

Abstract
Quantitative real-time PCR (QRT-PCR) has been widely implemented for clinical viral load testing, but a lack of standardization and relatively poor precision have hindered its usefulness. Digital PCR offers highly precise, direct quantification without requiring a calibration curve. Performance characteristics of real-time PCR were compared to those of droplet digital PCR (ddPCR) for cytomegalovirus (CMV) load testing. Tenfold serial dilutions of the World Health Organization (WHO) and the National Institute of Standards and Technology (NIST) CMV quantitative standards were tested, together with the AcroMetrix CMV tc panel (Life Technologies, Carlsbad, CA) and 50 human plasma specimens. Each method was evaluated using all three standards for quantitative linearity, lower limit of detection (LOD), and accuracy. Quantitative correlation, mean viral load, and variability were compared. Real-time PCR showed somewhat higher sensitivity than ddPCR (LODs, 3 log10 versus 4 log10 copies/ml and IU/ml for NIST and WHO standards, respectively). Both methods showed a high degree of linearity and quantitative correlation for standards (R2 ≥ 0.98 in each of 6 regression models) and clinical samples (R2 = 0.93) across their detectable ranges. For higher concentrations, ddPCR showed less variability than QRT-PCR for the WHO standards and AcroMetrix standards (P < 0.05). QRT-PCR showed less variability and greater sensitivity than did ddPCR in clinical samples. Both digital and real-time PCR provide accurate CMV load data over a wide linear dynamic range. Digital PCR may provide an opportunity to reduce the quantitative variability currently seen using real-time PCR, but methods need to be further optimized to match the sensitivity of real-time PCR.
INTRODUCTION
Over the past several years, viral load testing has evolved from a highly complex and labor-intensive procedure to a routine part of patient care. Such methods are now integral to a diverse range of clinical practice settings and diagnostic and treatment guidelines. These include their implementation in patients with human immunodeficiency virus (HIV) infection or hepatitis and posttransplant monitoring for cytomegalovirus (CMV), Epstein-Barr virus, adenovirus, and BK virus, among others. Quantitative values have been used to follow the efficacy of antiviral therapy and to help determine changes in that therapy. Rising viral burden has been used as a trigger for preemptive treatment, to prevent symptomatic infection. CMV testing is the archetype for the latter application; viral load testing by real-time PCR in particular has changed the epidemiology of CMV disease in transplant patients and is now a standard part of posttransplant care (1–6).
Despite these advances, challenges in viral load testing remain, relate primarily to intrinsic limitations of the current methodology, and center on the issues of accuracy, standardization, and precision (7–10). Quantitative determinations by real-time PCR are indirect, depending on the relationship of the cycle threshold (CT) of a test sample to a calibration curve. The latter, in turn, is typically generated by testing a series of known standards across the linear range of the assay. However, marked variation in assay performance characteristics and in materials used as calibration standards may prevent agreement between different laboratories, even when testing identical material. The use of international standards, such as those that have been made available by the World Health Organization (WHO) (11–13), has helped mitigate this issue but has not resolved the problem. Standards are available only for a few of the most common target analytes. Even when available, they may not be fully commutable and thus may behave differently, depending on the assay system utilized (14, 15).
Perhaps even more problematic is the poor reproducibility often seen among quantitative molecular tests. Within- and between-lab precision, even when using an identical methodology, can be surprisingly poor (7, 8, 16). Variations in any aspect of these complex methods are magnified by the multistep nature of the process and by the reliance on calibration curves, which themselves may vary over time. The very nature of relying on the measure of a dynamic process, such as the rate of target amplification, carries with it intrinsic fluctuations that one could not expect to fully eliminate. This suggests that a more direct method of quantification may be of value.
Droplet digital PCR (ddPCR) is such a direct method (17–22). It relies on limiting partition of the PCR volume, such that a positive result in any of a large number of microreactions (in this case, 20,000) indicates the presence of a single target molecule in a given reaction. The number of positive reactions, together with Poisson's distribution, can then be used to produce a direct, high-confidence measurement of the original target concentration. This methodology removes both the reliance on rate-based measurements (CT values) and the need for the use of calibration curves. Studies targeting low-copy-number genes, typically in the field of molecular oncology, have demonstrated a high degree of sensitivity and precision of digital PCR (dPCR) compared to quantitative real-time PCR (QRT-PCR) (23–26). To date, limited data exist regarding the application of this new technology to viral load testing, but it promises to markedly improve our ability to reproducibly quantify viruses and to obviate developing a costly series of international standards.
Herein, we compared ddPCR to real-time PCR for quantitative detection of CMV in both artificially seeded samples and clinical plasma samples, with the former including recently developed international standard materials.
MATERIALS AND METHODS
Reference materials. (i) WHO international standard for hCMV.
WHO human CMV (hCMV) was purchased from the National Institute for Biological Standards and Control (NIBSC; Potters Bar, Hertfordshire, United Kingdom) and was prepared from whole virus of the hCMV Merlin strain. It was supplied in a lyophilized format containing a total of 6.7 log10 international units (IU; 5 × 106 IU). One vial of the WHO standard was reconstituted with 1.0 ml of nuclease-free water to a concentration of 6.7 log10 IU/ml (5 × 106 IU/ml). Dilutions were made in CMV-negative human plasma to obtain a 10-fold concentration gradient from 1 to 6 log10 IU/ml. For each concentration, 6 aliquots of 200 μl were extracted on a Qiagen EZ1 XL extractor using a Qiagen EZ1 virus minikit (version 2.0) (Qiagen, Inc., Valencia, CA) following the manufacturer's recommended protocol, which includes the addition of an internal control (IC; 7.2 μl). The specimens were eluted in 60 μl, pooled, aliquoted, and stored at −80°C.
(ii) NIST standard reference material.
NIST hCMV was purchased from the National Institute of Standards and Technology (NIST; Gaithersburg, MD) and prepared from a bacterial artificial chromosome of CMV TowneΔ147 containing the genome of the Towne strain of CMV. The standard was supplied in a unit of three component DNA materials, with each component containing 150 μl of purified DNA solution at a different concentration of CMV genome per μl. NIST component C (127 μl, with a given concentration of 19,641 copies/μl [4.29 log10 copies/μl]) was diluted with 373 μl of TE (Tris-EDTA) for a concentration of 6.7 log10 copies/ml (5 × 106 copies/ml). Dilutions were made in TE to obtain a 10-fold concentration gradient from 1 to 6 log10 copies/ml and stored at −80°C. As the NIST was received as purified nucleic acid, it was not extracted prior to amplification.
(iii) AcroMetrix CMV tc panel.
The AcroMetrix CMV tc panel was purchased from AcroMetrix (Life Technologies, Carlsbad, CA) and contains intact, encapsulated viral particles of hCMV strain AD169. The panel consisted of a normal human plasma sample (which tested nonreactive for CMV DNA) and four members over a 10-fold concentration gradient from 2.5 to 5.5 log10 IU/ml (2.9 × 102 to 2.9 × 105 IU/ml) in human plasma. For each concentration, 200 μl was extracted and eluted in 60 μl on the Qiagen EZ1 XL extractor using the Qiagen EZ1 virus minikit (version 2.0; Qiagen, Inc., Valencia, CA) following the manufacturer's recommended protocol. The IC (7.2 μl) was added preextraction. Six aliquots of 200 μl from each concentration were processed, and extracts were pooled, aliquoted, and stored at −80°C until usage.
Patient specimens.
Fifty deidentified human plasma specimens were used in the study. Samples were collected from residual material, originally collected for clinical testing at Emory University Hospital from June through August 2011. Samples were chosen to represent a wide spectrum of CMV concentrations, based on clinical test results. For each clinical specimen, three aliquots of 200 μl were extracted on the Qiagen EZ1 XL extractor using the Qiagen EZ1 virus minikit (version 2.0; Qiagen, Inc., Valencia, CA) following the manufacturer's recommended protocol, which includes the addition of an IC (7.2 μl). The specimens were eluted in 60 μl, pooled, aliquoted, and stored at −80°C. This protocol was approved by the Emory University Institutional Review Board (IRB).
ddPCR.
A laboratory-developed test (LDT) for CMV detection was used, together with a QX100 droplet digital PCR system (Bio-Rad, Pleasanton, CA). The ddPCR reaction mixture consisted of 10 μl of a 2× ddPCR master mix (Bio-Rad), 2 μl of CMV primer/probe mix (artus CMV PCR analyte-specific reagent; Qiagen, Inc., Valencia, CA), and 5 μl of sample nucleic acid solution in a final volume of 20 μl. The entire reaction mixture was loaded into a disposable plastic cartridge (Bio-Rad) together with 70 μl of droplet generation oil (Bio-Rad) and placed in the droplet generator (Bio-Rad). After processing, the droplets generated from each sample were transferred to a 96-well PCR plate (Eppendorf, Germany). PCR amplification was carried out on a T100 thermal cycler (Bio-Rad) using a thermal profile of beginning at 95°C for 10 min, followed by 40 cycles of 94°C for 30 s and 60°C for 60 s, 1 cycle of 98°C for 10 min, and ending at 12°C. After amplification, the plate was loaded on the droplet reader (Bio-Rad) and the droplets from each well of the plate were read automatically at a rate of 32 wells per hour. ddPCR data were analyzed with QuantaSoft analysis software (Bio-Rad), and the quantification of the target molecule was presented as the number of copies per μl of PCR mixture.
Quantitative real-time PCR.
Quantitative CMV PCR testing was performed using an LDT incorporating artus CMV PCR analyte-specific reagents (Qiagen, Inc., Valencia, CA) on a Qiagen Rotor-Gene instrument. For each amplification reaction, 20 μl of purified nucleic acid was added to 30 μl of a reaction mixture of master mix and magnesium. The thermocycler parameters were as follows: hold for 10 min at 95°C, followed by 15 s at 95°C, 30 s at 65°C, and 20 s at 72°C for 45 cycles. A four-point standard curve as well as a positive and a negative control were included on all runs. The laboratory-determined limit of detection (LOD) in this assay using plasma was 2.3 log10 IU/ml.
Statistical analysis.
For each standard's dilution series, the concentration was log transformed as log10(concentration + 1) for purposes of subsequent statistical analyses. Digital PCR measurements were transformed as log10(number of copies/ml + 1), and real-time PCR measurements were transformed as log10(IU/1.38 + 1) for purposes of subsequent statistical analyses. The factor of 1.38 was used in the transformation of real-time PCR measurements to convert IU/ml to the number of copies/ml units reported by digital PCR. One was added prior to log transformation to avoid obtaining undefined values by taking the log of 0 for negative samples. For each technology and standard, the LOD was defined as the smallest nonzero concentration at which all replicates gave a positive qualitative result. For each standard and technology, the log-transformed measurements at or above the LOD were regressed against the log-transformed concentrations by simple least squares. Bartlett's test (27) was used to compare the standard deviations (SDs) of log-transformed results across technologies for each concentration and standard.
For clinical samples, the mean log-transformed measurement was computed for each technology and each clinical sample. Simple least-squares regression was used to evaluate the quantitative agreement of the mean values of the two technologies. For each sample, the number of repeats giving a positive result was determined for each technology. The number of samples with a difference in the number of positive results was determined. A binomial test was used to determine the significance of the number of samples with fewer positive results by digital PCR than by real-time PCR. Similarly, the standard deviation of the results was computed for each sample and technology. A binomial test was used to determine the statistical significance of the number of samples with a smaller standard deviation by real-time PCR than by digital PCR.
RESULTS
Evaluation using quantitative standards.
The three quantitative standards (WHO, NIST, AcroMetrix) were tested as unknowns by both real-time PCR and ddPCR to generate analytical operating characteristics for each method. Each concentration of each standard was run in triplicate on three separate runs (total of nine results per sample). Analysis was performed to determine the limit of detection, quantitative linearity, quantitative agreement compared both to the nominal concentration and to results from real-time PCR, and reproducibility of results using each methodology.
As shown in Table 1, each method had the same LOD when using AcroMetrix standards (2.48 log IU/ml). However, real-time PCR demonstrated a lower LOD by 1-log-unit dilution for both the WHO and NIST standards (10-fold greater sensitivity than ddPCR). dPCR detected 100% of samples at log 3 IU/ml using WHO material and at log 4 copies/ml using NIST material.
Table 1.
Results for standardsa
| Standard | Nominal log10 concnb | Real-time PCR |
Digital PCR |
Variability (P value) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of positive specimens/total no. of attempts | Mean viral load (log10 no. of copies/ml) | SD | % CV | No. of positive specimens/total no. of attempts | Mean viral load (log10 no. of copies/ml) | SD | % CV | |||
| AcroMetrix | 2.48 | 9/9 | 2.11 | 0.34 | 16.11 | 7/7c | 2.23 | 0.24 | 10.76 | 0.409 |
| AcroMetrix | 3.48 | 9/9 | 3.15 | 0.12 | 3.81 | 8/8c | 3.06 | 0.08 | 2.61 | 0.248 |
| AcroMetrix | 4.48 | 9/9 | 4.19 | 0.12 | 2.86 | 8/8c | 4.16 | 0.03 | 0.72 | 0.003 |
| AcroMetrix | 5.48 | 9/9 | 5.25 | 0.07 | 1.33 | 9/9 | 5.19 | 0.03 | 0.58 | 0.048 |
| NIST | 1 | 0/9 | 0 | 0 | NA | 1/9 | 0.27 | 0.82 | 303.7 | NA |
| NIST | 2 | 7/9 | 1.09 | 0.67 | 61.47 | 3/9 | 0.83 | 1.24 | 149.4 | 0.104 |
| NIST | 3 | 9/9 | 2.35 | 0.23 | 9.79 | 7/9 | 2.1 | 1.2 | 57.14 | <0.001 |
| NIST | 4 | 9/9 | 3.29 | 0.09 | 2.74 | 9/9 | 3.66 | 0.1 | 2.73 | 0.637 |
| NIST | 5 | 9/9 | 4.34 | 0.05 | 1.15 | 9/9 | 4.63 | 0.07 | 1.51 | 0.348 |
| NIST | 6 | 9/9 | 5.35 | 0.06 | 1.12 | 9/9 | 5.6 | 0.05 | 0.89 | 0.394 |
| NIST | 6.7 | 9/9 | 6.1 | 0.11 | 1.8 | 9/9 | 6.21 | 0.08 | 1.29 | 0.345 |
| WHO | 1 | 6/9 | 1.17 | 0.91 | 77.78 | 1/7c | 0.29 | 0.76 | 262.07 | 0.660 |
| WHO | 2 | 9/9 | 2.18 | 0.38 | 17.43 | 3/7c | 0.91 | 1.14 | 125.27 | 0.008 |
| WHO | 3 | 9/9 | 3.16 | 0.11 | 3.48 | 9/9 | 2.71 | 0.1 | 3.69 | 0.727 |
| WHO | 4 | 9/9 | 4.14 | 0.11 | 2.66 | 8/8c | 3.77 | 0.04 | 1.06 | 0.007 |
| WHO | 5 | 9/9 | 5.13 | 0.07 | 1.36 | 9/9 | 4.75 | 0.02 | 0.42 | 0.004 |
| WHO | 6 | 9/9 | 5.99 | 0.05 | 0.83 | 8/8c | 5.72 | 0.02 | 0.35 | 0.020 |
The number of positive specimens/number of attempts, the mean log-transformed measurements, and standard deviation of the log-transformed measurements are given for each technology at each concentration of each standard. Bartlett's test was used to compare the variability of the two technologies, based on the SD. A small-variability P value indicates that there is significant evidence that the two technologies do not have equal standard deviation in their measurements. CV, coefficient of variation; NA, not available.
Nominal concentrations in IU/ml for AcroMetrix and WHO standards and number of copies/ml for NIST standards.
Decreased replicates due to failure of the instrument (either failed to generate or read the droplet).
Both methods showed excellent linearity above the LOD. For each technology and standard, the estimated slope coefficients were close to 1 (0.93 to 1.04) and R2 values were very close to 1 (≥0.98), indicating both linearity and correlation between nominal and measured values (Fig. 1). Quantitative correlation was also shown between methods (Fig. 2). Correlation was reduced, particularly for ddPCR, at the low end of each dilution series, corresponding to analyte concentrations with less than 100% detection. Regression model equations for Fig. 1 and 2 are shown in Tables S1 and S2 in the supplemental material, respectively.
Fig 1.

Scatter plots with regression lines between nominal and measured concentrations of each reference material using ddPCR (A) and real-time PCR (B). All values are log10 transformed. Log10 transformation was defined as y = log10(x + 1), as described in Materials and Methods. Table S1 in the supplemental material provides the regression model equations for the fitted lines.
Fig 2.
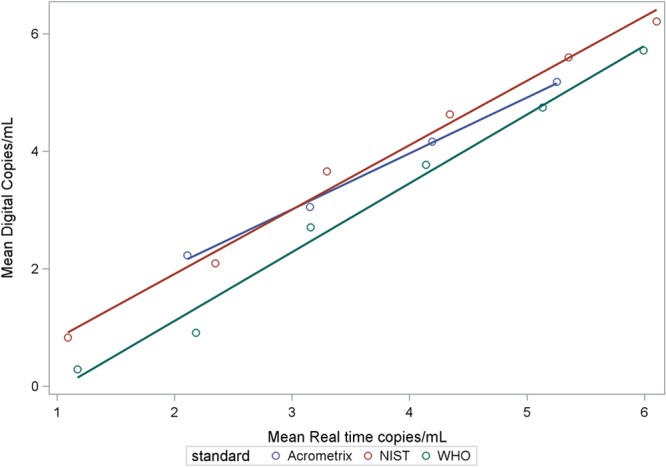
Quantitative correlation of mean values for each concentration of standard between droplet digital PCR and real-time PCR methods. Table S2 in the supplemental material provides the model equations for the fitted lines.
The lower sensitivity of ddPCR may be largely attributed to use of a 4-fold smaller input volume. When we plotted the probability of detection against the nominal total number of input viral copies (number of copies/ml × input volume, in ml), the two technologies showed very similar performance (see Fig. S1 in the supplemental material). This indicated that the sensitivity difference was largely attributable to the input volume differences.
Table 1 shows the standard deviation and mean log10-transformed number of copies for each technology, standard, and concentration. Only matched concentrations of each standard run with both methods are reported. Overall, the methods produced similar results. The maximum differences between mean measurement values of the two technologies for all concentrations tested were 0.12, 0.37, and 1.27 log10 copies/ml for the AcroMetrix, NIST, and WHO standards, respectively. Among concentrations above the LOD for both methods, the maximum differences were 0.12, 0.37, and 0.45, respectively. Digital PCR showed significantly less variability than real-time PCR for AcroMetrix log10 concentrations of ≥4.48 and for WHO log10 concentrations of ≥4. Real-time PCR showed significantly less variability than digital PCR at an NIST log10 concentration of 3 and a WHO log10 concentration of 2. The two technologies did not show significantly different variability for the other concentrations of the standards.
Evaluation using clinical samples.
Clinical samples were each tested in duplicate on three separate PCR runs (total of six results per sample; see Table S3 in the supplemental material). Mean viral load and result variability was compared for each method. Figure 3 shows linear regression and Bland-Altman plots comparing the mean quantitative values for ddPCR with those for real-time PCR, demonstrating close agreement between the two systems (mean difference = −0.247 log10 copies/ml; SD = 0.336 log10 copies/ml).
Fig 3.
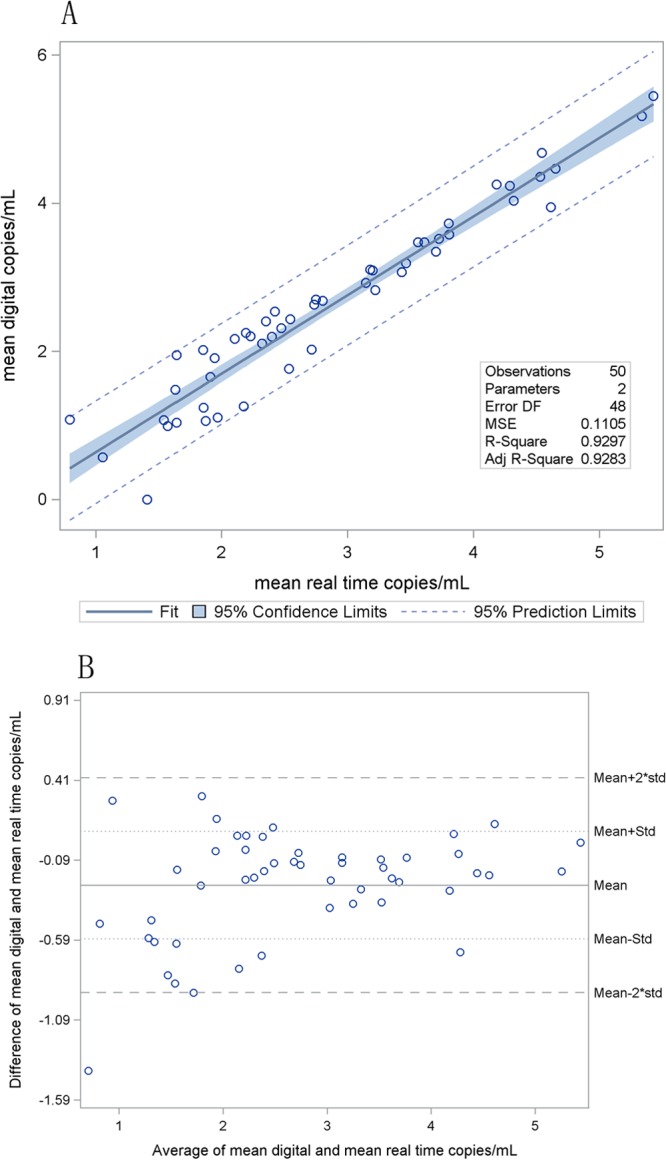
(A) Linear regression for 50 clinical samples (intercept = −0.43, slope = 1.06). The confidence limits are interval estimates for the regression line, and the prediction limits provide intervals that will include approximately 95% of all observations. The mean squared error is the vertical variance of the observed points from the fitted line. R2 is the proportion of variability in y that is attributable to x, and the adjusted R2 adjusts for the number of model parameters. DF, degrees of freedom; MSE, mean squared error; Adj, adjusted. (B) Bland-Altman plots comparing the quantitative values of ddPCR with those of real-time PCR for 50 clinical samples (mean difference = −0.247 log10 copies/ml; SD [Std] = 0.336 log10 copies/ml).
Digital PCR had a significantly lower positivity rate than real-time PCR. Digital PCR obtained fewer positive results among its six repeated replicates than did real-time PCR for 14 samples, but the converse was not true for any samples (P = 0.0001). Additionally, the variability of real-time results was less than that of digital PCR results for 34 of 50 (68%) samples (95% confidence interval = 53% to 81%; P = 0.015).
DISCUSSION
This study offers one of the earliest published assessments of digital PCR as a viable means for quantitative detection of DNA viremia in clinical samples. Previous work with this relatively new technology has focused on its application in molecular oncology (21, 23). However, the utility of a direct (rather than relative) measure of quantification for the field of clinical molecular virology cannot be overstated, particularly if demonstrated to be accurate, sensitive, and precise. Findings here show ddPCR to have such potential for the measurement of CMV. While the current assay showed somewhat lower sensitivity than the real-time methodology, other operating characteristics were comparable. Quantitative accuracy was high, and the assay generated viral loads that matched well with nominal values of international and commercial quantitative standards. Linearity and quantitative correlation with real-time PCR results were high, and precision matched or exceeded that of real-time PCR, when assessed within the analytical measurement range of the test. Furthermore, our data suggest that the lower sensitivity and greater variability of this ddPCR may be attributed to a lower input volume; however, our experiment was not designed to definitively determine the impact of input volume on sensitivity. Another experiment designed explicitly for that purpose may be necessary to confirm our interpretation. Clinical samples showed greater variability with ddPCR than with real-time PCR, while the converse was true for reference materials. Samples with values near the LOD of either method showed greater variability. Because the LOD of ddPCR was somewhat higher than that of real-time PCR, the results of clinical samples disproportionately fell close to the LOD of ddPCR, potentially accounting for the increased variability (since clinical samples tended to have a lower viral load overall, while reference materials had their concentrations evenly distributed along the linear range of the assays).
The disadvantages of real-time PCR for such measurements have been well documented. Several authors have now shown a high degree of quantitative variability among such assays, not only for those targeting CMV but also for those targeting other blood-borne viruses (7–9, 28). Laboratory-developed assays are particularly prone to such variability, and a wide range of factors has been shown to affect both the accuracy and precision of these tests (7). The development of international standards for CMV and other such viruses promises to mitigate such problems (particularly that of accuracy), but numerous issues remain. These include the difficulty of generating widely commutable standards and other aspects of assay design that impinge on amplification efficiency, all integrally related to the generation of reproducible results when measurement depends on relative rates of amplification, as with real-time methods. In addition, gains seen in the development of international standards and automated, FDA-cleared assays are unlikely to be duplicated for the wide range of viruses currently of clinical interest and may be limited only to those with a strong commercial market.
The clinical import of improved assay characteristics is multifold. Present methods for measuring CMV load may show quantitative variability among laboratories exceeding 4 orders of magnitude (7, 8). Quantitative accuracy and precision are crucial to assay interpretation and to the ability to set meaningful, universal treatment thresholds, both for clinical disease attribution and for preemptive therapeutic strategies. In addition, the portability of results can have direct implications for patient care. Currently, ongoing treatment of patients who move from one institution to another may be challenging and require new baseline viral loads. Comparability of clinical study results that rely on such measurements is a final issue of importance fundamental to advancing patient care. While the recent availability of an FDA-cleared, commercial assay may improve the situation for CMV testing, its impact remains uncertain. Digital PCR, utilizing endpoint PCR for direct measurement of viral loads and potentially improving accuracy and precision over those of current, real-time methods, may address all of these issues.
Since it was initially described in the 1990s, other authors have shown digital PCR to have advantages in several early reports of studies focused primarily on research applications in molecular genetics and oncology. Specifically, it has been used for mutational analysis, assessment of allelic imbalance, cancer detection, and allelic expression analysis (17, 21, 23). Its ability to detect and accurately quantify minority mutations in a predominating background of normal sequence has proven advantageous over other PCR methodologies. This capability has also been exploited in the area of prenatal diagnostics, where its use for the detection of fetal aneuploidies and single-gene Mendelian disorders using maternal plasma samples has also been described (29). Few authors, however, have yet investigated the application of this technology to infectious disease diagnostics. Experimental systems have demonstrated proof of principle for quantification of adenoviral genome copies, GB virus in transfected cell culture lines, serial dilutions of hepatitis C virus and HIV RNA, and purified, serially diluted HIV RNA from two clinical samples (19, 30, 31). The latter studies demonstrated a 4-log10-unit linear range and variable agreement with real-time PCR methods. None of these studies examined a commercially available digital PCR system amenable to routine clinical use, and none utilized large numbers of clinical samples in a clinical laboratory setting. This report supports the clinical applicability of ddPCR for routine use in clinical diagnostic molecular virology. The system used here has a relatively small footprint (requiring less than 2 linear feet of bench space for both the droplet generator and reader together). It can be used at roughly the same cost as real-time PCR (as most reagents used here were identical in the two systems) and has scalable, rapid throughput (6.5 h for 96 reactions and 2.2 h for 8 reactions, compared to 4 h for 33 real-time PCRs). Hands-on time for ddPCR was 25 min for 8 reactions and 120 min for 96 reactions, while for real-time PCR it was 85 min for 33 reactions. This now makes digital PCR practical for routine use in clinical laboratories, adding relevance to the results shown above.
The conclusions here are, of course, limited on the basis of the number and genetic heterogeneity of clinical samples available. In addition, the conclusions presented here are drawn on the basis of a comparison using only one assay design targeting only one of a number of viruses for which such a method may have clinical utility. Indeed, ongoing work by our group seeks to extend the present study by looking at other comparator assays and other viruses, particularly those with RNA genomes. In addition, the development and use of other digital platforms will be crucial to building upon this work. The somewhat higher LOD, compared to that of real-time PCR, seen can be accounted for entirely by the small total sample volume compared to that for the real-time system used (5 μl for ddPCR compared to 20 μl for real-time PCR; statistical analysis not shown). An increased reaction volume for ddPCR, currently limited by system design, might also lead to an improved quantitative correlation between methods at low concentrations of analyte and might extend the precision advantage of ddPCR already seen at higher concentrations. Finally, future studies comparing ddPCR to the recently released FDA-approved test for CMV load testing (and others, as they become available) may prove valuable in assessing the future roles for each technology.
The data shown here confirm earlier work in experimental oncology and show proof of principle for the application of ddPCR for routine use in clinical, molecular virology. The advantages of direct quantification by endpoint, limiting-partition PCR could obviate development and purchase of costly quantitative calibrators. This paradigm shift could reduce testing costs and improve the accuracy and precision of quantitative viral load assays. Such improvements will have direct clinical utility, particularly in diagnostic microbiology and virology laboratories serving immunocompromised patients, where the number of viruses of importance continues to outstrip our ability to develop methods for quantitative detection and where our present diagnostic tools remain severely limited in utility due to suboptimal operating characteristics. These findings support both the continued development of this technology and further work to explore its value for routine use in clinical diagnostic testing for infectious pathogens.
Supplementary Material
ACKNOWLEDGMENTS
Artus CMV RG PCR reagents were graciously provided by Qiagen, Inc. (Valencia, CA).
This work was supported in part by the Anderson Charitable Foundation, the American Lebanese Syrian Associated Charities (ALSAC), and the Emory Center for AIDS Research (P30 AI050409).
A. M. Caliendo is a coinvestigator on a CMV load clinical trial that is funded by Qiagen.
Footnotes
Published ahead of print 5 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.02620-12.
REFERENCES
- 1. Barker JN, Hough RE, van Burik JA, DeFor TE, MacMillan ML, O'Brien MR, Wagner JE. 2005. Serious infections after unrelated donor transplantation in 136 children: impact of stem cell source. Biol. Blood Marrow Transplant 11:362–370 [DOI] [PubMed] [Google Scholar]
- 2. Boeckh M, Ljungman P. 2009. How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood 113:5711–5719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ljungman P. 2010. Molecular monitoring of viral infections after hematopoietic stem cell transplantation. Int. J. Hematol. 91:596–601 [DOI] [PubMed] [Google Scholar]
- 4. Ljungman P, Hakki M, Boeckh M. 2011. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol. Oncol. Clin. North Am. 25:151–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mori T, Kato J. 2010. Cytomegalovirus infection/disease after hematopoietic stem cell transplantation. Int. J. Hematol. 91:588–595 [DOI] [PubMed] [Google Scholar]
- 6. Zhang LF, Wang YT, Tian JH, Yang KH, Wang JQ. 2011. Preemptive versus prophylactic protocol to prevent cytomegalovirus infection after renal transplantation: a meta-analysis and systematic review of randomized controlled trials. Transpl. Infect. Dis. 13:622–632 [DOI] [PubMed] [Google Scholar]
- 7. Hayden RT, Yan X, Wick MT, Rodriguez AB, Xiong X, Ginocchio CC, Mitchell MJ, Caliendo AM. 2012. Factors contributing to variability of quantitative viral PCR results in proficiency testing samples: a multivariate analysis. J. Clin. Microbiol. 50:337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pang XL, Fox JD, Fenton JM, Miller GG, Caliendo AM, Preiksaitis JK. 2009. Interlaboratory comparison of cytomegalovirus viral load assays. Am. J. Transplant. 9:258–268 [DOI] [PubMed] [Google Scholar]
- 9. Preiksaitis JK, Pang XL, Fox JD, Fenton JM, Caliendo AM, Miller GG. 2009. Interlaboratory comparison of Epstein-Barr virus viral load assays. Am. J. Transplant. 9:269–279 [DOI] [PubMed] [Google Scholar]
- 10. Wolff DJ, Heaney DL, Neuwald PD, Stellrecht KA, Press RD. 2009. Multi-site PCR-based CMV viral load assessment-assays demonstrate linearity and precision, but lack numeric standardization: a report of the Association for Molecular Pathology. J. Mol. Diagn. 11:87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fryer JFH, Heath AB, Anderson R, Minor PD, the Collaborative Study Group 2010. Collaborative study to evaluate the proposed 1st WHO international standard for human cytomegalovirus (HCMV) for nucleic acid amplification (NAT)-based assays. National Institute for Biological Standards and Control, Potters Bar, United Kingdom [Google Scholar]
- 12. Holden MJ, Madej RM, Minor P, Kalman LV. 2011. Molecular diagnostics: harmonization through reference materials, documentary standards and proficiency testing. Expert Rev. Mol. Diagn. 11:741–755 [DOI] [PubMed] [Google Scholar]
- 13. Madej RM, Davis J, Holden MJ, Kwang S, Labourier E, Schneider GJ. 2010. International standards and reference materials for quantitative molecular infectious disease testing. J. Mol. Diagn. 12:133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caliendo AM, Shahbazian MD, Schaper C, Ingersoll J, Abdul-Ali D, Boonyaratanakornkit J, Pang XL, Fox J, Preiksaitis J, Schonbrunner ER. 2009. A commutable cytomegalovirus calibrator is required to improve the agreement of viral load values between laboratories. Clin. Chem. 55:1701–1710 [DOI] [PubMed] [Google Scholar]
- 15. Vesper HW, Miller WG, Myers GL. 2007. Reference materials and commutability. Clin. Biochem. Rev. 28:139–147 [PMC free article] [PubMed] [Google Scholar]
- 16. Kraft CS, Armstrong WS, Caliendo AM. 2012. Interpreting quantitative cytomegalovirus DNA testing: understanding the laboratory perspective. Clin. Infect. Dis. 54:1793–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhat S, Herrmann J, Armishaw P, Corbisier P, Emslie KR. 2009. Single molecule detection in nanofluidic digital array enables accurate measurement of DNA copy number. Anal. Bioanal. Chem. 394:457–467 [DOI] [PubMed] [Google Scholar]
- 18. Kiss MM, Ortoleva-Donnelly L, Beer NR, Warner J, Bailey CG, Colston BW, Rothberg JM, Link DR, Leamon JH. 2008. High-throughput quantitative polymerase chain reaction in picoliter droplets. Anal. Chem. 80:8975–8981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kreutz JE, Munson T, Huynh T, Shen F, Du W, Ismagilov RF. 2011. Theoretical design and analysis of multivolume digital assays with wide dynamic range validated experimentally with microfluidic digital PCR. Anal. Chem. 83:8158–8168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinheiro LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S, Emslie KR. 2012. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 84:1003–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pohl G, Shih I-M. 2004. Principle and applications of digital PCR. Expert Rev. Mol. Diagn. 4:41–47 [DOI] [PubMed] [Google Scholar]
- 22. Vogelstein B, Kinzler KW. 1999. Digital PCR. Proc. Natl. Acad. Sci. U. S. A. 96:9236–9241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diehl F, Diaz LA., Jr 2007. Digital quantification of mutant DNA in cancer patients. Curr. Opin. Oncol. 19:36–42 [DOI] [PubMed] [Google Scholar]
- 24. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang S, Stumbo DP, Hodges SP, Romine S, Milanovich FP, White HE, Regan JF, Karlin-Neumann GA, Hindson CM, Saxonov S, Colston BW. 2011. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 83:8604–8610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanders R, Huggett JF, Bushell CA, Cowen S, Scott DJ, Foy CA. 2011. Evaluation of digital PCR for absolute DNA quantification. Anal. Chem. 83:6474–6484 [DOI] [PubMed] [Google Scholar]
- 26. Whale AS, Huggett JF, Cowen S, Speirs V, Shaw J, Ellison S, Foy CA, Scott DJ. 2012. Comparison of microfluidic digital PCR and conventional quantitative PCR for measuring copy number variation. Nucleic Acids Res. 40:e82 doi:10.1093/nar/gks203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bartlett MS. 1937. Properties of sufficiency and statistical tests. Proc. R. Soc. Lond. Ser. A 160:268–282 [Google Scholar]
- 28. Hayden RT, Hokanson KM, Pounds SB, Bankowski MJ, Belzer SW, Carr J, Diorio D, Forman MS, Joshi Y, Hillyard D, Hodinka RL, Nikiforova MN, Romain CA, Stevenson J, Valsamakis A, Balfour HH, Jr, U.S. EBV Working Group 2008. Multicenter comparison of different real-time PCR assays for quantitative detection of Epstein-Barr virus. J. Clin. Microbiol. 46:157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zimmermann BG, Grill S, Holzgreve W, Zhong XY, Jackson LG, Hahn S. 2008. Digital PCR: a powerful new tool for noninvasive prenatal diagnosis? Prenat. Diagn. 28:1087–1093 [DOI] [PubMed] [Google Scholar]
- 30. Shen F, Sun B, Kreutz JE, Davydova EK, Du W, Reddy PL, Joseph LJ, Ismagilov RF. 2011. Multiplexed quantification of nucleic acids with large dynamic range using multivolume digital RT-PCR on a rotational SlipChip tested with HIV and hepatitis C viral load. J. Am. Chem. Soc. 133:17705–17712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. White RA, III, Quake SR, Curr K. 2012. Digital PCR provides absolute quantitation of viral load for an occult RNA virus. J. Virol. Methods 179:45–50 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.