The lysate was collected, sonicated, diluted in HMM, and plated for H. capsulatum CFU on HMM-agarose plates at 37°C. CFU were counted 14 days later.
Microscopic analysis of intracellular growth.
A total of 2 × 105 BMDMs per well were seeded in 24-well tissue-culture treated dishes containing 12-mm glass coverslips. Approximately 18 h later, these BMDMs were infected with yeast cells at an MOI of 0.1, centrifuged for 5 min at 800 × g, and incubated for 2 h. The media was then removed from the infected BMDMs, the monolayers were washed twice with DMEM, and 500 μl of BMM was added to each well. The infected macrophages were then incubated at 37°C with 5% CO2. The media was changed at 48 hpi and every day thereafter. At various time points postinfection the medium was removed from each well and the monolayers were fixed with 1 ml of 3.7% formaldehyde in 100% ethanol for 1 min. The fixative was then removed, and the cells were washed twice with PBS before being stored in 1 ml of PBS at 4°C. Coverslips were then stained with periodic acid-Schiff reagent (to highlight fungal cells) and methyl green (to counterstain macrophage nuclei) as previously described (12). Microscopic images were taken with the Leica DM 1000 microscope.
Bioinformatics analysis.
The protein sequence of the predicted gene that was disrupted in mutant FE6-C3 was searched against the nonredundant (nr) protein database with the BLASTP program (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (13). Upon identification as an HMG CoA lyase homolog, the corresponding H. capsulatum protein, designated Hcl1, was aligned using CLUSTAL W2 (http://www.clustal.org/clustal2/) (14) with its homologs in Aspergillus fumigatus, Pseudomonas aeruginosa, Pseudomonas mevalonii, Bacillus subtilis, Brucella melitensis, and Homo sapiens.
Analysis of growth in leucine.
To monitor growth in the presence of leucine on solid medium, stationary-phase yeast cells from a 4-day culture were washed and resuspended in PBS. Portions (2 μl) of 10-fold serial dilutions, in PBS, were plated on agarose plates containing either HMM, 3M with glucose, or 3M without glucose supplemented with 10 mM leucine. Plates were then incubated at 37°C and 5% CO2 for 16 days. Images were taken with a Nikon digital camera. To monitor growth in liquid culture, yeast cells were washed, pelleted, and resuspended to an OD600 of 0.01 to 0.05 in either HMM, 3M (with glucose), 3M-glu, or 3M-glucose+leucine. Samples (500 μl) were removed from each culture, vortexed for 30 s to break up clumps, and analyzed to determine their OD600 using the Molecular Devices Spectramax Plus 384 plate reader. Of note, as cultures go from early logarithmic growth to stationary phase, an increase in OD600 is observed with both the Spectramax plate reader and the Eppendorf Biophotometer described above. However, the absolute OD600 numbers differ for the two instruments, with stationary phase registering as an OD of 12 on the Biophotometer and an OD of approximately 2 to 3 on the plate reader.
In vitro pH modulation.
To monitor the ability of the H. capsulatum strains to modulate the pH of culture broth, stationary-phase yeast cells from a 4-day culture were washed and resuspended in PBS. Cells were diluted to an OD600 of 2 in 30 ml of pH-HMM (HMM without HEPES, containing 40 μg of bromocresol purple/ml [pH 4.5]). Cultures were incubated at 37°C with 5% CO2 in an orbital shaker. At each time point, three 1-ml samples were removed from each culture, and the pH was measured using a digital pH meter. The OD of each culture was measured using the Molecular Devices Spectramax Plus 384 plate reader.
Monitoring of phagosomal pH.
Acidification of the phagosome was assessed by use of the acidotropic dye LysoSensor Green DND-189 (Invitrogen; pKa = 5.2). In a 12-well plate, 2 × 105 BMDMs per well were infected in duplicate with mid-logarithm-phase H. capsulatum strains (wild-type, the hcl1 mutant, the complemented strain, or fixed wild-type cells [the wild-type strain fixed with 4% paraformaldehyde]) at an MOI of 2 in the presence or absence of 50 nM bafilomycin A1 (Sigma), an inhibitor of the vacuolar ATPase. Cells were slowly centrifuged at 1,200 rpm for 5 min at room temperature. At 1 hpi, extracellular yeast cells were removed by washing. At 30 min prior to each time point (3, 6, 12, and 24 hpi), infected BMDMs were loaded with LysoSensor (1 μM) at 37°C. After this 30-min incubation, macrophages were washed with PBS and fixed in 4% paraformaldehyde for 30 min. Fixed macrophages were blocked with 2% FBS and 0.2% saponin in PBS for 1 h at room temperature and then stained at 37°C for 1 h with rabbit anti-Hc (a kind gift from Joe Wheat), which detects H. capsulatum yeast cells, and rat anti-LAMP-1 (Invitrogen, catalog no. 1D4B). The cells were then washed and stained with rabbit Alexa Fluor 405 and rat Alexa Fluor 546 secondary antibodies for 1 h at 37°C. Cells were mounted in Gel-Mount (Biomedia) and imaged using a Nikon TiE inverted microscope with Yokogawa spinning disk CSU-X1. Images were taken using Nikon Perfect Focus and unbiased gridding of 50 points per samples of 0.11 μm/pixel. Images were brightened in Adobe Photoshop by applying the identical adjustments to all images.
Mouse infections.
Nine-week-old female C57BL/6 mice (Charles River Laboratories) were anesthetized with isoflurane and infected intranasally with wild-type H. capsulatum, the hcl1 mutant, or the complemented stain. In preparation for infection, mid-logarithmic-phase cultures of these H. capsulatum strains were washed once with PBS, sonicated for 3 s on setting 2 using a Fisher Scientific Sonic Dismembrator Model 100, and counted by using a hemacytometer to determine the cell concentration. The CFU before and after sonication were enumerated to determine that sonication did not affect yeast cell viability (data not shown). To monitor mouse survival, 15 mice were infected intranasally with 1.25 × 106 yeast cells in approximately 25 μl of PBS. At 4 hpi, the lungs were harvested and homogenized from 5 mice infected with each strain. These homogenates were plated for H. capsulatum CFU on brain-heart infusion agar (BHI) plates and incubated at 30°C. The remaining mice were monitored daily for symptoms of disease (i.e., weight loss, lack of activity/response to stimulus, panting, lack of grooming). Mice were sacrificed after they exhibited 3 days of sustained weight loss >15% of their maximum weight in conjunction with one other symptom of disease. To determine the fungal burden of infected mice when they succumbed to the infection, lungs and spleens were harvested from four mice infected with each H. capsulatum strain. In addition, lungs and spleens were harvested from three mice infected with the hcl1 mutant strain at 7 days postinfection to determine the fungal burden. Lung homogenate was plated for CFU on BHI plates at 30°C. Statistical analysis for survival and colonization experiments was performed using Prism (GraphPad Software, San Diego, CA). Survival curves were compared using the log rank (Mantel-Cox) test. The in vivo fungal burden of different H. capsulatum strains was compared using the Mann-Whitney-Wilcoxon test. Two-tailed P values were calculated.
All mouse experiments were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of California San Francisco.
RESULTS
The HMG CoA lyase HCL1 was identified in a genetic screen for lysis-defective mutants.
Wild-type H. capsulatum is able to replicate within the macrophage phagosome, ultimately triggering host cell lysis. We performed a genetic screen to identify H. capsulatum genes that are important for intracellular pathogenesis. A library of 14,000 H. capsulatum insertion mutants generated by Agrobacterium-mediated transformation (15) was screened to identify those mutants with defects in lysis of primary murine bone marrow-derived macrophages (BMDMs) and the macrophage-like cell line, J774. One of the mutants, FE6-C3, was unable to lyse both BMDMs and J774 cells and was subjected to further study.
To determine which H. capsulatum gene was disrupted in the FE6-C3 insertion mutant, we isolated genomic DNA from the mutant strain and performed inverse PCR from the left and right borders of the Agrobacterium T-DNA insertion. Sequence analysis of the resulting PCR products indicated that the H. capsulatum gene “HISTO_DA.Contig93.Fgenesh_histo.97.final_new” contained an insertion in the ORF, approximately 300 bp downstream from the predicted translational start site (Fig. 1A). A BLAST search of the corresponding protein sequence against the nr protein database revealed strong similarity to the HMG CoA lyase enzyme from multiple organisms (Fig. 2). We named the corresponding gene HCL1 (HMG CoA Lyase 1) and hereafter refer to mutant FE6-C3 as the hcl1 mutant. Quantitative reverse transcription-PCR (qRT-PCR) and Northern blot analysis of fungal cells grown in vitro confirmed the loss of HCL1 expression in the hcl1 mutant and the restoration of HCL1 expression when the wild-type gene was reintroduced into the insertion mutant (Fig. 1B and C).
Fig 1.

The FE6-C3 mutant contains an insertion in the HCL1 ORF. (A) Schematic of the HCL1 genomic locus showing the site of the TDNA insertion in the hcl1 mutant, the region of the HCL1 locus that was used to generate the complementation construct, and the region of the HCL1 gene that was used to probe transcript levels. Tick marks are 1 kb apart. (B and C) Total RNA was isolated from wild-type (WT), the hcl1 mutant, and the complemented strain (hcl1+HCL1) grown in broth culture at 37°C. HCL1 mRNA levels were monitored by quantitative RT-PCR (B) and Northern blot analysis (C). Primers for qRT-PCR analysis are described in Materials and Methods and located in the coding sequence of HCL1.
Fig 2.

Key catalytic residues are conserved in the H. capsulatum HMG CoA lyase homolog Hcl1. CLUSTAL W alignment of H. capsulatum Hcl1 with HMG CoA lyases from bacterial and mammalian species. Conserved catalytic residues are marked in red. The asterisk symbol indicates that the residues in that column are identical in all sequences in the alignment, the colon indicates that conserved substitutions between the sequences are observed, and the period indicates that semiconserved substitutions are observed.
HMG CoA lyase enzymes have been studied in several organisms, including humans. Biochemical characterization of the human HMG CoA lyase, HMGCL, has identified several active-site residues that are critical for enzymatic activity, including Arg-41, Asp-42, Glu-72, His-233, and Cys-266 (16–19). Using CLUSTAL W, we aligned the H. capsulatum Hcl1 sequence with the uncharacterized HMG CoA lyase for the fungus Aspergillus fumigatus (which shares 80% amino acid sequence identity with H. capsulatum Hcl1), as well as the functionally characterized enzyme from Pseudomonas aeruginosa (47%), Pseudomonas mevalonii (46%), Bacillus subtilis (36%), Brucella melitensis (42%), and Homo sapiens (44%) (numbers in parentheses indicate shared sequence identity with H. capsulatum Hcl1). We found that these catalytic active-site residues were conserved in all examined HMG CoA lyases, including H. capsulatum Hcl1 (Fig. 2).
The hcl1 mutant has severe lysis and intracellular growth defects in macrophages.
To quantify the severity of the lysis defect of the hcl1 insertion mutant, we infected BMDMs with wild-type H. capsulatum (WT), the hcl1 insertion mutant (hcl1), or the complemented strain (hcl1 +HCL1), where the wild-type HCL1 gene was reintroduced into the mutant strain. (The precise genotypes of these strains are described in Materials and Methods.) We then monitored the kinetics of host cell lysis by measuring release of the cytosolic enzyme lactate dehydrogenase (LDH) from host macrophages into the supernatant at the indicated time points after infection (Fig. 3A). Whereas a considerable amount of LDH was present in the culture supernatant of BMDMs infected with wild-type H. capsulatum and the complemented strain, no LDH release was observed in supernatants from macrophages infected with the hcl1 mutant.
Fig 3.

Hcl1 is required for macrophage colonization and lysis during H. capsulatum infection. (A) BMDMs were mock infected or infected with either wild-type (WT), the hcl1 mutant, or the complemented strain (hcl1+HCL1) at an MOI of 2. At 2, 24, 48, 72, 96, and 120 hpi, supernatants were removed from the infected monolayers and lactate dehydrogenase activity was assessed to monitor BMDM lysis. The average percent BMDM lysis of four measurements ± the standard deviation (from one representative experiment) is shown. (B) BMDMs were infected with wild-type (WT), the hcl1 mutant, or the complemented strain (hcl1+HCL1) at an MOI of 2. At 3, 12, 24, 36, 48, 72, and 144 hpi, BMDMs were osmotically lysed, and the lysates were plated for H. capsulatum CFU. Each measurement is the average of four platings (duplicate infections/duplicate platings) ± the standard deviation from one representative experiment. In some cases, the standard deviation is negligible and is obscured by strain symbols. (C) BMDMs were infected with either wild-type H. capsulatum or the hcl1 mutant at an MOI of 0.1. At 2 and 120 hpi, the infected monolayers were fixed and H. capsulatum yeast cells (arrowhead) were stained with periodic acid-Schiff (PAS) reagent. Macrophage nuclei were counterstained with methyl green. Representative images at ×100 magnification are shown. (D) In vitro growth of wild-type (WT), the hcl1 mutant, and the complemented strain (hcl1+HCL1) was examined in broth culture. Yeast cells were inoculated into HMM at a starting OD600 of 0.01 and subsequent OD600 was monitored over time. The average of three measurements ± the standard deviation is shown from one representative experiment. In some cases, the standard deviation is negligible and is obscured by strain symbols.
To further explore the role that HCL1 plays in H. capsulatum pathogenesis, we examined the intracellular growth of the hcl1 mutant during macrophage infection. BMDMs were infected with wild-type H. capsulatum, the hcl1 mutant, or the complemented strain. At various time points postinfection, intracellular yeasts were released from BMDMs by osmotic lysis and CFU were enumerated. Wild-type H. capsulatum and the complemented strain showed a 16-fold increase in the intracellular CFU within 48 h of infection, after which host cell lysis ensued. In contrast, the hcl1 mutant only grew a modest 4- to 5-fold over the entire 5-day infection, which was indicative of a strong intracellular growth defect (Fig. 3B). Microscopic examination of infected macrophages corroborated the growth defect observed by CFU enumeration. In these experiments, BMDMs were infected at a very low MOI to maximize the period of intracellular growth before wild-type cells lysed the macrophages. Macrophage monolayers were stained with periodic acid-Schiff (PAS) reagent at 2 h and 5 days postinfection to reveal intracellular H. capsulatum yeasts (Fig. 3C). At 5 days postinfection, macrophages infected with the hcl1 mutant contained significantly fewer yeast cells than macrophages infected with wild-type H. capsulatum. Notably, the growth kinetics of the hcl1 mutant were indistinguishable from that of wild-type H. capsulatum when it was cultured in vitro in standard medium (Fig. 3D).
HCL1 is required for growth when leucine is the major carbon source.
HMG-CoA lyases are metalloenzymes that catalyze the final step in the breakdown of leucine, cleaving HMG-CoA into acetyl-CoA and acetoacetate. Since fungi can then assimilate acetyl-CoA into carbohydrates through the glyoxylate cycle, leucine can be utilized as the sole carbon source for growth. Mutants that lack HMG-CoA lyase in several bacterial and fungal species, including Pseudomonas aeruginosa and Aspergillus nidulans, are unable to grow when leucine is the sole carbon source (20, 21). We tested the ability of the hcl1 mutant to utilize leucine by replacing glucose with leucine in minimal media (Fig. 4). The growth of the hcl1 mutant was comparable to that of wild-type H. capsulatum and the complemented strain when it was grown on rich medium (HMM) and on minimal medium (3M) where glucose is the main carbon source (Fig. 4A and B). In minimal medium that lacks glucose (3M-glu), cells of all three genotypes showed limited growth (Fig. 4C). However, only the hcl1 mutant showed severe growth restriction when leucine was substituted for glucose in minimal medium (Fig. 4A and D). Taken together, with the macrophage infection data, these in vitro data suggest that HCL1 could contribute to the ability of H. capsulatum to replicate in the glucose-poor nutritional environment of the phagosome.
Fig 4.

H. capsulatum HCL1 is required for growth on leucine as the major carbon source. (A) Four-day cultures of wild-type H. capsulatum (WT), the hcl1 mutant, and the complemented strain (hcl1+HCL1) were pelleted, washed with PBS, and resuspended in PBS. Tenfold serial dilutions were then made on the indicated medium. In the case of 3M-glu+leu, medium was supplemented with 10 mM leucine. Plates were incubated at 37°C and 5% CO2 for 16 days. For the HMM plates, the 10−3 to 10−6 dilutions are shown (due to the enhanced growth on this rich medium). On all other plates, undiluted, 10−1, and 10−2 dilutions are shown. (B to D) Four-day cultures of wild-type H. capsulatum (WT), the hcl1 mutant, and the complemented strain (hcl1+HCL1) were pelleted, washed with PBS, and resuspended in either minimal media (3M) (B), minimal medium without glucose (3M-glu) (C), or minimal media without glucose, supplemented with 5 mM leucine (3M-glu+leu). At the indicated time points, the OD600 was measured in triplicate to monitor growth. The average OD and standard deviation from one representative experiment are plotted, but the standard deviation is negligible and is obscured by strain symbols.
Hcl1 deficiency results in acidification of H. capsulatum culture medium and the H. capsulatum-containing phagosome.
In humans, HMG CoA lyase deficiency results in the accumulation of upstream acidic intermediates in the leucine catabolic pathway, causing concomitant metabolic acidosis and aciduria (22). Similar effects are observed in fungi, where acidic intermediates accumulate in culture supernatants of cells that lack HMG CoA lyase (21). We were particularly interested in this phenotype, since inhibition of phagosome acidification is thought to be vital for the survival of H. capsulatum within macrophages. Unlike many microbes, wild-type H. capsulatum is able to neutralize the pH of culture medium (23). We predicted that the hcl1 mutant would be able to neutralize acidic medium in vitro but might be unable to maintain neutral pH due to the accumulation of acidic species. To test this hypothesis, we transferred the wild-type, hcl1 mutant, and complemented strains into unbuffered, acidic medium (pH 4.5). We then allowed the cells to grow and monitored the pH over time (Fig. 5). Within 24 h, all three strains had neutralized the culture medium to pH 7, indicating that Hcl1 is not required for the establishment of neutral pH. Wild-type and complemented strains maintained a near-neutral pH (pH 6.2 to 6.5) for the duration of the experiment. However, the pH of the culture medium of the hcl1 mutant dropped to 5.6 after 120 h. The three strains displayed identical growth kinetics during this experiment, as determined by monitoring the optical density (data not shown). These data indicate that Hcl1 is required for maintenance of a neutral culture environment by H. capsulatum.
Fig 5.

Hcl1 is required for maintenance of neutral pH in culture. Four-day cultures of wild-type H. capsulatum (WT), the hcl1 mutant, and the complemented strain (hcl1+HCL1) were pelleted, washed, resuspended in PBS, and diluted into 30 ml of unbuffered pH-HMM at OD600 of 2.0. The pH of the culture medium was measured at the indicated time points. The average of three pH measurements ± the standard deviation is shown from one representative experiment. In most cases, the standard deviation is negligible and is obscured by strain symbols.
Wild-type H. capsulatum is known to inhibit phagolysosome acidification during infection of macrophages (4, 5). Acidification of the phagolysosome is thought to be important to activate lysosomal hydrolases, which have potent degradative and antimicrobial activities. Since the hcl1 mutant acidifies its surroundings, we hypothesized that the macrophage phagolysosome containing the mutant cells might also acidify with time. To monitor acidification of the phagolysosome during infection, we used the acidotropic dye LysoSensor Green DND-189, which has a pKa of 5.2 and accumulates within acidic compartments. We first determined that mid-logarithmic cells from the wild-type, hcl1 mutant, and complemented strains did not stain with LysoSensor Green (data not shown). We then infected BMDMs with fixed wild-type cells or live, mid-logarithmic wild-type, hcl1 mutant, or complemented cells and assessed acidification of the phagosome at 3, 6, 12, and 24 hpi. At each time point, the macrophages were pretreated for 30 min with LysoSensor Green and then washed, fixed, permeabilized, and subjected to confocal immunofluorescence microscopy to detect internalized H. capsulatum, the lysosomal protein LAMP-1, and LysoSensor Green accumulation (Fig. 6A). By 3 hpi, we observed that virtually all internalized H. capsulatum cells (in all samples) localized to a LAMP1-positive compartment. In the case of fixed wild-type cells, 75% colocalized with LysoSensor Green by 3 hpi (Fig. 6B), indicating that they were present in an acidic compartment. By 6 hpi, nearly 100% of fixed wild-type cells colocalized with LysoSensor Green, indicating that, as expected, fixed H. capsulatum cells could not counter the ability of macrophages to acidify the phagolysosome. (At subsequent time points, fixed wild-type cells were degraded by macrophages and could no longer be assessed.) In contrast to fixed wild-type H. capsulatum cells, only 25% of live wild-type H. capsulatum cells colocalized with LysoSensor Green at 3 and 6 hpi. By 12 and 24 hpi, this number dropped to ca. 10%. Notably, 60% of the hcl1 mutant cells, but not the complemented cells, colocalized with LysoSensor Green by 3 hpi, and upward of 85 to 90% of the mutant cells colocalized with LysoSensor Green in subsequent time points. LysoSensor Green appeared to accumulate within and around the hcl1 mutant cells. These data indicate that the hcl1 mutant cells were present in an acidified environment within the macrophage, whereas wild-type cells were not. Since the hcl1 mutant can accumulate acidic species in culture (Fig. 5), we suspected that acidification of phagosomes containing the hcl1 mutant was driven by the mutant itself rather than by the host vacuolar ATPase, which promotes phagosome acidification. Treatment of macrophages with bafilomycin A1, an inhibitor of the host vacuolar ATPase, abrogated acidification of the phagosomes containing fixed H. capsulatum cells but only had a mild effect on the percentage of hcl1 mutant cells that colocalized with LysoSensor Green (Fig. 6B). Similarly, the ura5 mutant parental strain lacking a complementing URA5 gene did not grow in macrophages (data not shown) and accumulated in acidic phagosomes over time, but, unlike phagosomes containing the hcl1 mutant, phagosome acidification was inhibited by treatment with bafilomycin (Fig. 6C). These data indicate that acidification of the vacuole containing hcl1 mutant cells was not dependent on the host vacuolar ATPase and strongly suggest that the hcl1 mutant itself was promoting vacuolar acidification.
Fig 6.
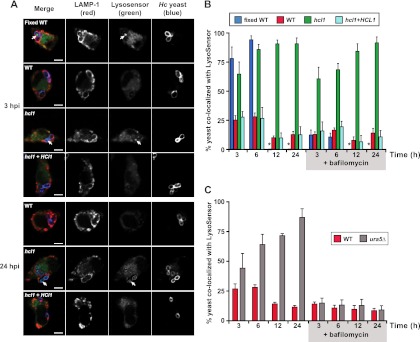
Hcl1 is required for maintenance of near-neutral pH in the macrophage phagosome. (A and B) BMDMs were infected with either wild-type (WT), fixed wild-type cells, the hcl1 mutant, or the complemented strain (hcl1+HCL1) at an MOI of 2. BMDMs were incubated with LysoSensor Green DND-189 30 min prior to fixation at 3, 6, 12, and 24 hpi. Control experiments were performed in the presence of 50 nM bafilomycin A1. (A) At 3 hpi, the acidic pH of the phagosomes was revealed by bright green fluorescence (LysoSensor; white arrows), which surrounded and impregnated the yeast cells (blue). LAMP-1 staining is shown in red. hcl1 mutant cells, but not wild-type cells, maintained localization with LysoSensor at later time points such as 24 hpi. Scale bars, 5 μm. (B) Quantification of the percentage of yeast cells that colocalized with LysoSensor fluorescence within LAMP-1-positive phagosomes. Fixed wild-type cells were degraded by the macrophage after 6 hpi and thus could not be quantified after that time point. Their absence is indicated by an asterisk. At least 200 yeast cells were counted per sample and standard deviation is shown. (C) BMDMs were infected with either the wild-type strain (WT) or the WU15 G217B ura5Δ strain at an MOI of 2 in the absence or presence of bafilomycin as described above. Quantification of the percentage of yeast cells that colocalized with LysoSensor fluorescence within LAMP-1-positive phagosomes was performed. At least 200 yeast cells were counted per sample and the standard deviation is shown.
HCL1 is required for maximal virulence in the mouse model of histoplasmosis.
To determine whether HCL1 is required for virulence of H. capsulatum in the mouse model of histoplasmosis, we infected female C57BL/6 mice intranasally with a lethal dose (1.25 × 106 yeast cells/mouse) of wild-type H. capsulatum, the hcl1 mutant, or the complemented strain (Fig. 7A). Mice were monitored daily for symptoms of disease, including weight loss, panting, and lack of grooming. Mice infected with the wild-type or complemented strains became symptomatic by day 3, and the majority succumbed to infection between days 7 and 8. In contrast, mice infected with the hcl1 mutant strain started to show mild symptoms at day 7, and the majority of the mice succumbed to infection by days 10 and 11. The overall median survival times for each group were 7.5 days (wild type), 7 days (complemented strain), and 10 days (hcl1 mutant), yielding a statistically significant difference in virulence for the hcl1 mutant compared to wild-type (P < 0.0001). CFU were enumerated from the lungs (Fig. 7B) and spleens (Fig. 7C) of mice infected with the wild-type, hcl1 mutant, and complemented strains at day 7. Surprisingly, even though the hcl1-infected mice were only mildly symptomatic, their pulmonary fungal burden was not significantly different from mice infected with the wild-type strain at day 7 (P = 0.4). At day 10, when mice infected with the hcl1 mutant were succumbing to infection, the fungal burden was not significantly different from the fungal burden in wild-type mice at time of death (day 7, P = 0.5), or hcl1-infected mice at day 7 (P = 0.4). These data confirmed that the hcl1 mutant was able to grow in vivo despite its severe restriction within BMDMs in vitro and suggested that some other aspect of infection, such as host response, might contribute to the decreased virulence of the hcl1 mutant. In sum, these data highlight that Hcl1 function is required for full virulence in the mouse model of H. capsulatum infection.
Fig 7.
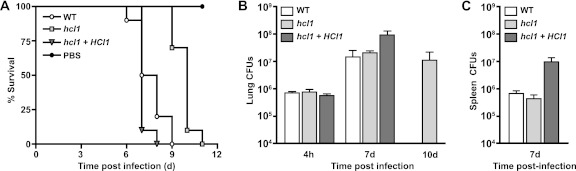
The hcl1 mutant exhibits decreased virulence in vivo. Female C57BL/6 mice were infected intranasally with 1.25 × 106 CFU of either the wild-type (WT) (n = 15), the hcl1 mutant (hcl1) (n = 15), or the complemented strain (hcl1+HCL1) (n = 15) strain. (A) Kaplan-Meir plots/survival curve. P < 0.0001 (log rank test, WT versus hcl1). (B) In the case of all three cohorts of mice, lungs were harvested from infected mice and lung homogenates were plated to enumerate H. capsulatum CFU at 4 hpi and at 7 days postinfection, which was time of death for mice infected with the wild-type strain. Lung CFU were also enumerated at time of death (10 days postinfection) for mice infected with the hcl1 mutant. Average CFU/group is plotted. (C) Spleen CFU were enumerated from all three cohorts of mice at 7 days postinfection. The average CFU/group is plotted.
DISCUSSION
H. capsulatum is an intracellular fungal pathogen that colonizes the macrophage phagolysosome, ultimately triggering host cell lysis. Here, we used a forward genetic approach to identify an enzyme, HMG CoA lyase, which is required for growth within macrophages and host cell lysis. Interestingly, due to the metabolic defects that ensue in response to HMG CoA lyase deficiency, it is indirectly required for maintenance of phagolysosomal pH. Studies in other organisms have shown that HMG CoA lyase catalyzes the final step in leucine catabolism; thus, HMG CoA lyase deficiency results in the inability of microbes to grow when leucine is the sole carbon source. In addition, both microbes and humans with HMG CoA lyase deficiency accumulate acidic species that arise due to the block in leucine metabolism. In the case of H. capsulatum, we observed that Hcl1 is dispensable for growth in standard laboratory medium supplemented with glucose but required for growth on minimal medium when leucine is substituted for glucose as the major carbon source. In addition, growth of H. capsulatum in unbuffered medium revealed that Hcl1 is required to prevent the accumulation of acidic species in the culture supernatant. During colonization of the phagolysosome of macrophages, the hcl1 mutant is present in an acidified compartment, suggesting that these acidic species can lower the pH of the phagolysosome, which, in turn, could potentially activate lysosomal hydrolases and restrict microbial growth.
These studies highlight the challenges faced by intracellular microbes that grow within the phagosome of host cells (reviewed in reference 1). The initiation of phagocytosis of microbes by macrophages is normally accompanied by a superoxide burst catalyzed by the NADPH complex. Subsequently, the normal course of phagosome maturation involves transient interactions between the phagosome and a variety of intracellular organelles, ultimately culminating in fusion with lysosomes. As this maturation process occurs, phagosomal pH drops and the phagosome acquires lysosomal hydrolases that are active at acidic pH. H. capsulatum counters normal phagosome function and maturation in at least two key ways. First, H. capsulatum fails to trigger a superoxide burst in resting macrophages (3) and produces an extracellular superoxide dismutase for protection against the reactive oxygen species generated upon phagocytosis by activated macrophages and polymorphonuclear leukocytes (24). Second, although the H. capsulatum phagosome undergoes fusion with the lysosome (in murine macrophages), the fungus uses unknown means to block acidification of phagolysosomes (4, 5). It has been assumed that the ability of H. capsulatum to inhibit phagolysosome acidification is critical for survival of the fungus within host cells. Our data demonstrate that the hcl1 mutant is competent to neutralize acidic pH in vitro but cannot maintain neutral pH as it grows, presumably due to the production of acidic species. Correspondingly, in the macrophage we observed that the hcl1 mutant, unlike wild-type cells, is present in an acidified phagosome, suggesting that the phagosome containing the mutant cells may be more hydrolytically competent than the phagosome containing wild-type H. capsulatum. Consistent with this model, the hcl1 mutant is clearly restricted for growth within macrophages. We hoped to determine whether neutralization of the phagosome with the weak base chloroquine (7) could rescue the intracellular growth defect of the hcl1 mutant; if so, these results would strongly suggest that the intracellular growth deficiency of the mutant is due to acidification of the phagosome. However, these experiments were not possible in BMDMs for technical reasons (data not shown).
The inability of the hcl1 mutant to grow within macrophages may also reflect the nutritional environment of the phagosome. Studies of other phagosomal pathogens such as Leishmania major and Mycobacterium tuberculosis have shown that enzymes required for gluconeogenesis or the glyoxylate shunt, respectively, are required for colonization or persistence in macrophages and mice, suggesting that the phagosome is a glucose-poor environment (25, 26). Similarly, the fungal pathogen Candida albicans upregulates its expression of isocitrate lyase, a key glyoxylate cycle enzyme, after phagocytosis by host cells and requires this enzyme for full virulence in the mouse model of pathogenesis (27). HMG CoA lyase in other organisms is required for growth when leucine is the sole carbon source, since HMG CoA lyase is a key enzyme in leucine catabolism. A similar result was observed for H. capsulatum, since the hcl1 mutant could not grow in vitro when leucine was substituted for glucose. The levels of leucine or other amino acids in the Histoplasma-containing phagolysosome is unknown, although the inability of the hcl1 mutant to grow within macrophages leads us to speculate that the phagolysosome is a glucose-poor, leucine-replete environment. Interestingly, leucine catabolism generates acetyl-CoA, which can be used as a carbon source when assimilated through the glyoxylate cycle (bypassing the catabolic steps of the standard tricarboxylic acid cycle). Thus, the requirement for Hcl1 in macrophage colonization and virulence may correlate with its link to carbohydrate metabolism.
Whether Hcl1 affects other metabolic processes in the H. capsulatum cell is unknown. In vertebrates, HMG CoA lyase plays additional roles in energy transfer within and between cells. For example, HMG CoA lyase is required to link fatty acid oxidation in the mitochondria to ketogenesis, which is a critical mode of energy transport in higher eukaryotes (28). Fatty acid oxidation produces acetoacetyl CoA, which is then converted into HMG CoA by mitochondrial HMG CoA synthase. HMG CoA lyase then converts HMG CoA into acetoacetate, which is a “ketone body” that is produced in the liver and used to transport energy to other organs when glucose is not available. However, lower eukaryotes lack mitochondrial HMG synthase, and thus there is no known direct link between fatty acid oxidation and HMG CoA lyase-dependent metabolism in fungi.
Despite the profound growth defect of the hcl1 mutant in BMDMs and the J774 macrophage-like cell line, the mutant was able to replicate in the mouse model of pathogenesis. However, mice infected with the hcl1 mutant took significantly longer to succumb to infection than wild-type mice, indicating that the mutant is partially attenuated for virulence. Interestingly, the fungal burden in the lungs of mice infected with the wild-type strain at time of death (day 7, Fig. 7B) was indistinguishable from the fungal burden in the lungs of mice infected with the hcl1 mutant at the same day postinfection (Fig. 7B), but mice in the hcl1-infected cohort did not succumb to infection for another 3 days (Fig. 7A). In addition, there was no significant increase in CFU between day 7 and day 10 for mice infected with the hcl1 mutant (Fig. 7B). Finally, fungal burden in the spleens of mice infected with either wild-type H. capsulatum or the hcl1 mutant at day 8 was not significantly different (P = 0.4, Fig. 7C), indicating that the mutant does not have a gross dissemination defect. These data evoke the hypothesis that an aspect of infection other than fungal burden, such as the host inflammatory response, could differ in mice infected with the wild-type versus mutant strains. Alternatively, it could be the case that in vivo growth kinetics of the wild-type and mutant strains differ at earlier time points in infection not examined in this study, which could affect subsequent symptomatology and disease progression. In addition, these data suggest that despite its profound growth defect in BMDMs, the hcl1 mutant replicates well within an unknown cell type in vivo, such as alveolar macrophages and/or inflammatory monocytes. Future studies of the role in Hcl1 in fungal pathogenesis will continue to utilize the mutant as an informative tool to dissect the nutritional and metabolic requirements of life in the Histoplasma phagosome, as well as the relationship between intracellular growth and immune response during infection of the host.
ACKNOWLEDGMENTS
We thank L. Joseph Wheat and Patti Connolly for the gracious gift of the anti-Histoplasma antibody. We are grateful to Daniel Portnoy, Denise Monack, Jeffery Cox, Alexander Johnson, and members of the Sil laboratory for useful discussion as this study progressed. We are indebted to Davina Hocking Murray for assistance with figure preparation. We thank Davina Hocking Murray and Johnny Tse for technical assistance. We thank Sinem Beyhan, Sarah Gilmore, and Mark Voorhies for comments on the manuscript.
This study was supported by Microbial Pathogenesis and Host Defense Training Grant (NIH T32 A1060537) support to D.T.I., a UCSF NIGMS fellowship to D.T.I. (1R25GM56847), an NSF Graduate Research Fellowship to A.C., an A. P. Giannini Medical Foundation Fellowship to N.V.P., NIH grants (R01AI066224, PO1AI063302, and RO1AI093640), and an HHMI Early Career Scientist Award (http://www.hhmi.org/research/ecs/) to A.S.
Footnotes
Published ahead of print 26 November 2012
REFERENCES
- 1. Russell DG. 2011. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol. Rev. 240:252–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cano MV, Hajjeh RA. 2001. The epidemiology of histoplasmosis: a review. Semin. Respir. Infect. 16:109–118 [DOI] [PubMed] [Google Scholar]
- 3. Wolf JE, Kerchberger V, Kobayashi GS, Little JR. 1987. Modulation of the macrophage oxidative burst by Histoplasma capsulatum. J. Immunol. 138:582–586 [PubMed] [Google Scholar]
- 4. Eissenberg LG, Goldman WE, Schlesinger PH. 1993. Histoplasma capsulatum modulates the acidification of phagolysosomes. J. Exp. Med. 177:1605–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Strasser JE, Newman SL, Ciraolo GM, Morris RE, Howell ML, Dean GE. 1999. Regulation of the macrophage vacuolar ATPase and phagosome-lysosome fusion by Histoplasma capsulatum. J. Immunol. 162:6148–6154 [PubMed] [Google Scholar]
- 6. Lane TE, Wu-Hsieh BA, Howard DH. 1991. Iron limitation and the gamma interferon-mediated antihistoplasma state of murine macrophages. Infect. Immun. 59:2274–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Newman SL, Gootee L, Brunner G, Deepe GS., Jr 1994. Chloroquine induces human macrophage killing of Histoplasma capsulatum by limiting the availability of intracellular iron and is therapeutic in a murine model of histoplasmosis. J. Clin. Invest. 93:1422–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Worsham PL, Goldman WE. 1988. Quantitative plating of Histoplasma capsulatum without addition of conditioned medium or siderophores. J. Med. Vet. Mycol. 26:137–143 [PubMed] [Google Scholar]
- 9. Nguyen VQ, Sil A. 2008. Temperature-induced switch to the pathogenic yeast form of Histoplasma capsulatum requires Ryp1, a conserved transcriptional regulator. Proc. Natl. Acad. Sci. U. S. A. 105:4880–4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hwang L, Hocking-Murray D, Bahrami AK, Andersson M, Rine J, Sil A. 2003. Identifying phase-specific genes in the fungal pathogen Histoplasma capsulatum using a genomic shotgun microarray. Mol. Biol. Cell 14:2314–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hwang LH, Mayfield JA, Rine J, Sil A. 2008. Histoplasma requires SID1, a member of an iron-regulated siderophore gene cluster, for host colonization. PLoS Pathog. 4:e1000044 doi:10.1371/journal.ppat.1000044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Webster RH, Sil A. 2008. Conserved factors Ryp2 and Ryp3 control cell morphology and infectious spore formation in the fungal pathogen Histoplasma capsulatum. Proc. Natl. Acad. Sci. U. S. A. 105:14573–14578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. CLUSTAL W and CLUSTAL X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 15. Sullivan TD, Rooney PJ, Klein BS. 2002. Agrobacterium tumefaciens integrates transfer DNA into single chromosomal sites of dimorphic fungi and yields homokaryotic progeny from multinucleate yeast. Eukaryot. Cell 1:895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roberts JR, Mitchell GA, Miziorko HM. 1996. Modeling of a mutation responsible for human 3-hydroxy-3-methylglutaryl-CoA lyase deficiency implicates histidine 233 as an active site residue. J. Biol. Chem. 271:24604–24609 [DOI] [PubMed] [Google Scholar]
- 17. Roberts JR, Narasimhan C, Miziorko HM. 1995. Evaluation of cysteine 266 of human 3-hydroxy-3-methylglutaryl-CoA lyase as a catalytic residue. J. Biol. Chem. 270:17311–17316 [DOI] [PubMed] [Google Scholar]
- 18. Tuinstra RL, Miziorko HM. 2003. Investigation of conserved acidic residues in 3-hydroxy-3-methylglutaryl-CoA lyase: implications for human disease and for functional roles in a family of related proteins. J. Biol. Chem. 278:37092–37098 [DOI] [PubMed] [Google Scholar]
- 19. Tuinstra RL, Wang CZ, Mitchell GA, Miziorko HM. 2004. Evaluation of 3-hydroxy-3-methylglutaryl-coenzyme A lyase arginine-41 as a catalytic residue: use of acetyldithio-coenzyme A to monitor product enolization. Biochemistry 43:5287–5295 [DOI] [PubMed] [Google Scholar]
- 20. Chavez-Aviles M, Diaz-Perez AL, Reyes-de la Cruz H, Campos-Garcia J. 2009. The Pseudomonas aeruginosa liuE gene encodes the 3-hydroxy-3-methylglutaryl coenzyme A lyase, involved in leucine and acyclic terpene catabolism. FEMS Microbiol. Lett. 296:117–123 [DOI] [PubMed] [Google Scholar]
- 21. Rodriguez JM, Ruiz-Sala P, Ugarte M, Penalva MA. 2004. Fungal metabolic model for type I 3-methylglutaconic aciduria. J. Biol. Chem. 279:32385–32392 [DOI] [PubMed] [Google Scholar]
- 22. Pie J, Lopez-Vinas E, Puisac B, Menao S, Pie A, Casale C, Ramos FJ, Hegardt FG, Gomez-Puertas P, Casals N. 2007. Molecular genetics of HMG-CoA lyase deficiency. Mol. Genet. Metab. 92:198–209 [DOI] [PubMed] [Google Scholar]
- 23. Berliner MD. 1973. Histoplasma capsulatum: effects of pH on the yeast and mycelial phases in vitro. Sabouraudia 11:267–270 [DOI] [PubMed] [Google Scholar]
- 24. Youseff BH, Holbrook ED, Smolnycki KA, Rappleye CA. 2012. Extracellular superoxide dismutase protects histoplasma yeast cells from host-derived oxidative stress. PLoS Pathog. 8:e1002713 doi:10.1371/journal.ppat.1002713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naderer T, Ellis MA, Sernee MF, De Souza DP, Curtis J, Handman E, McConville MJ. 2006. Virulence of Leishmania major in macrophages and mice requires the gluconeogenic enzyme fructose-1,6-bisphosphatase. Proc. Natl. Acad. Sci. U. S. A. 103:5502–5507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr, Russell DG. 2000. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406:735–738 [DOI] [PubMed] [Google Scholar]
- 27. Lorenz MC, Fink GR. 2001. The glyoxylate cycle is required for fungal virulence. Nature 412:83–86 [DOI] [PubMed] [Google Scholar]
- 28. Mitchell GA, Wang SP, Ashmarina L, Robert MF, Bouchard G, Laurin N, Kassovska-Bratinova S, Boukaftane Y. 1998. Inborn errors of ketogenesis. Biochem. Soc. Trans. 26:136–140 [DOI] [PubMed] [Google Scholar]